
Abstract
Exogenous microglia pass through the blood—brain barrier and migrate to ischemic hippocampal lesions when injected into the circulation. We investigated the effect of exogenous microglia on ischemic CA1 pyramidal neurons. Microglia were isolated from neonatal mixed brain cultures, labeled with the fluorescent dye PKH26, and injected into the subclavian artery of Mongolian gerbils subjected to ischemia reperfusion neuronal injury. PKH26-labeled microglia migrated to the ischemic hippocampal lesion, resulting in increased numbers of surviving neurons compared with control animals, even when injected 24 h after ischemia. Interferon-γ stimulation of isolated microglia enhanced the neuroprotective effect. Administration of exogenous microglia resulted in normal performance in a passive avoidance-learning task. Additionally, administration of exogenous microglia increased the expression of brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in the ischemic hippocampus, and thus might have induced neurotrophin-dependent protective activity in damaged neurons. Peripherally injected microglia exhibited a specific affinity for ischemic brain lesions, and protected against ischemic neuronal injury in vivo. It is possible that administration of exogenous microglia can be developed as a potential candidate therapy for central nervous system repair after transitory global ischemia.
Keywords
Introduction
Microglia arise from the monocyte/macrophage lineage, and are ubiquitously distributed in the central nervous system, representing up to 20% of the total glial cell population in the brain (Lawson et al, 1990). In accordance with del Rio Hortega's early teaching (Hortega, 1932), the current view is that resident microglia are of mesodermal origin, derived from bone marrow precursor cells (Ling and Wong, 1993). These cells invade the central nervous system at an early embryonic stage to give rise to typical process-bearing microglia (Ling and Wong, 1993). Therefore, microglia may have high affinity for the brain.
Many reports describe neuron—microglia interactions after cerebral ischemia and mechanical injury (for review, see Gehrmann et al, 1995). It remains controversial, however, whether activated microglia promote neuronal death or neuronal survival. Many investigators maintain that microglia induce a neurotoxic effect by secreting nitric oxide (Chao et al, 1992) and toxic cytokines (Sawada et al, 1989). Moreover, microglia may contribute to the pathogenesis of neurodegeneration, such as in multiple sclerosis (Diemel et al, 1998), Alzheimer's disease (Barger and Harmon, 1997), and acquired immunodeficiency syndrome-associated dementia (Giulian et al, 1996). Conversely, other studies show that microglia protect neurons in the damaged brain by secreting cytokines, such as interleukin-1β (Giulian et al, 1986), interleukin-6 (Sawada et al, 1995), transforming growth factor-β (Suzumura et al, 1993), basic fibroblast growth factor (Shimojo et al, 1991), hepatocyte growth factor (Hamanoue et al, 1996), nerve growth factor (Mallat et al, 1989), and tumor necrosis factor α (Sawada et al, 1989).
To determine whether microglia are neurotoxic or neuroprotective, they must be analyzed with in vivo models of neuronal injury. The role of microglia may be clarified by injecting isolated microglia at the site of neuronal injury in animal models. Direct injection of microglia into the brain, however, induces undesirable events, such as the entry of blood cells to the site of injury and immunologic responses, which complicate the analysis of the role of microglia.
Our previous studies showed that microglia isolated from rodent mixed-brain cultures retain the ability to enter the normal brain from the circulation (Imai et al, 1997, 1999; Sawada et al, 1998). Hence, microglia can be introduced into the central nervous system by injection into the bloodstream in animal models of neuronal injury. Using this system, the roles of microglia in various pathologic conditions can be analyzed.
The purpose of the present study was to evaluate whether microglia have neurotrophic or neurotoxic effects on in vivo brain ischemia, in which CA1 pyramidal neurons are specifically damaged. Microglia isolated from a primary culture of mixed glial cells from neonatal Mongolian gerbils were injected into the subclavian arteries of host animals subjected to ischemia reperfusion neuronal injury. We examined the number of CA1 pyramidal neurons in the brain at various time points up to 8 days. In another group of animals, we tested the effect of microglia injection on performance in a passive avoidance-learning task. We also investigated changes in the expression of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) in the ischemic hippocampus after microglia injection.
Materials and methods
Animals and Induction of Global Forebrain Ischemia
Adult male Mongolian gerbils (n=300; 7 to 8 weeks old; ≈60 g; Seac Yoshitomi, Ltd, Fukuoka, Japan) were used in the study. Animals were housed individually, with food and water available ad libitum. All procedures were performed in accordance with the Guidelines for Animal Experimentation of the Fujita Health University, School of Medicine.
Ischemia was induced as described by Tone et al (1987). In brief, anesthesia was induced and maintained with a mixture of 2.5% halothane and nitrous oxide/oxygen (1:1). Body temperature was maintained at 37°C with a heating pad. When the spinal reflex was absent, both common carotid arteries were exposed through a ventral cervical incision and occluded with aneurysm clips for 5 mins. The clips were then released to resume normal flow to the forebrain (ISCH). Gerbils operated on without occlusion of the carotid arteries were used as controls (sham ISCH). Postoperative rectal temperature was measured three times daily (at 0700, 1400, and 2200) immediately after the induction of anesthesia with a mixture of 2.5% halothane and nitrous oxide/oxygen (1:1).
Duration of Survival Period
Ischemia was confirmed by examination of Nissl-stained brain sections 7 days after ischemia. Delayed neuronal death was observed in CA1 pyramidal neurons in all animals (n=20). Approximately 2% of the animals did not survive the ischemic episode. All animals that survived were included in the data analysis. There were no differences in survival between groups.
Cell Culture
Microglia were prepared using postnatal day 1 Mongolian gerbils (n=200), as described previously (Suzumura et al, 1984). In brief, the meninges were removed, and the brain was dissociated by passing it through a 320-μm-pore nylon mesh. After washing with Hank's balanced salt solution, the cell suspension was triturated and plated in 75-cm2 culture flasks (Falcon 3024, Becton-Dickinson, Franklin Lakes, NJ, USA) at a density of one brain per flask in 10 mL Eagle's minimal essential medium, supplemented with 10% fetal bovine serum, 5 μg/mL bovine insulin, and 0.2% glucose. Microglia were isolated on the 14th day by the shaking off method (Suzumura et al, 1984). In some experiments, purified microglia were stimulated by incubation with human interferon-γ (IFNγ; Shionogi Co., Osaka, Japan) at a concentration of 5 × 103 U in 10 mL in a plastic dish at 37°C for 24 h.
Macrophages were collected by injecting 10 mL of cold (4°C) phosphate-buffered saline into the peritoneal cavities of a separate group of gerbils. Peritoneal fluid was withdrawn three times with a 21-gauge needle attached to a plastic syringe. The cells were maintained at 4°C, centrifuged at 1,000 g for 5 mins, and seeded onto plastic dishes containing Eagle's minimal essential medium with 10% fetal bovine serum, where they adhered for 1 h at 37°C in 5% CO2.
PKH26 Labeling
PKH staining provides stable, clear, intense, accurate, and reproducible fluorescent labeling of live cells for an extended period of time, with no apparent toxic effects (Horan and Slezak, 1989). PKH is an aliphatic reporter molecule that is inserted into the cell membrane lipid bilayer by selective partitioning. It is effective for a variety of cell types and exhibits no significant leaking or transfer between cells. We used PKH26 to label the exogenous microglia within the brain, using a PKH26 Red Fluorescent Cell Linker Kit for Phagocytic Cell Labeling (PKH26-PCL; Sigma Chemical Co., St Louis, MO, USA). PKH26-PCL labels phagocytic cells with excitation (551 nm) and emission (567 nm) wavelengths compatible with rhodamine or phycoerythrin detection systems. The labeling occurs through the formation of aggregates of particles. The aggregation significantly inhibits the uptake of dye by nonphagocytic cells, such as lymphocytes, but facilitates dye uptake by phagocytic cells. Labeled cells appear patchy or spotted because the dye localizes in phagocytic compartments of the cells. The dye appears to be resistant to metabolic breakdown and labels the cells for at least 21 days in vivo. Use of PKH26 can be combined with immunocytochemistry (Silverman et al, 2000), but is not suitable for double staining, because it is easily abolished by the fixation required for immunocytochemistry.
Labeling of microglia and macrophages with PKH26 was performed using the standard phagocytosis procedure of microscopic particles, according to the manufacturer's instructions. In brief, cells in a 10-cm plastic dish were incubated with PKH26 staining solution containing 10 μmol/L PKH26, 50% Diluent B (a phagocytic cell-labeling solution, Sigma Chemical Co.), and 50% culture medium for 15 mins, then washed three times in 10 mL of serum-containing medium. PKH26-stained cells were harvested using a rubber policeman in 2 mL of ice-cold phosphate-buffered saline (pH 7.2) and washed three times with 5 mL of ice-cold phosphate-buffered saline (pH 7.2) by centrifugation at 1,000 g for 3 mins. The cells were then resuspended in the culture medium for injection into the host.
Injection of Cells
Intraarterial cell injection was performed as described previously (Ishihara et al, 1993). Cells were injected either 24 h before or 24 h after ischemia was induced. In a subset of experiments, microglia were injected 48 h after ischemia induction. In brief, sham ISCH and ISCH gerbils were anesthetized with a mixture of 2.5% halothane and nitrous oxide/oxygen (1:1). When the spinal reflex was absent, a transverse incision was made under the left clavicle. The left subclavian artery was exposed and dissected from the surrounding tissues. After temporary occlusion of the subclavian artery with an aneurysm clip, a small hole was made in the distal side of the artery with a 27-gauge needle, and a polyethylene tube (Becton Dickinson, Tokyo, Japan) was inserted proximally into the hole. After the aneurysm clip was released, PKH26-labeled microglia or macrophages (0.5 mL of final suspension media estimated to contain 1 × 106 cells) were injected as a bolus for 30 secs into the subclavian artery through the polyethylene tube.
Tissue Preparation
Gerbils were deeply anesthetized with ketamine hydrochloride (60 mg/kg, intraperitoneally) and killed by transcardial perfusion with isotonic saline solution. Brains were removed, frozen in liquid nitrogen, and embedded in OTC compound (Tissue Tek, Elkhart, IN, USA). Coronal sections (8 μm thick) at the hippocampal level were cut with a cryostat, mounted on slides, and dried. To preserve PKH26 labeling, the sections were lightly fixed with 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.2, for 10 mins. To determine the location of exogenous microglia relative to ischemic CA1 pyramidal neurons, an immunofluorescent study was performed using terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuracil nucleotides-biotin nick-end labeling (TUNEL), Griffonia simplifolia B4 isolectin (IB4), and an antibody to microtubule-associated protein 2 (MAP2). To localize BDNF and GDNF expression, immunohistochemistry using the avidin—biotin complex method was performed.
TUNEL staining was used to detect DNA fragmentation in cell nuclei. The hippocampal sections were pretreated with or without 20 μg/mL proteinase K (Sigma Chemical Co.) at room temperature (RT) for 15 mins. After treatment with 0.3% H2O2 in methanol for 30 mins, sections were incubated with 100 U/mL TdT and 10 nmol/L per mL biotinylated 16-2′-dUTP (Boehringer-Mannheim-Yamanouchi, Tokyo, Japan) in TdT buffer (100 mmol/L sodium cacodylate, pH 7.0, 1 mmol/L cobalt chloride, and 50 μg/mL gelatin) in a humid atmosphere at 37°C for 60 mins. Further incubation with fluorescein isothiocyanate-conjugated avidin (Nichirei, Tokyo, Japan) was performed for 60 mins at RT.
Microglial cells were identified by IB4 staining. Cryosections were incubated with 20 μg/mL biotinylated IB4-fluorescein isothiocyanate conjugate (Sigma Chemical Co.) in 0.01 mol/L phosphate-buffered 0.15 mol/L saline (pH 7.2) containing 0.1% Triton X-100 at RT for 2 h (Streit, 1990).
Hippocampal CA1 pyramidal neurons were visualized using a monoclonal antibody to MAP2 (Chemicon, Temecula, CA, USA). Sections were incubated with 10% normal goat serum for 30 mins at RT. They were then incubated with a mouse monoclonal anti-MAP2 antibody, diluted 1:100, for 1 day at 4°C. Further incubations were performed with fluorescein isothiocyanate-conjugated anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), diluted 1:500, at RT for 60 mins.
We visualized BDNF epitopes using a rabbit polyclonal anti-BDNF antibody (diluted 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and GDNF epitopes were visualized using a rabbit polyclonal anti-GDNF antibody (diluted 1:250; Santa Cruz Biotechnology). Sections were then incubated in secondary antibody, biotinylated goat anti-rabbit IgG, followed by avidin—biotin complex (Elite kit: Vector Laboratories, Burlingame, CA, USA), and visualized after chromogenic reaction with 3,3′-diaminobenzidine.
The sections were analyzed with an Olympus BX51 microscope equipped with bright-field and fluorescent light sources (Tokyo, Japan). Both bright-field and fluorescent images were captured from the same field using a Penguin 600 CL camera (Pixera Corporation, Los Gatos, CA, USA).
Cell Counts
Six serial, coronal frozen sections (8 μm thick) at the CA1 hippocampal level were examined. Cells were counted if they were within a randomly selected 1-mm linear length of CA1. To determine the time course of exogenous microglia migration to the ischemic hippocampus, PKH26-labeled microglia were counted in animals injected with nonstimulated or IFNγ-stimulated microglia 24 h before ischemia, and at 2, 3, 5, and 7 days after ischemia. To determine whether exogenous microglia reduced ischemic damage caused by transient global ischemia, surviving pyramidal neurons were counted in ISCH or sham ISCH animals infused with vehicle (ISCH/VEH, SHAM/VEH), nonstimulated microglia (ISCH/Mi, SHAM/Mi), or IFNγ-stimulated microglia (ISCH/γMi, SHAM/γMi), 24 h before (PRE) or 24 h after (POST) ischemia. All neurons (cresyl violet-positive cells) with an intact morphologic appearance were counted in each animal either 7 or 14 days after ischemia. A total of 10 animals were examined for each of the 12 conditions.
Passive Avoidance Task
A step-through type passive avoidance apparatus was used to evaluate memory, as described previously (Nanri et al, 1998). The apparatus comprised two compartments: one light and one dark (each 16.5 × 16.0 × 14.7 cm), separated by a guillotine door. The dark compartment had a stainless-steel grid floor. A scrambled electric shock was delivered through the grid floor by a shock generator (Daiichi Kikai Inc., Tokushima, Japan). The passive avoidance learning trial was performed either 7 or 14 days after inducing ischemia. At the beginning of the training session, each animal was placed in the light compartment. When the animal stepped into the dark compartment, the door between compartments was closed and a foot shock (0.6 mA, 3 secs) was delivered through the floor. The door was then opened and the animal was allowed to cross back into the light compartment. Each time the animal stepped into the dark compartment, a foot shock was delivered. Eventually, the animal remained in the light compartment. The learning trial lasted 8 mins.
The retention test was performed 24 h after the learning trial. In the retention test, the gerbil was placed in the light compartment, and the latency to enter the dark compartment was measured. If the gerbil did not enter the dark compartment within 180 secs, a score of 180 secs was assigned. Performance on the passive avoidance task was evaluated in the SHAM/VEH-(PRE/POST), ISCH/VEH-(PRE/POST), ISCH/Mi-(PRE/POST), and ISCH/γMi-(PRE/POST) groups. Ten animals were used for each of the eight conditions.
Enzyme-Linked Immunosorbent Assay
Changes in BDNF and GDNF levels were measured in the hippocampus in SHAM/VEH, ISCH/VEH, ISCH/Mi, or ISCH/γMi animals using the BDNF and GDNF Emax Immunoassay System Kit (Promega, Madison, WI, USA). In the hippocampus of ISCH/VEH-PRE or ISCH/Mi-PRE gerbils, each neurotrophic factor level was measured 2 h, 1 day, 3 days, and 7 days after inducing ischemia. In the hippocampus of ISCH/VEH-POST and ISCH/Mi-POST animals, each neurotrophic factor level was measured 26 h, 2 days, 4 days, and 8 days after ischemia. As a control, we used the hippocampus from SHAM/VEH-PRE animals, which was dissected 6 h after the treatment (Marmigere et al, 2003). Neurotrophic factor levels were also measured in the rest of the brain after the hippocampus was removed.
Results
Recruitment of Exogenous Microglia into CA1 Hippocampal Lesions
Figure 1 shows fluorescent staining in the hippocampal CA1 field of ISCH animals injected with PKH26-labeled exogenous microglia 24 h before ischemia. PKH26-labeled cells accumulated near TUNEL-positive cells (Figures 1A–1D). PKH26-labeled cells were also located in the vicinity of MAP2-positive pyramidal neurons 3 days after ischemia (Figure 2A). IB4 staining of adjacent sections revealed that all PKH26-labeled cells were IB4-positive (Figures 2B–2D).
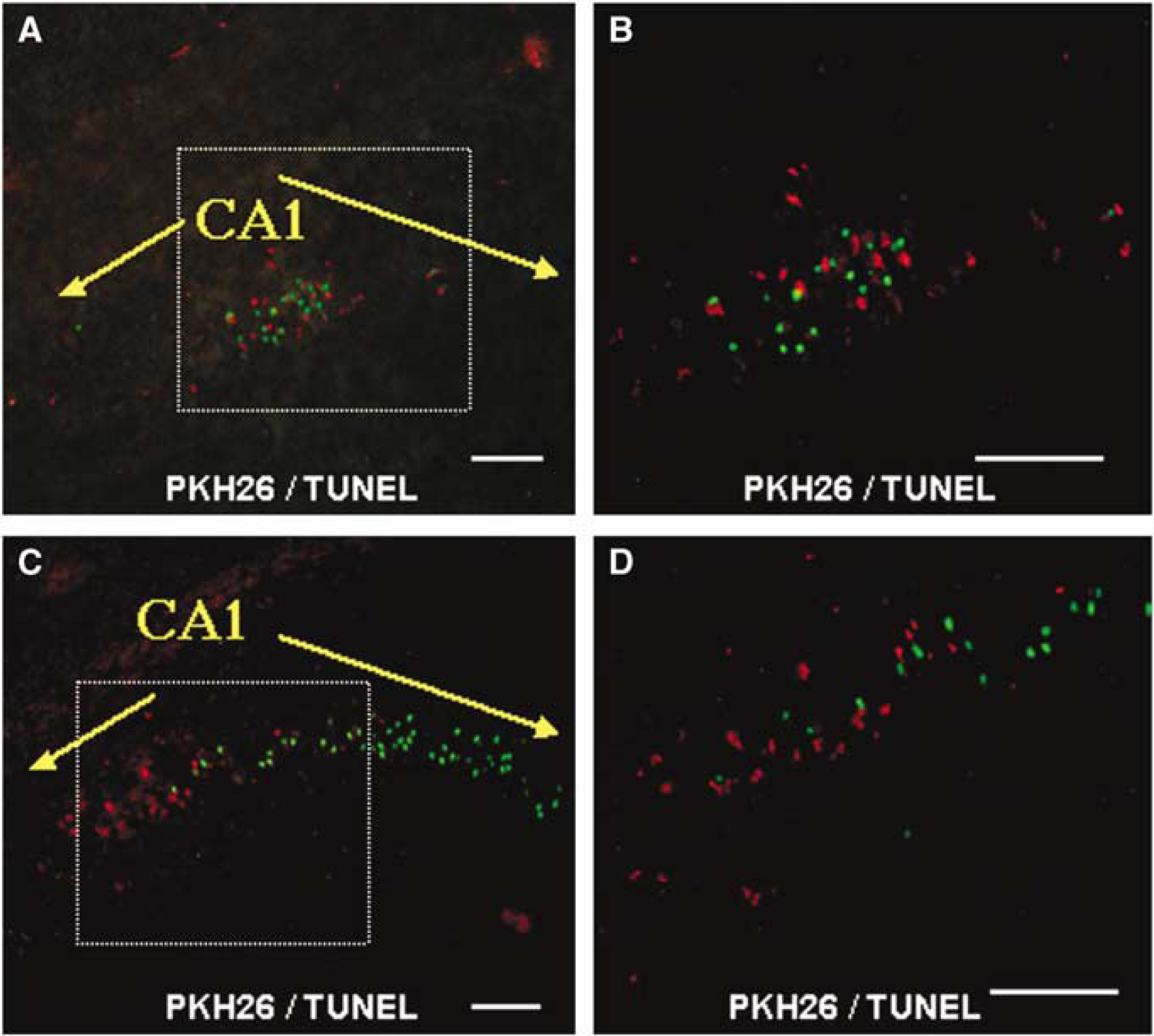
Migration of PKH26-labeled cells to TUNEL-positive CA1 pyramidal neurons after transient global ischemia. The panels show fluorescent staining in hippocampal CA1 sections obtained from ISCH animals injected with PKH26-labeled exogenous microglia 24 h before ischemia. (
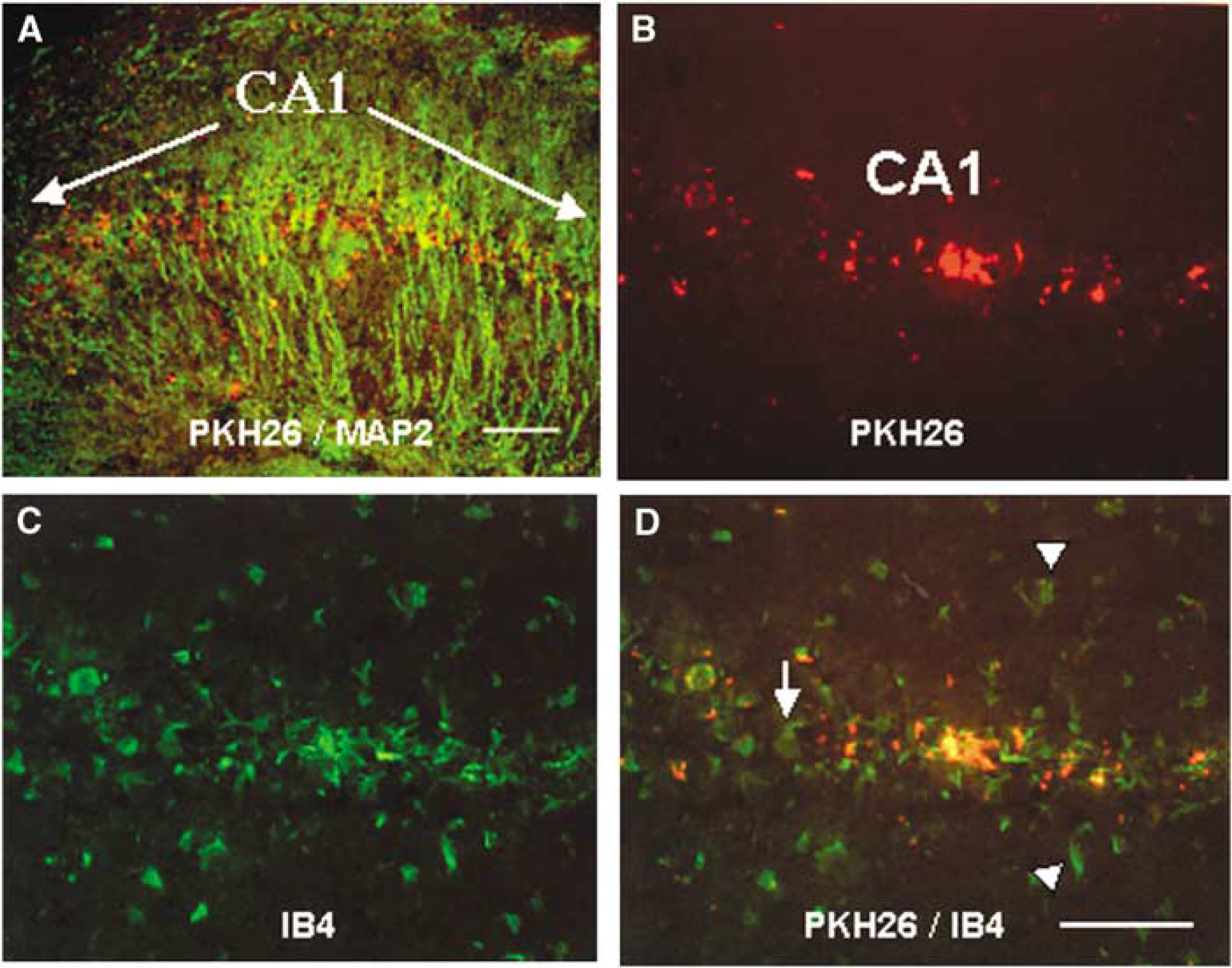
Migration of exogenous microglia to hippocampal CA1 lesions after transient global ischemia. The panels show fluorescent staining in hippocampal CA1 sections obtained from ISCH animals injected with PKH26-labeled exogenous microglia 24 h before ischemia. (
PKH26-labeled cells that reacted with IB4 were identified as microglia. Most of the microglia that accumulated in the CA1 field were both PKH26- and IB4-positive exogenous microglia, whereas there were only a few PKH26-negative and IB4-positive endogenous microglia (Figures 2B–2D). The TUNEL-positive cells were probably pyramidal neurons, because they were in the CA1 pyramidal cell layer and were not labeled with PKH26 (Figures 1A–1D). Neither TUNEL-positive pyramidal neurons nor PKH26-labeled microglia were detected in CA1 1 day after ischemia (data not shown). Two days after ischemia, only a few TUNEL-positive pyramidal neurons were detected in a portion of CA1, and PKH26-labeled microglia had accumulated in or around the TUNEL-positive neurons (Figures 1A and 1B). Three days after ischemia, a few TUNEL-positive pyramidal neurons were detected in CA1 (data not shown) and PKH26-labeled microglia were scattered throughout the CA1 field (Figure 2A). Five days after ischemia, many TUNEL-positive pyramidal neurons and PKH26-labeled microglia were distributed throughout CA1. Many of these cells had accumulated near the CA1–CA2 border (Figures 1C and 1D). The number of TUNEL-positive pyramidal neurons and PKH26-labeled microglia in the CA1 layer increased in a time-dependent manner until 7 days after ischemia (Figures 1A–1D and 3A). Stimulation with IFNγ increased the number of migrating exogenous microglia in the ischemic CA1 pyramidal cell layer (Figure 3A). The temporal profile of exogenous microglia recruitment was the same if the microglia were injected after ischemia onset (data not shown). In a separate set of experiments, PKH26-labeled macrophages did not enter the brain from the circulation (data not shown).
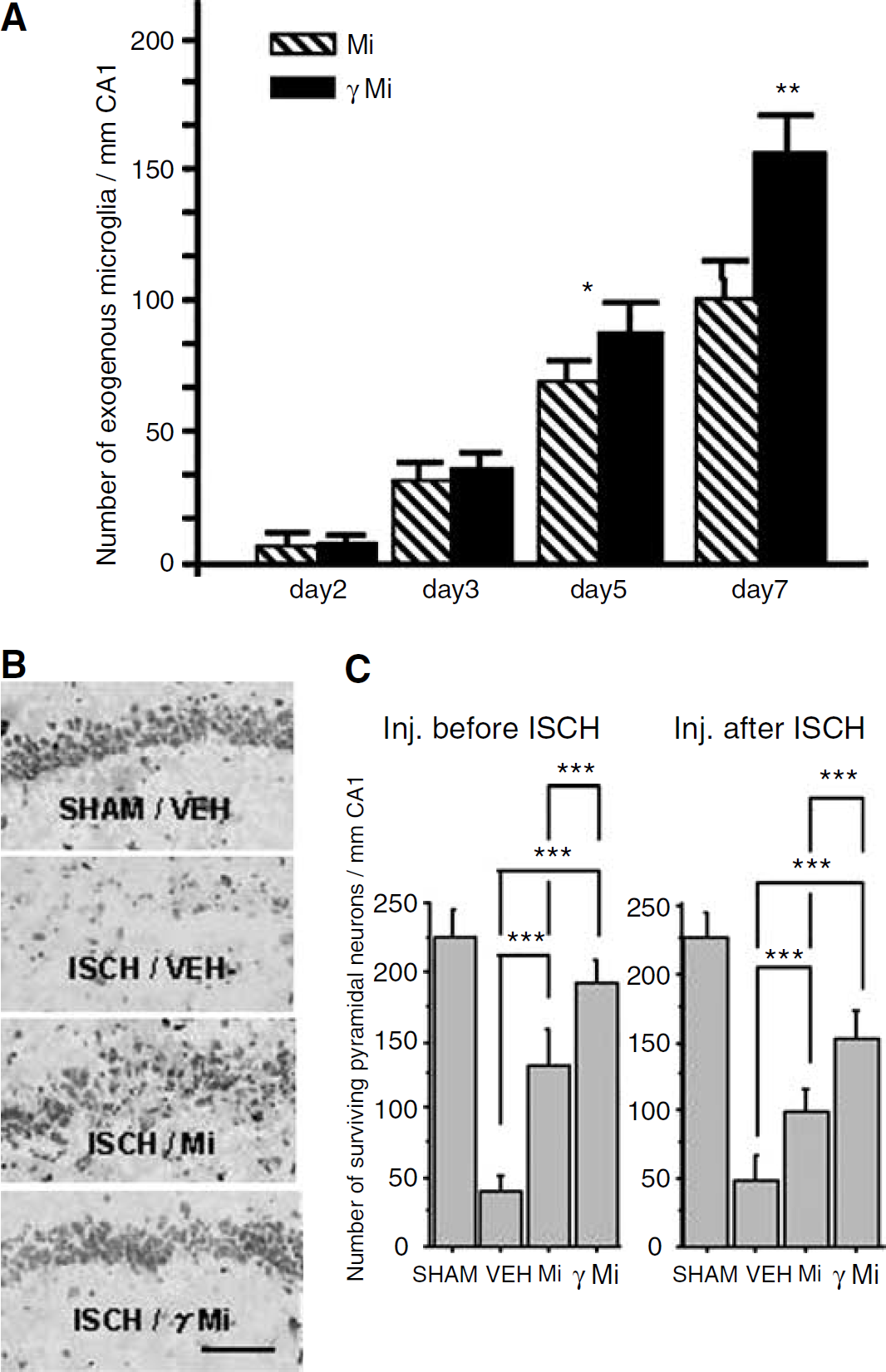
Neuroprotective effects of exogenous microglia. (
Reduction of Ischemic Damage by Microglia Injection
Figure 3B shows the hippocampus from SHAM/VEH, ISCH/VEH, ISCH/Mi, and ISCH/γMi animals 7 days after ischemia. Most of the pyramidal neurons in the CA1 field had degenerated in ISCH/VEH animals. Microglial infusion (ISCH/Mi) increased the number of surviving pyramidal neurons, and IFNγ stimulation (ISCH/γMi) enhanced the microglial neuroprotective effect.
Figure 3C shows the number of surviving pyramidal neurons in the CA1 field 7 days after ischemia in SHAM/VEH, ISCH/VEH, ISCH/Mi, and ISCH/γMi animals. SHAM/VEH animals were used as controls (n=10). There was no effect of injection of nonstimulated microglia or IFNγ-stimulated microglia on the number of surviving CA1 pyramidal neurons in SHAM/VEH animals (data not shown). Ischemia significantly reduced the number of surviving pyramidal neurons. Intraarterial injection of microglia (either PRE or POST) significantly increased the number of surviving neurons (P<0.001). Stimulation of the microglia with IFNγ further enhanced neuron survival (P<0.001). The injection of vehicle, nonstimulated microglia, or IFNγ-stimulated microglia had no effect on postoperative body temperature throughout the survival period (data not shown). The number of surviving CA1 neurons at 14 days after ischemia was not different from that at 7 days after ischemia in each study (data not shown). Injection of microglia 48 h after ischemia had no significant effect on pyramidal neuron death (data not shown). The injection of PKH26-labeled macrophages into ISCH animals had no effect on the number of surviving CA1 pyramidal neurons (data not shown).
Prevention of Ischemia-Induced Learning Impairment by Microglia Injection
The passive avoidance task was used to determine whether exogenous microglia prevent ischemia-induced learning impairment. Figure 4 shows behavioral responses from gerbils 7 days after ischemia. The mean retention latency was shorter for ISCH/VEH animals than for SHAM/VEH animals (P<0.01). The mean retention latency of the ISCH/Mi-PRE animals was longer than that of the ISCH/VEH-PRE animals (P<0.01), although the mean retention latency of the ISCH/Mi-POST animals was not different from the ISCH/VEH-POST animals. The mean retention latency was significantly longer in the ISCH/γMi-PRE (P<0.001) and ISCH/γMi-POST (P<0.01) animals (Figure 4) compared with the ISCH/VEH (PRE/POST) animals. The microglia-induced increase in the mean retention latency at 14 days after ischemia was not statistically different from the mean retention latency at 7 days after ischemia in each study (data not shown). The injection of macrophages into ISCH animals had no effect on retention latency (data not shown).
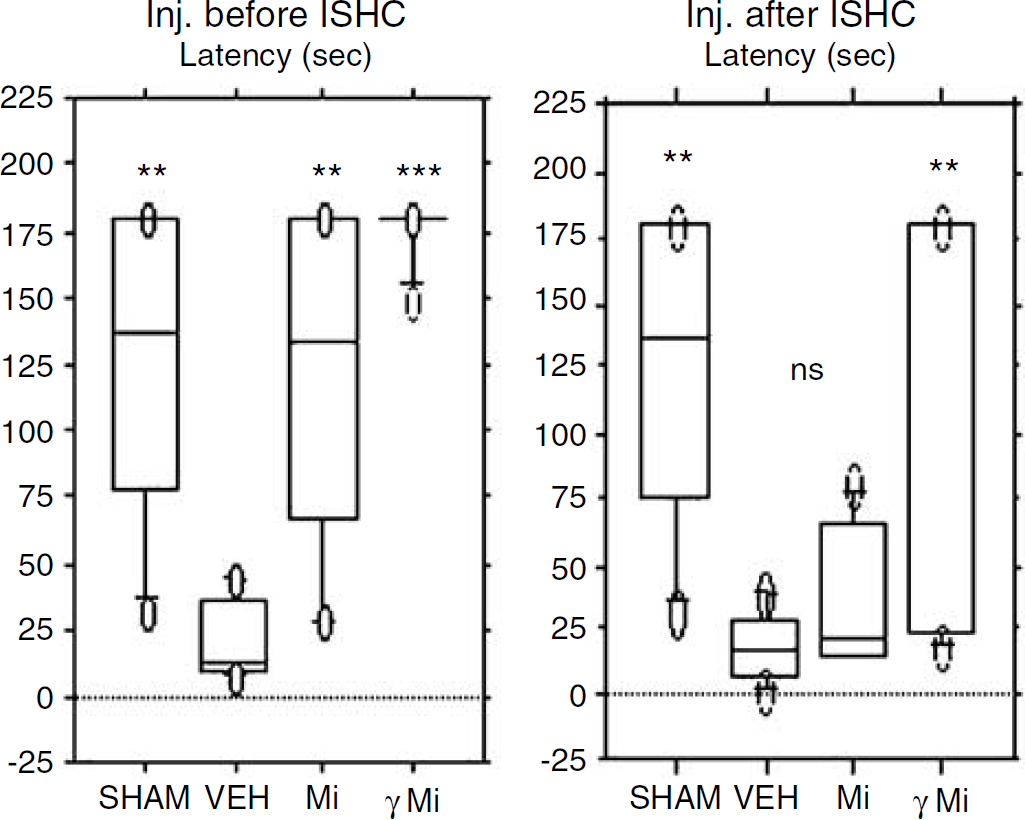
The effects of microglia on the latency of the transient ischemic gerbils in the step-through passive avoidance task. The graphs show the results of passive avoidance task 7 days after ischemia. The mean retention latency of ISCH/Mi-PRE and ISCH/γMi-PRE animals was longer than that of ISCH/VEH-PRE animals. The mean retention latency of ISCH/γMi-POST animals was significantly longer than that of ISCH/VEH-POST animals, whereas that of ISCH/Mi-POST was not significantly longer. ∗∗P<0.01, ∗∗∗P<0.001, significantly different from ISCH/VEH with Scheffe's post hoc test for multiple pairwise comparisons (n=10). Inj. before ISCH: intraarterial injection 24 h before ischemia treatment; Inj. after ISCH: intraarterial injection 24 h after ischemia treatment; SHAM: injection of culture medium into sham-ischemia treated animals; VEH: injection of culture medium into ischemia-treated animals; Mi: injection of nonstimulated microglia into ischemia-treated animals; γMi: injection of IFNγ-stimulated microglia into ischemia-treated animals.
BDNF and GDNF Expression in the ISCH Hippocampus
The time course of hippocampal BDNF and GDNF expression was examined in SHAM and ISCH animals, and the effects of exogenous microglia on the expression of these neurotrophic factors was investigated. The time course of hippocampal BDNF and GDNF expression after ischemia was measured by enzyme-linked immunosorbent assay (Figure 5). In the hippocampus of ISCH animals, GDNF expression levels decreased significantly 2 h after ischemia and gradually increased during the next 7 days. In contrast, animals that received microglial injections 24 h before ischemia showed no decrease in BDNF or GDNF levels. Expression levels of BDNF remained constant until 3 days after ischemia, at which time they approximately doubled. Expression of GDNF in microglia-injected animals showed a rapid increase 2 h after ischemia and continued to increase during the course of 7 days to approximately 600% of the baseline level. There was no difference in BDNF or GDNF expression levels in animals injected with IFNγ-stimulated microglia, compared with nonstimulated microglia. Injection of microglia 24 h after ischemia induced a similar increase in BDNF and GDNF expression (Figure 5). Microglia-induced increases in neurotrophic factors were not observed except in the hippocampus (data not shown). The injection of macrophages into ISCH animals did not change BDNF or GDNF expression in the hippocampus (data not shown).
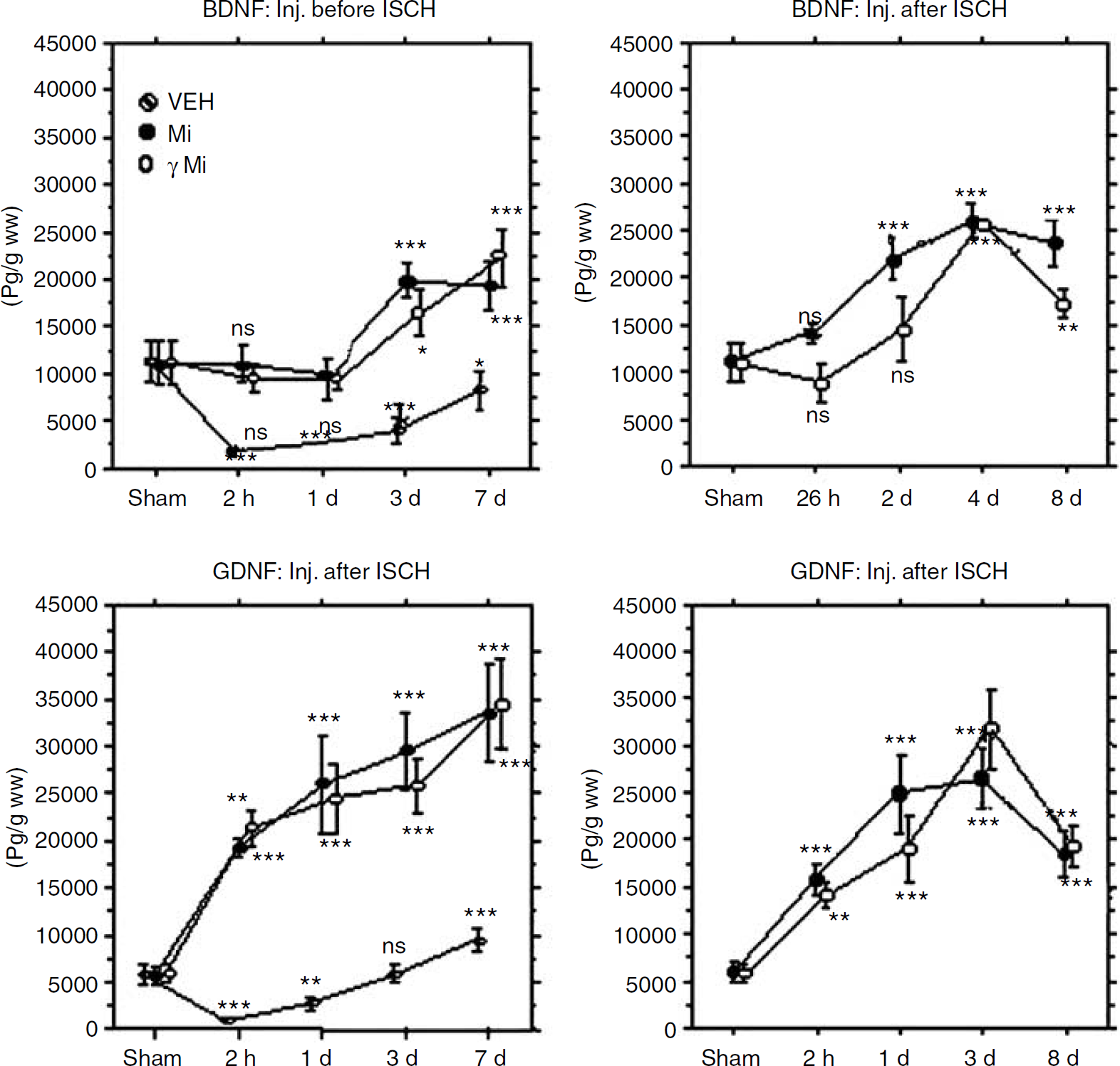
The effects of exogenous microglia on the time course of hippocampal BDNF and GDNF expression after transient global ischemia. Graphs indicate the BDNF and GDNF expression level in the hippocampus detected by enzyme-linked immunosorbent assay. Results are expressed as means of picograms of BDNF or GDNF normalized to gram of wet tissue weight (pg/g ww). As a control, we used SHAM/VEH hippocampus, which was dissected 6 h after the treatment. In SHAM/VEH hippocampus, BDNF and GDNF expression was observed. In ISCH/VEH-PRE animals, however, the levels were less than 20% of the baseline level (SHAM/VEH-PRE level) 2 h after ischemia. Injection of microglia prevented the decrease of both BDNF and GDNF and then induced an increase in both factors above baseline levels. In ISCH/Mi-PRE hippocampus, the neurotrophin levels were measured 2 h, 1 day, 3 days, and 7 days after ischemia. In ISCH/Mi-POST hippocampus, neurotrophin levels were measured 26 h, 2 days, 4 days, and 8 days after ischemia. Vertical bars represent the mean±s.d. (n=6). ∗P<0.05, ∗∗P<0.01, ∗∗∗P<0.001, significantly different from SHAM control with Bonferroni post hoc test. Abbreviations are as in Figure 2. Hatched circles (VEH) indicate values of BDNF or GDNF expression level in the ischemia-treated hippocampus injected with culture medium, black circles (Mi) indicate those injected with nonstimulated microglia, and open circles (γMi) indicate those injected with IFNγ-stimulated microglia.
To determine which cells expressed BDNF and GDNF in our ischemia model, we examined the hippocampus using immunohistochemical analysis in SHAM/VEH, ISCH/VEH-PRE, and ISCH/Mi-PRE gerbils (Figure 6). In SHAM animals, hippocampal CA1 neurons and endogenous microglia-like cells expressed both GDNF and BDNF (Figures 6A and 6E). Two hours after ischemia, most of the CA1 neuron-like cells did not express BDNF and GDNF, and BDNF-positive endogenous microglia-like cells were scattered throughout CA1 (Figures 6B and 6F). Three days after ischemia, most CA1 neurons weakly expressed BDNF, whereas GDNF levels recovered (Figures 6C and 6G). Seven days after ischemia, BDNF immunoreactivity was detected in the CA1 neuron-like cell bodies, endogenous microglia, and also in some cell bodies and processes of reactive astrocyte-like cells near the ischemic CA1 field (Figure 6D).
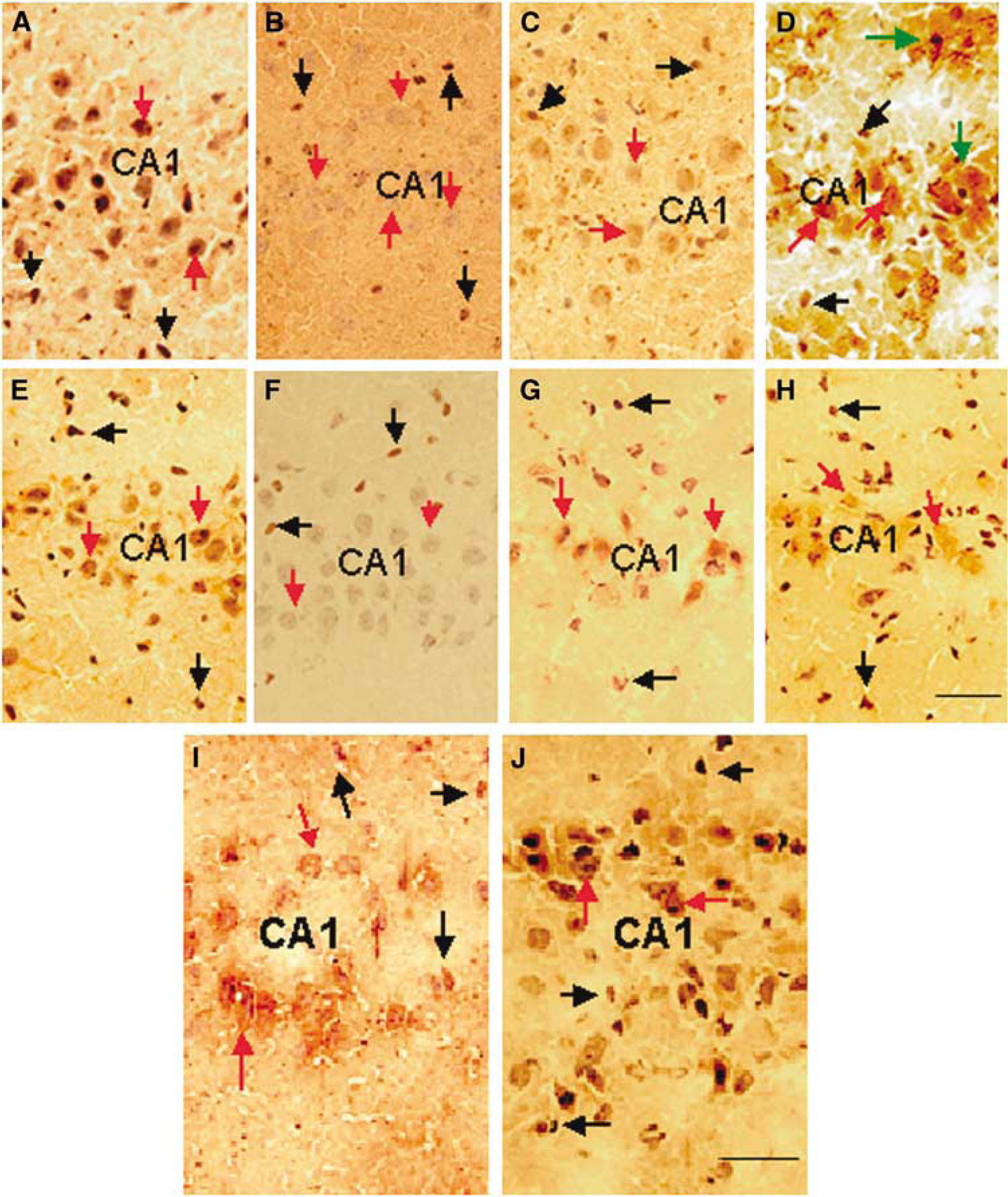
Immunohistochemical staining for BDNF and GDNF after transient global ischemia. (
At 7 days, damaged CA1 neuron-like cells expressed GDNF and many GDNF-positive, microglia-like cells were observed throughout CA1, whereas GDNF-positive, astrocyte-like cells were not detected (Figure 6H). Most of the CA1 neuron-like cells expressing BDNF or GDNF seemed to undergo degeneration because the cytoplasm was enlarged and the nucleus was not visible. Injection of microglia 24 h before ischemia greatly increased staining of BDNF- and GDNF-like immunoreactivity in CA1 neurons.
Discussion
In this study, we show that systemically injected microglia provide significant neuroprotection to the hippocampus after an ischemic insult. In addition to promoting CA1 cell survival, injection of microglia either before or 24 h after ischemia improved performance in a passive-avoidance learning task. There was also an increase in BDNF and GDNF expression in the hippocampus.
Previous studies indicate that endogenous microglia migrate to the CA1 pyramidal cell layer and protect ischemic neurons after reperfusion neuronal injury (Nitatori et al, 1995; Pinteaux et al, 2006; Neumann et al, 2006). In the present study, exogenous microglia migrated to areas that suffered ischemic damage, indicated by the presence of PKH26-positive cells around TUNEL-positive neurons in the CA1 pyramidal cell layer. Most of the cells were both PKH26-positive and IB4-positive exogenous microglia, although there were also a few PKH26-negative, IB4-positive cells. PKH26-negative, IB4-positive cells are either endogenous microglia/macrophage lineage cells or vascular endothelial cells.
Activated microglia share several characteristics with peripheral macrophages in vitro, but they display different phenotypes in vivo (Ling and Wong, 1993; Sawada et al, 1995, 1990, 1992). Several studies suggest a possible contribution of the invasion of blood-borne macrophages to delayed neuronal death after transient global ischemia (Lees, 1993). Our earlier studies showed that macrophages do not enter undamaged brain from the circulation (Imai et al, 1997). They might, however, enter the brain from the site of delayed neuronal death, where neovascularization and blood—brain barrier breakdown occur after ischemia (Kataoka et al, 2000). In our ischemia model, PKH26-labeled peritoneal macrophages did not enter the brain from the circulation. Therefore, IB4-positive, PKH26-negative cells migrating to the ischemic CA1 layer are likely to be endogenous microglia. Most of the migrating cells in the CA1 area were not endogenous microglia, but rather exogenous microglia. It is possible that isolated microglia from the mixed brain cultures are already activated (Streit, 1993) and so migrate to the ischemic lesion more rapidly than endogenous microglia.
Injection of exogenous microglia increased the number of surviving CA1 neurons after transient global ischemia, even when microglia were injected after the ischemic insult. The microglial neuroprotective effect also prevented an ischemia-induced learning impairment. In this and previous studies, we have shown that microglia isolated from a newborn mixed brain culture express both BDNF and GDNF, and intraarterial injection of microglia increases the presence of these neurotrophic factors in the ischemic hippocampus (Suzuki et al, 2001). Beyer et al (2000) reported that phagocytosis of Latex beads induced a microglial amoeboid morphology but did not increase immunologically relevant molecules. Microglia that phagocytose PKH26 would have similar properties as those that phagocytose Latex beads. PKH26 phagocytosis labeling did not affect microglial expression of neurotrophic factors and neuroprotection (data not shown). Upregulation of both neurotrophic factors in damaged brain is crucial for protection from neurodegeneration (Ebadi et al, 1997). The association between exogenous microglia at the lesion site and decreased cell death, increased neurotrophic factor expression, and improved learning ability after ischemic injury leads us to conclude that microglia have a protective effect rather than a toxic effect, on neurons under ischemic conditions.
We examined the time course of hippocampal BDNF and GDNF expression after transient global ischemia, and investigated the effect of exogenous microglia on the expression of these neurotrophic factors. In normal CA1 neurons, the expression of both neurotrophic factors was preserved; however, their immunoreactivity was reduced 2 h after ischemia and then increased in a time-dependent manner. Seven days after ischemia, the main sources of BDNF were damaged CA1 neurons, endogenous microglia, and reactive astrocytes. Reactive astrocytes were excluded as the source of GDNF because they do not express GDNF in our model. The BDNF results are consistent with previous studies (Kokaia et al, 1996; Lee et al, 2002), whereas the GDNF results are inconsistent with a study by Miyazaki et al (2001). They reported that GDNF expression in the hippocampal CA1 region increased between 3 and 24 h after ischemia, and then declined to baseline levels. In their model, in which rats are subjected to a transient global ischemia induced by four-vessel occlusion, reactive astrocytes express GDNF. The discrepancy in the results between the two studies is possibly because of the use of different animal models of ischemia. In the present study, injection of microglia prevented the decrease in BDNF and GDNF expression in the hippocampus immediately after ischemia, and later increased the expression of these trophic factors. CA1 neurons, rather than microglia or astrocytes, were the main source of the later high expression of BDNF and GDNF in the hippocampus observed 7 days after ischemia. Injections of microglia into SHAM animals did not significantly increase BDNF or GDNF expression in the brain, although a large number of PKH26-labeled microglia enter the brain from the circulation (Imai et al, 1997; Sawada et al, 1998). Astrocytes expressing these neurotrophic factors were not detected in ischemic hippocampus injected with microglia. The maintenance of BDNF and/or GDNF expression in injured neurons might be one mechanism of neuroprotection in the ischemic hippocampus.
Intraventricular administration of a Sendai virus vector carrying GDNF or nerve growth factor increases the expression of these neurotrophic factors in the hippocampus and prevents delayed neuronal death induced by transient global ischemia in gerbils, even when administered 6 h after ischemia (Shirakura et al, 2004). The time course for increased GDNF expression was similar to that in the present experiment, except that it took less time for exogenous microglia to express GDNF than it took for the Sendai virus vector. This might be one reason why we observed neuroprotection even when the microglia were injected 24 h after ischemia. Intrahippocampal administration of BDNF in adult rats improves performance in a spatial memory task (Cirulli et al, 2004). Administration of GDNF and BDNF to the site of ischemia might be one of the mechanisms underlying microglial neurotrophic effects on ischemic CA1 neurons.
Lee et al (2002) reported that BDNF mRNA increases 3 h after ischemia and returns to sham-operated level in the CA1 pyramidal neurons 1 day after ischemia, whereas BDNF protein decreases 3 h after ischemia and recovers to some extent at 7 days after ischemia. This expression disparity between neurotrophin mRNA and protein was reported to be because of a translational and/or posttranslational dysfunction of protein synthesis, release, or transport immediately after cerebral injury (Lee et al, 2002). In our study, the recovery of BDNF and GDNF expression was observed in degenerated neurons. The expression of neurotrophic factors might not enhance the survival of ischemic pyramidal neurons, however, because most degenerating cells do not express BDNF or GDNF receptors after ischemic insult (Ferrer et al, 1998; Wang et al, 2004). Although pretreatment of microglia with IFNγ significantly enhanced microglial neuroprotective effects on ischemic CA1 pyramidal neurons, it did not increase hippocampal BDNF or GDNF expression, compared with nontreated microglia. Nevertheless, pretreatment with IFNγ increased the number of microglia migrating to the ischemic hippocampus. It is possible that microglia pretreated with IFNγ induce expression of neurotrophic factor receptors in ischemic pyramidal neurons, thus providing a neuroprotective effect. We are now investigating the mechanism.
Cytokines secreted by microglia might also affect ischemic CA1 neurons. We did not examine cytokine activity in this study because neurons and microglia interact with each other and produce complicated effects in vivo. For example, activated microglia release factors capable of generating oxidative damage, such as superoxide anions (O2−) and nitric oxide (Saud et al, 2005), whereas transient global ischemia increases transforming growth factor-β1 expression in CA1 pyramidal neurons (Zhu et al, 2000), which could eliminate microglial O2− and nitric oxide production (Herrera-Molina and von Bernhardi, 2005).
Microglial activation, after neuronal injury, appears to serve a neuroprotective function. A strong inflammatory response, however, could induce microglia to be hyperactive, which allows them to escape endogenous control and become toxic to neurons (Polazzi and Contenstabile, 2002). Interleukin-1β secreted by microglia activates p38 mitogen-associated protein kinase and cyclic adenosine monophosphate-response element binding protein to suppress long-term potentiation, which is thought to be an important underlying mechanism of learning and memory (Srinivasan et al, 2004). In our preliminary data, however, systemic injection of exogenous microglia in rabbits subjected to transient global ischemia prevented the ischemia-induced suppression of long-term potentiation (unpublished data). Microglia also produce interleukin-10 (Wu et al, 2005; Broderick et al, 2000), which reverses lipopolysaccharide-induced increases in signaling in the hippocampus (Lynch et al, 2004). Exogenous microglia introduced to the site of ischemia might reverse ischemia-induced disruption of endogenous control by secreting antiinflammatory cytokines such as interleukin-10 (Wu et al, 2005; Ooboshi et al, 2005; Broderick et al, 2000). Further studies are necessary to verify the mechanism of microglial neuroprotective effects on ischemic CA1 neurons.
It is also possible that injection of nonautologous microglia induced an immune reaction that is responsible for the present findings. We believe that this is unlikely because there is no evidence in the literature for a nonspecific immune response to be neuroprotective and/or reverse behavioral deficits. This will be examined in the future.
In conclusion, peripherally injected exogenous microglia exhibit a specific affinity for ischemic brain lesions and protect against neuronal damage in our ischemia model, suggesting that one role of microglia is to protect damaged neurons after transient global ischemic insult. To continue the exploration of the uses of exogenous microglia, we are currently isolating microglia from bone marrow (Ono et al, 1999; Tanaka et al, 2003). In the future, the administration of microglia might be a potential tool for cell or gene therapy in the treatment of brain disease.
Footnotes
Acknowledgements
We express our gratitude to Zlokovic Berislav, who belongs to the University of Rochester Medical Center, Rochester, NY, USA, for helpful discussion and critical review of the manuscript. The authors have no conflicting financial interests.