
Abstract
We tested the hypothesis that astrocytic matrix metalloproteinase-9 (MMP-9) mediates hemorrhagic brain edema. In a clinical case of hemorrhagic stroke, MMP-9 co-localized with astrocytes and neurons in peri-hematoma areas. In a mouse model where blood was injected into striatum, MMP-9 was colocalized with astrocytes surrounding the hemorrhagic lesion. Because MMP-9 is present in blood as well as brain, we compared four groups of wild type (WT) and MMP-9 knockout (KO) mice: WT blood injected into WT brain, KO blood into KO brain, WT blood into KO brain, and KO blood into WT brain. Gel zymography showed that MMP-9 was elevated in WT hemorrhagic brain tissue but absent from KO hemorrhagic brain tissue. Edematous water content was elevated when WT blood was injected into WT brain. However, edema was ameliorated when MMP-9 was absent in either blood or brain or both. To further assess the mechanisms involved in astrocytic induction of MMP-9, we next examined primary mouse astrocyte cultures. Exposure to hemoglobin rapidly upregulated MMP-9 in conditioned media within 1 to 24 h. Hemoglobin-induced MMP-9 was reduced by the free radical scavenger U83836E. Taken together, these data suggest that although there are large amounts of MMP-9 in blood, hemoglobin-induced oxidative stress can trigger MMP-9 in astrocytes and these parenchymal sources of matrix degradation may also be an important factor in the pathogenesis of hemorrhagic brain edema.
Introduction
Intracerebral hemorrhage (ICH) accounts for up to 15% of all strokes (Ariesen et al, 2003), and it has been estimated that 30% to 40% of ischemic strokes will undergo some degree of hemorrhagic conversion (Lyden and Zivin, 1993). The prognosis after ICH is poor and treatment options are limited. Yet, in comparison to ischemic stroke, pathogenic mechanisms of ICH remain relatively unclear.
In experimental models, recent data suggest that neuronal injury and cell death evolves in peri-hematoma areas via multifactorial mechanisms. These pathways include damaging cytokines and inflammatory mediators (Castillo et al, 2002; Hua et al, 2000; Peeling et al, 2001a, 2001b), apoptosis (Felberg et al, 2002; Gong et al, 2001; Matsushita et al, 2000; Matz et al, 2000), and microglial activation (Wang et al, 2003; Wang and Tsirka, 2005a, 2005b). Blood flow reductions have also been documented in some models of ICH (Kobari et al, 1988; Yang et al, 1994), although the question of whether perfusion deficits truly reach ischemic thresholds remains to be confirmed (Rosand et al, 2002; Zazulia et al, 2001). As these molecular pathways become better defined, new therapeutic opportunities for preventing neuronal death after ICH may eventually be found.
In the meantime, one of the biggest challenges from a clinical perspective are related to the brain edema that accompanies ICH (Hoff and Xi, 2003). Edema and increases in intracranial pressure typically occur between 24 to 48 h post-ictus, and are, suggested to be a major cause of clinical deterioration and death. Emerging data suggest an important role for extracellular proteases that are abundant in blood that has leaked into brain (Gingrich and Traynelis, 2000). For example, thrombin amplifies edema adjacent to ICH (Lee et al, 1996; Xi et al, 1998), and treatment with thrombin inhibitors can reduce edema (Kitaoka et al, 2002). More recently, the large family of matrix metalloproteinases (MMPs) have also been implicated. Matrix metalloproteinases have already been shown to mediate blood—brain barrier (BBB) injury after cerebral ischemia (Asahi et al, 2001a, 2001b; Gasche et al, 2001; Rosenberg et al, 1998). Since MMPs are also upregulated after ICH (Power et al, 2003; Wang and Tsirka, 2005a, 2005b), these proteases may also trigger peri-hematoma edema via similar effects on the neurovascular matrix and BBB.
In this study, we specifically ask whether MMP-9 mediates brain edema after ICH using a combination of clinical case material, knockout (KO) mouse models, and cell culture systems. Our data show that MMP-9 may indeed be upregulated in reactive astrocytes surrounding an ICH, and although large amounts of MMP-9 are already present in blood, these endogenous astrocytic sources of MMP-9 may still play a significant role in mediating brain edema that accompanies ICH.
Materials and methods
Clinical Intracerebral Hemorrhage Case
Brain tissue was obtained from a patient who suffered a massive hemorrhagic stroke involving the right hemisphere, and died approximately 3 h after stroke onset. Sampling was performed within 6 h after onset. Computed tomography (CT) scans obtained at admission were used to guide tissue sampling during autopsy. Blood mass was removed and adjacent parenchyma was obtained along with matching contralateral tissue. All samples were snap-frozen and stored at −80°C until immunohistochemistry was performed. This study was approved by the ethics and human research committee of the Vall d'Hebron Hospital in Barcelona, Spain. Informed consent was obtained from relatives before autopsy.
Mouse Intracerebral Hemorrhage Model
All experiments were performed following an institutionally approved protocol in accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals. Matrix metalloproteinase-9 KO and background-matched wild-type (WT) mice (male, 24 to 30 g, 10 to 12 weeks old) were used. Mice were anesthetized with halothane (1% to 1.5%) under spontaneous respiration in a nitrous oxide/oxygen mixture and placed in a stereotactic frame. A midline scalp incision was made, and a craniotomy was performed. A 30-gauge needle was lowered into the center of striatum (from bregma: 1 mm anterior, 2 mm lateral, 3.5 mm depth), and 20 μL of whole blood was infused into over a 10 mins period. The needle was left in place for 10 mins after completion of the infusion to prevent leakage of blood out of the brain, and then removed slowly. The craniotomy was sealed with bone wax, the scalp sutured closed, and mice were allowed to recover.
Immunohistochemistry
At 48 h after ICH, mice (n=4) were transcardially perfused with ice-cold phosphate-buffered saline (PBS), pH 7.4, followed with ice-cold 4% paraformaldehyde in PBS (pH 7.4). Mouse brains were removed, immersed with 4% paraformaldehyde in PBS overnight at 4°C, and then cryoprotected in 30% sucrose in PBS at 4°C. Frozen coronal sections (20 μm) were prepared using a cryostat. The following antibodies were used: rabbit anti-MMP-9 polyclonal antibody (1:200; gift from Dr Robert Senior, Washington University, St Louis, MO, USA), fluorescein isothiocyanate-conjugated goat anti-rabbit immunoglobulin (IgG) (1:100; Jackson Immuno Research), and anti-grail fibrillary acidic protein (GFAP) antibody (1:100; Chemicon, USA). Negative control sections received identical treatment except for the primary antibody. Immunostainings were analyzed with a fluorescence microscope interfaced with a digital charge-coupled device camera and an image analysis system.
Measurement of Brain Edema
A standard wet—dry method was used for the measurement of total brain water content as an indicator of edema. At 48 h after ICH, mice were euthanized and brains were removed immediately, without fixation. A 2 mm thick coronal slice located at the center of the hemorrhagic lesion was cut then divided into ipsilateral and contralateral sides. Tissue samples were immediately weighed to obtain the wet weight (WW), dried in a dessicator oven at 100°C for 24 h, then weighed again to obtain the dry weight (DW). Total water contents (%) was calculated as follows: (WW−DW)/WW × 100. To assess the role of MMP-9 in the development of ICH brain edema in our mouse model, we studied four different sets of mice (n=8 per group): WT blood injected into WT brain, KO blood injected into KO brain, WT blood injected into KO brain, and KO blood injected into WT brain.
Primary Culture of Mouse Cortical Astrocytes
Primary astrocyte cultures were prepared from cerebral cortices of 1-day-old neonatal mouse after standard procedures. Briefly, dissociated cortical cells were suspended in Dulbecco's modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA, USA) containing 25 mmol/L glucose, 4 mmol/L glutamine, 1 mmol/L sodium pyruvate, and supplemented with 10% fetal bovine serum. Isolated cells were plated into 25 cm2 flasks at a density of one cortex per flask. Monolayers of type I astrocytes were obtained at 12 to 14 days after initial seeding. Astrocytes were dissociated by trypsinization and then reseeded into 6-, 12-, or 24-well plates or slide chambers at a density of 20,000 cells/cm2. After cells reached confluence (7 to 8 days post-seeding), cultures were switched to serum-free DMEM medium, and experiments were initiated 24 h later. In this system, more than 95% of the cells were type I astrocytes identified by GFAP and 4′-6-Diamidino-2-phenylindole (DAPI) double staining (data not shown). Purified (clinical grade endotoxin-free) human hemoglobin was obtained from Hemosol Inc., Canada. The combination of rodent or murine cell cultures to human hemoglobin has been previously validated (Wang et al, 1999, 2002).
Gelatin Zymography
Matrix metalloproteinase-2 and -9 levels in conditioned media were assessed with standard gelatin zymography. After treatments, the culture medium was collected and centrifuged at 14,000 r.p.m. for 5 mins at 4°C to remove cells and debris. The cleared medium was concentrated 10-fold using Microcon (Millipore, Billerca, MA, USA) with a 10 kDa pore diameter cutoff, then each sample was mixed with equal amounts of sodium dodecyl sulfate (SDS) sample buffer (Invitrogen) and electrophoresed on 10% SDS-polyacrylamide gels (Invitrogen) containing 1 mg/ml gelatin as the protease substrate. After electrophoresis, gels were placed in 2.7% Triton X-100 for 1 h to remove SDS, and then incubated for 20 h at 37°C in developing buffer (50 mmol/L Tris base, 40 mmol/L HCl, 200 mmol/L NaCl, 5 mmol/L CaCl2, and 0.2% Briji 35; Invitrogen) on a rotary shaker. After incubation, gels were stained in 30% methanol, 10% acetic acid, and 0.5% w/v Coomassie brilliant blue for 2 h followed by destaining. Gelatinolytic activity was manifested as horizontal white bands on a blue background. Matrix metalloproteinase bands were identified after standard techniques (Arai et al, 2003; Asahi et al, 2001a, 2001b; Wang et al, 2000), using molecular weight criteria and the loading of mouse MMP-2 and MMP-9 purchased from Chemicon. To further confirm that active forms of MMP-9 were involved, we also incubated mouse MMP-9 standards with trypsin to achieve proteolytic cleavage/activation. The multiple lower MW bands of MMP-9 that emerged were consistent with previously published results (Fisher et al, 2002). Relative gelatinolytic activity (MMP-2 and MMP-9) was quantified via measurement of optical density using the NIH image analysis software.
Statistical Analysis
Data were analyzed using analysis of variance followed by Tukey's honestly significant difference tests between individual groups. Data were expressed as mean±s.d. Differences at P<0.05 were considered significant.
Results
Matrix Metalloproteinase-9 is Upregulated in Peri-Hematomal Astrocytes
Brain samples were rapidly obtained in a clinical case of hemorrhagic stroke within 6 h of death. This patient suffered from a large spontaneous ICH in the right hemisphere. Computed tomography scans revealed a massive hematoma with leakage into the ventricles and gross midline shift (Figure 1A). Hematoxylin—eosin histopathology showed that at this relatively early timepoint, no neutrophil infiltration in peri-hematomal areas were detectable (Figure 1B). Brain sections from areas adjacent to the hematoma showed clear signs of upregulated MMP-9. Although multiple cell types were involved, the strongest signals appeared to come from GFAP-positive astrocytes and NeuN-positive neurons (Figure 1C).
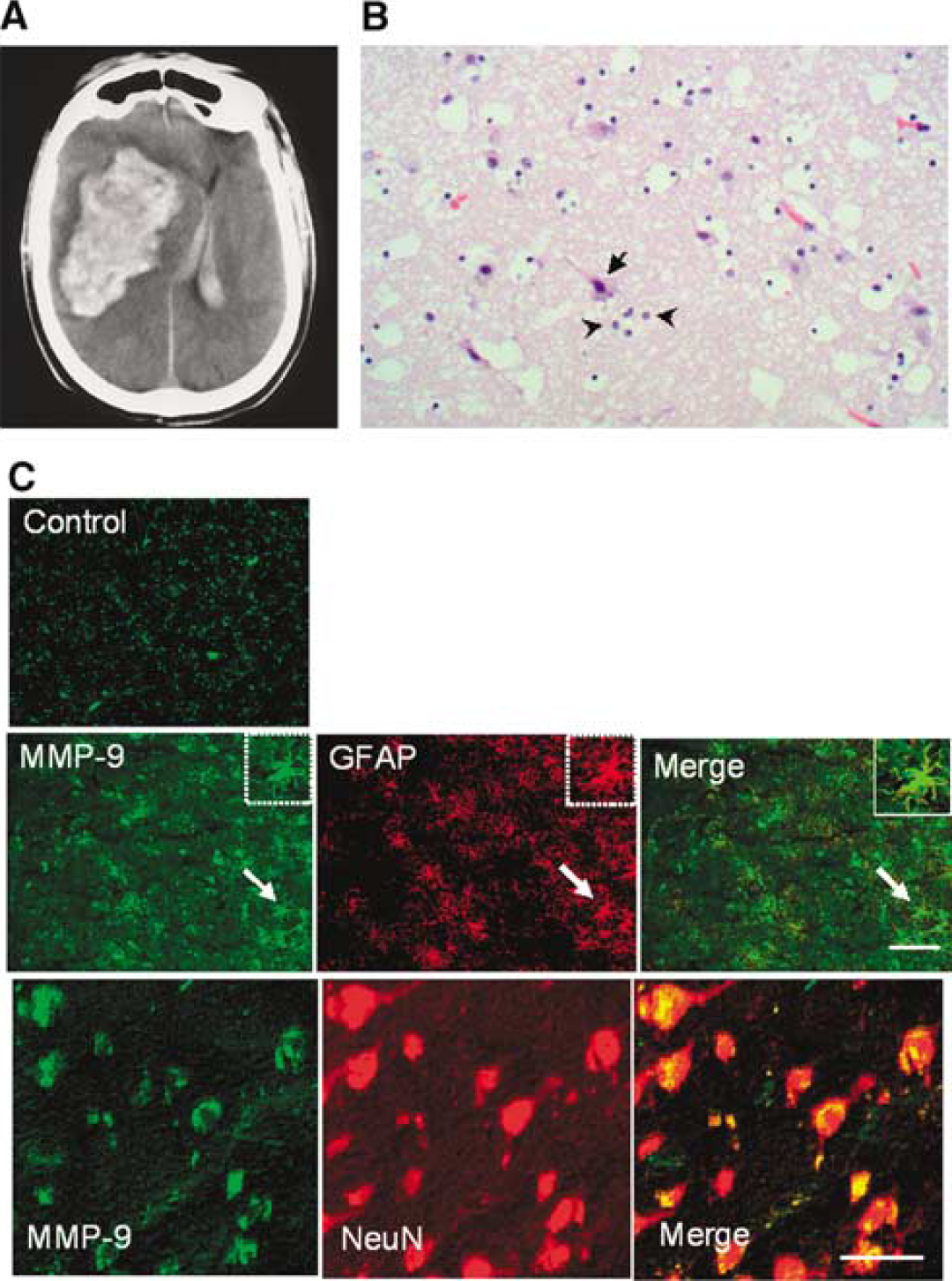
Clinical case of spontaneous hemorrhagic stroke in a 74 year old male. (
To further assess this idea of MMP-9 upregulation after ICH, we turned to an experimental mouse model where 20 μL of homologous blood was stereotactically injected into the striatum. Brain sections obtained at 48 h post-ICH showed that MMP-9 signals were primarily colocalized with GFAP-positive astrocytes (Figure 2A). Gel zymograms showed that MMP-9 levels were increased in hemorrhagic brain tissue when WT blood was injected into WT brain (Figure 2B). The MW position of the bands suggested that active forms of MMP-9 may be involved. No MMP-9 bands were detected when KO blood was injected into KO brain (Figure 2B).
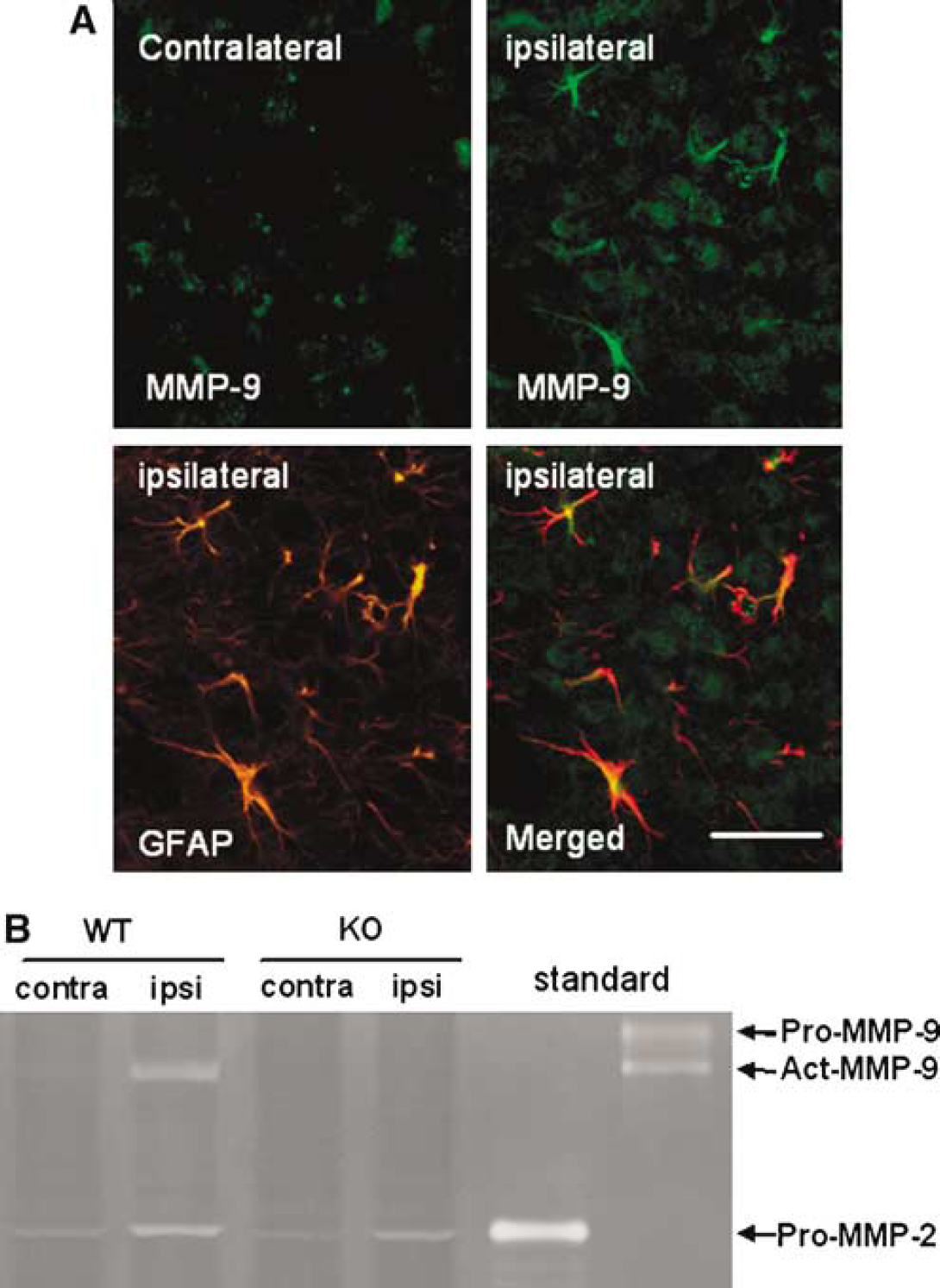
MMP-9 in a mouse model of ICH. (
Edema After Intracerebral Hemorrhage is Reduced by Matrix Metalloproteinase-9 Gene Knockout in Blood or Brain
To assess the role of MMP-9 in our model of ICH, we compared four groups of WT and MMP-9 KO mice: WT blood injected into WT brain, KO blood injected into KO brain, WT blood injected into KO brain, and KO blood injected into WT brain. At 48 h post-ICH, brains were removed and edema was quantified as the total water content using the wet/dry weight assay. In all groups, water content in contralateral hemisphere was within normal range (76% to 77%) compared with sham operated controls. In the ipsilateral hemisphere, edema was clearly induced when WT blood was injected into WT brain (79.5%±0.2%). However, when MMP-9 was missing from either blood or brain or both compartments, the elevation in ipsilateral brain water content was significantly ameliorated (Figure 3). Nevertheless, there were no differences between the three groups (KO blood into KO brain, WT blood into KO brain, or KO blood into WT brain), regardless of whether MMP-9 was deleted from blood or brain or both.
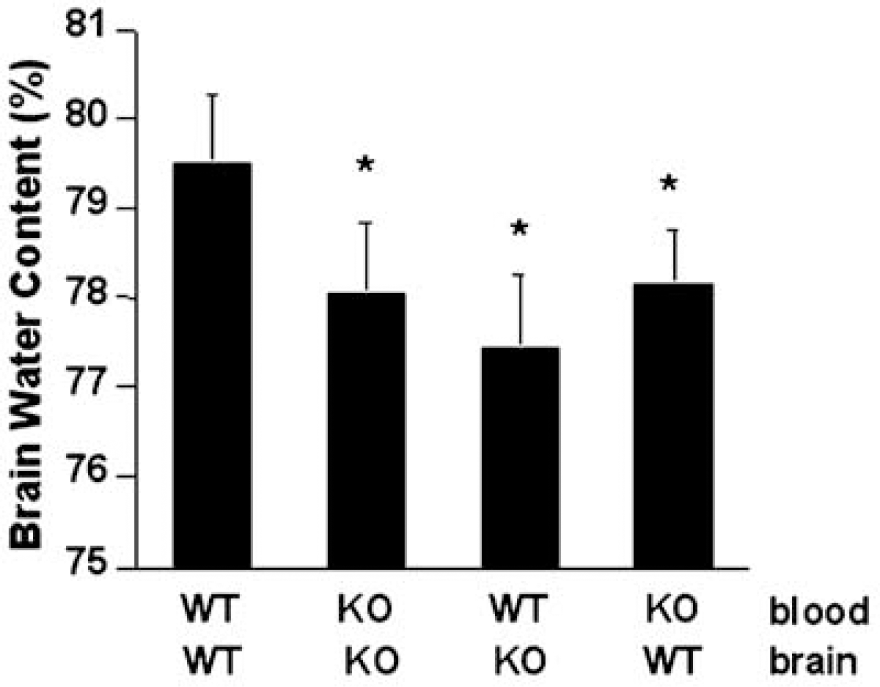
Brain edema in ipsilateral hemipsheres after ICH in the mouse model. WT blood injected into WT brain resulted in a 2% increase in water content, compared with normal values in contralateral brain (76% to 77%). Deletion of MMP-9 gene in either blood or brain or both compartments significantly reduced edema. ∗P<0.05 versus mice with WT blood injected into WT brains, n=8 per group.
Hemoglobin Induces Matrix Metalloproteinase-9 in Astrocytes via Oxidative Stress
To further assess the role of astrocytes as a source for MMP-9 after ICH, we turned to a cell culture system. Primary mouse cortical astrocytes were exposed to hemoglobin. Matrix metalloproteinase-9 levels in conditioned media were examined at 1 to 24 h. Hemoglobin (0.5 to 10 μmol/L) clearly upregulated MMP-9 (5- to 20-fold) in a concentration-dependent manner (Figure 4A). This response in MMP-9 was rapid, occurring within 1 h and was sustained for up to 24 h (Figure 4B). No clear changes in MMP-2 were detected in our system.
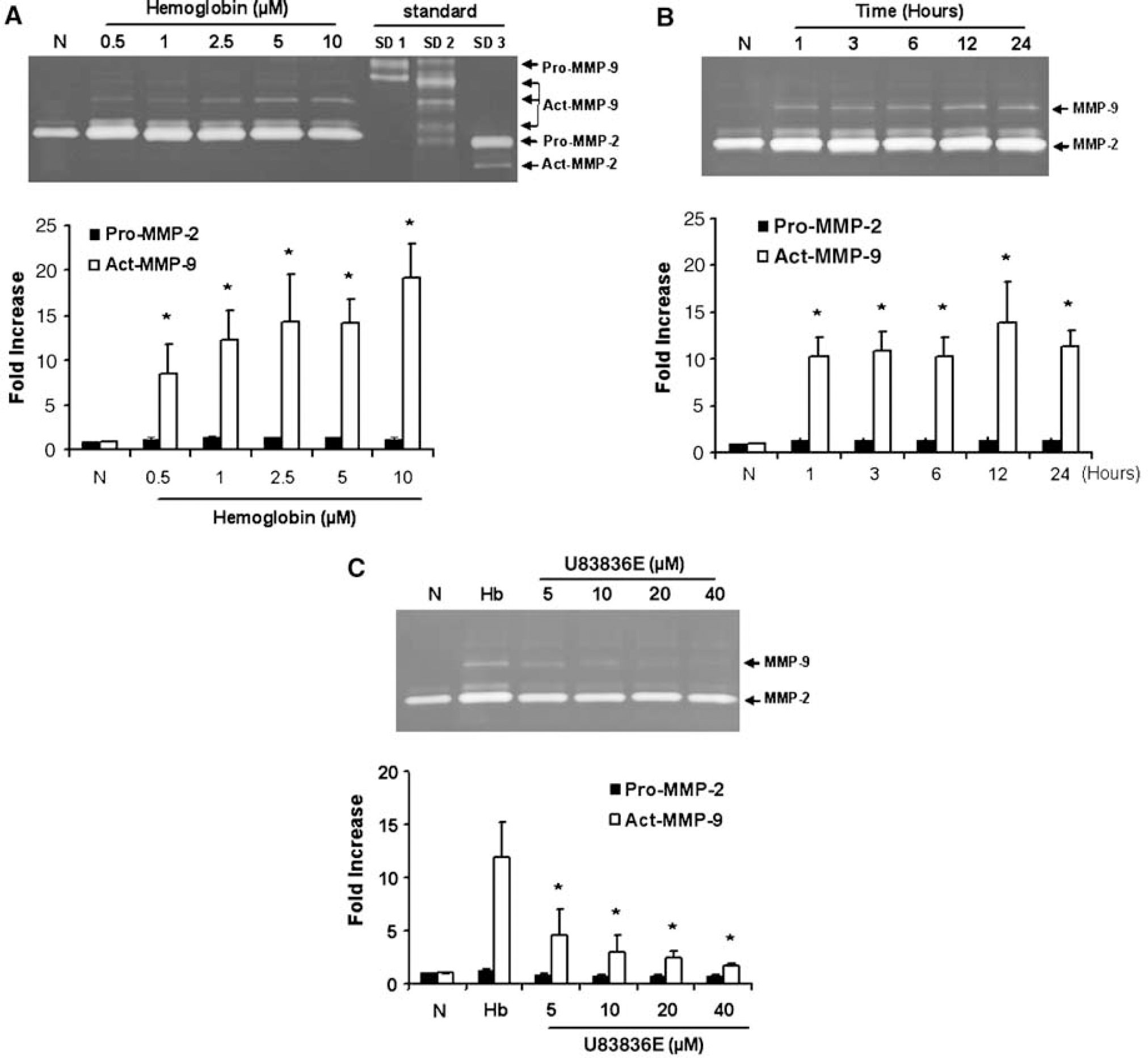
Induction of MMP-9 by hemoglobin in primary mouse astrocytes. Upper panels show representative gelatin zymograms. Lower panels show quantified densitometry. (
As MMP-9 can be induced by oxidative stress and hemoglobin is a potent trigger of oxidative stress, we sought to pharmacologically validate our results with a free radical scavenger. Co-incubating the astrocytes with the lazaroid compound U83836E (5 to 40 μmol/L) significantly reduced the hemoglobin-induced MMP-9 response (Figure 4C). No effects on MMP-2 were detected.
Discussion
Matrix metalloproteinases comprise a large family of zinc endopeptidases that can collectively modify almost all components of extracellular matrix in brain (Yong et al, 2001). The gelatinase MMP-9, in particular, has been implicated in central nervous system (CNS) injury (Lo et al, 2002; Rosenberg, 2002; Yong et al, 2001). Matrix metalloproteinase-9 is upregulated in human stroke (Clark et al, 1997) and many animal models of cerebral ischemia (Asahi et al, 2000, 2001a, 2001b; Fujimura et al, 1999; Gasche et al, 1999; Heo et al, 1999; Mun-Bryce and Rosenberg, 1998; Romanic et al, 1998; Rosenberg et al, 1996, 2001) and CNS trauma (Noble et al, 2002; Wang et al, 2000). By degrading homeostatic cell-matrix signaling, MMP-9 can induce anoikis-like neuronal death (Gu et al, 2002; Lee and Lo, 2004; Lee et al, 2004). Hence, MMP inhibitors have been shown to be neuroprotective in animal models of CNS ischemia, hemorrhage and trauma (Asahi et al, 2000; Gu et al, 2005; Jiang et al, 2001; Lee et al, 2004; Romanic et al, 1998; Wells et al, 2003). By degrading the integrity of extracellular matrix, especially within neurovascular basal lamina, MMP-9 can also disrupt the BBB thus causing edema and hemorrhagic conversion in models of cerebral ischemia (Aoki et al, 2002; Asahi et al, 2001a, 2001b; del Zoppo and Mabuchi, 2003; Gasche et al, 2001; Sumii and Lo, 2002). In the present study, we found that MMP-9 was increased in reactive astrocytes in a clinical case study, as well as a mouse model of ICH. Deletion of the MMP-9 gene significantly ameliorated edema in mouse ICH. And our astrocyte cultures showed that hemoglobin potently and rapidly upregulates MMP-9 in these cells, consistent with the in vivo findings. Taken together, the combination of clinical, in vivo and cell culture evidence suggest that MMP-9 is an important mediator of brain edema in ICH.
An important point to be discussed here involves the opposite results obtained by Zhang and co-workers recently, where hemorrhagic severity and mortality was increased in MMP-9 KO mice compared to WT mice (Tang et al, 2004). In that study, bacterial collagenase was injected into mouse striatum to damage collagen and laminin substrates in the microvascular basal lamina, thus causing hemorrhage. The advantage of the collagenase model is that it is highly reproducible, with basically spherical hematoma developing over 3 to 4 h. Furthermore, it replicates the primary vascular trauma and rupture that occurs in clinical ICH. However, the caveat with this model is the presence of bacterial collagenase, which may enhance inflammatory cascades and modify ICH pathology in subtle ways. In our present study, we simply infused homologous blood into the mouse striatum. The drawback here is that lesions tend to be more variable, and there is no primary vascular wall damage. But the advantage is that we do not have potentially deleterious agents other than blood in the model system. In contrast to the Tang et al study, we find that MMP-9 gene KO was beneficial by significantly ameliorating brain edema. Although we cannot unequivocally determine the reasons for these different results, a critical factor may ultimately involve the ICH model used.
The finding that deletion of MMP-9 reduced edema in our mouse ICH models may not be surprising per se. After all, we have previously shown that BBB damage was reduced in MMP-9 KO mice after cerebral ischemia (Asahi et al, 2001a, 2001b). What was somewhat unexpected however, was that it did not seem to matter whether we deleted MMP-9 from the infused blood or the recipient brain. After ICH, a whole host of activated blood components and cytokines extravasate into brain parenchyma. For MMPs, this may be a reasonable source for the pathogenesis of edema, since activated leukocytes can produce large amounts of this protease (Gidday et al, 2005; Justicia et al, 2003). Matrix metalloproteinase-9 levels are low in normal brain but relatively higher in blood anyway. So when blood-borne MMP-9 diffuses into brain after ICH, neurovascular proteolysis may ensue with the resulting effect of BBB leakage and spread of edema beyond the primary confines of the initial hemorrhagic lesion. Additionally, our data suggest that brain is also an important source of MMP-9 in ICH. Perihematomal astrocytes are MMP-9-positive and deletion of only brain MMP-9 also reduced edema. A limitation here is that we cannot determine whether blood or brain MMP-9 plays a more quantitatively important role. We were also unable to detect any additive effect of brain versus blood MMP-9 deletion. It is likely that there are close interactions and signaling feedback loops between blood and tissue as the ICH evolves. Nevertheless, this overall link between MMP-9 and hemorrhagic edema is consistent with emerging clinical findings. In humans, increased expression of plasma MMP-9 within the first 24 h of ICH has been reported (Alvarez-Sabin et al, 2004). Moreover, plasma MMP-9 measured at arrival among ICH patients has been shown to be correlated with peri-hematomal edema volume measured on CT scans, as well as progressive hematoma enlargement over the next 48 h (Abilleira et al, 2003).
Taken together, our clinical, animal and cell culture data suggest a significant role for astrocytic MMP-9. However, there are several caveats to keep in mind. First, we acknowledge that our in vivo mouse model does not fully replicate the clinical condition of ICH. Translating these experimental findings into a clinical context should be performed with caution. A second limitation here is that we cannot unequivocally define the cellular mechanisms involved. It has been suggested that both intracellular (presumably cytotoxic) and extracellular (presumably vasogenic) edema occurs after ICH. Which component comprises the rate-limiting step here is unknown because we measure edema using a relatively gross end point, that is, total brain water content. Nevertheless, it is generally accepted that the wet/dry weight assay is most reflective of overall edema in animal models. Furthermore, it is acknowledged that MMP-9 signals can also be detected in neurons, endothelium, and microglia as well. To examine cell cultures in all of these would be outside the scope of our study. We only focus on astrocytes but mechanisms of response in other cells remain to be fully dissected. Thirdly, although our pharmacologic data in cell culture suggest that oxidative stress from hemoglobin may be the trigger of MMP-9 in astrocytes, we do not directly measure free radical formation nor do we have evidence for this pathway in vivo. However, others have shown that oxidative stress is involved in ICH (Peeling et al, 2001a, 2001b), so our cell culture data is at least consistent with these other findings. A fourth caveat involves the question of timing. How rapidly does MMP-9 go up, and how does this compare with the time course of edema progression? In the clinical case, MMP-9 was detected within 6 h, whereas our mouse model data is limited to 48 h measurements. In animal models of cerebral ischemia and trauma, MMP-9 can be rapidly elevated within just a few hours. Clinical edema may peak by 24 to 48 h. Although there is no direct proof, the timing suggests that MMPs may precede the development of edema. A fifth question is related to the significance of edema in our admittedly artificial mouse model. Are the rather small changes in water content, in the range of 2% to 3%, of any biologic significance? A review of the literature shows that the wet/dry brain weight assay of water content typically yields numbers in this range for rodent models, so our data are consistent with known data. Even with massive brain swelling in rodents, wet/dry weight assays reveal water content elevations of only a few percent. But how do these changes affect neuronal cell death per se? Others have shown that neuronal apoptosis can be reduced by MMP inhibitors (Gu et al, 2002, 2005), so there is relevance to parenchymal damage. Of course, the potential confound here then is that we may not be able to say whether reductions in edema are directly caused by MMP-9 gene KO or whether this is a secondary effect of decreased neuronal injury. Ultimately, future studies will have to carefully examine how these improvements in edema relate to clinical outcomes. A sixth caveat is that our study is only focused on MMP-9, whereas others have shown that many others within the large MMP family may be dysregulated after ICH (Power et al, 2003). Further studies are needed to dissect the network effects of these other proteases when a particular member is deleted or blocked. And finally, we acknowledge that our focus here on acute stages of brain injury may be oversimplified. Whereas neurovascular proteolysis via MMPs may be damaging in the acute phase, emerging data now suggest that during delayed phases of stroke, MMPs may mediate potentially beneficial pathways of neurogenesis and plasticity (Lee et al, 2006; Zhao et al, 2006). How MMPs may be involved in the resolution and recovery phase after ICH needs to be carefully examined.
In conclusion, our data suggest that although blood itself may be an obvious source of damaging proteases, endogenous astrocytic MMP-9 responses that are triggered by oxidative stress after ICH may also play a significant role in the pathogenesis of edema. Further studies are warranted to test whether specific blockade of MMPs or blunting of astrocytic or other parenchymal reactions to injury, perhaps via antioxidants, may offer new ways of ameliorating acute edema after ICH.
Footnotes
Acknowledgements
We will always be grateful to Dr Robert Senior of Washington University St Louis for kindly allowing us to use his MMP-9 mice and reagents for so many years. We thank Dr Arantxa Ortega-Aznar for help with the neuropathology.