
Abstract
Central neurons are extremely vulnerable to hypoxic/ischemic insult, which is a major cause of neurologic morbidity and mortality as a consequence of neuronal dysfunction and death. Our recent work has shown that δ-opioid receptor (DOR) is neuroprotective against hypoxic and excitotoxic stress, although the underlying mechanisms remain unclear. Because hypoxia/ischemia disrupts ionic homeostasis with an increase in extracellular K+, which plays a role in neuronal death, we asked whether DOR activation preserves K+ homeostasis during hypoxic/ischemic stress. To test this hypothesis, extracellular recordings with K+-sensitive microelectrodes were performed in mouse cortical slices under anoxia or oxygen–glucose deprivation (OGD). The main findings in this study are that (1) DOR activation with [D-Ala2, D-Leu5]-enkephalinamide attenuated the anoxia- and OGD-induced increase in extracellular K+ and decrease in DC potential in cortical slices; (2) DOR inhibition with naltrindole, a DOR antagonist, completely abolished the DOR-mediated prevention of increase in extracellular K+ and decrease in DC potential; (3) inhibition of protein kinase A (PKA) with N-(2-[p-bromocinnamylamino]-ethyl)-5-isoquinolinesulfonamide dihydrochloride had no effect on the DOR protection; and (4) inhibition of protein kinase C (PKC) with chelerythrine chloride reduced the DOR protection, whereas the PKC activator (phorbol 12-myristate 13-acetate) mimicked the effect of DOR activation on K+ homeostasis. These data suggest that activation of DOR protects the cortex against anoxia- or ODG-induced derangement of potassium homeostasis, and this protection occurs via a PKC-dependent and PKA-independent pathway. We conclude that an important aspect of DOR-mediated neuroprotection is its early action against derangement of K+ homeostasis during anoxia or ischemia.
Introduction
Neurons in the central nervous system are extremely vulnerable to hypoxic/ischemic stress, which in turn is a major cause of neurologic morbidity and mortality as a consequence of neuronal dysfunction and death. Therapeutic strategies directed against such injury have had limited success, an indication of our limited understanding of the mechanisms that lead to hypoxic/ischemic injury and the factors that regulate neuronal adaptation to environmental stress.
Endogenous opioids are believed to be inhibitory neurotransmitters with three major classes of receptors (δ, μ, k) widely distributed throughout the central nervous system (Mansour et al, 1987; Hiller and Fan, 1996; Xia and Haddad, 2001). Of all the three major opioid receptors, δ-opioid receptor (DOR) has been proven more sensitive than others to stressful stimuli, such as hypoxia and ischemia (Mayfield et al, 1996; Boutin et al, 1999; Ma et al, 2005; Zhang et al, 2006). Our previous work has shown that activation of DOR is neuroprotective against hypoxic and excitotoxic stress (Zhang et al, 2000, 2002, 2006; Ma et al, 2005), whereas inhibition of DOR induces major injury in cortical neurons not only during hypoxia but also in normoxic conditions (Zhang et al, 2002, 2006). Others have also shown similar findings regarding neuroprotection via DOR (Borlongan et al, 2004; Lim et al, 2004). These observations suggest that DOR serves a neuroprotective function in the cortex and may be one of the factors that determine neuronal survival during hypoxic/ischemic stress. The mechanisms underlying DOR-mediated protection, however, remain unclear.
Because neuronal responses to stress vary depending on the duration of oxygen deprivation and survival time after hypoxia (Nieber, 1999; Lipton, 1999), DOR signals may trigger, depending on stress duration, different mechanisms at multiple levels to preserve proper functioning of surviving neurons. Short-term hypoxia to central neurons causes an immediate loss of ionic homeostasis, which is largely based on the functional response of inherent membrane proteins to the stress. Conversely, long-term hypoxia leads to major alterations in gene expression of intracellular elements, especially death/survival signal systems (Nieber, 1999; Lipton, 1999; Ma et al, 2005). δ-Opioid receptor-mediated responses may occur at various levels to protect neurons from short- or long-term stress. For example, it is possible that DOR signals maintain ionic homeostasis in the initial stage of hypoxia, while during prolonged hypoxia DOR then signals upregulation of the expression and function of benefit-signaling molecules and downregulates those involved in cell injury or death. Indeed, our recent studies show that DOR performs its neuroprotective role by enhancing the intracellular activity of the G protein–protein kinase C (PKC)–phosophorylated extracellular signal-regulated kinase (pERK)–B-cell lymphoma 2 (Bcl2) pathway and suppressing the phosphorylated p38 and cytochrome c death signals during long-term hypoxia (Ma et al, 2005). There is no information available, however, as to whether DOR regulates ionic homeostasis in response to hypoxic/ischemic stress.
The brain possesses homeostatic mechanisms that maintain constant ionic concentration in the cerebrospinal fluid and neurons (Hansen, 1985; Kofuji and Newman, 2004). There is only a transient and small range of fluctuation in ionic concentrations in the intracellular and extracellular compartments of neurons under physiologic conditions. Such homeostatic mechanisms can be disrupted in certain pathophysiologic conditions. For example, hypoxia or oxygen–glucose deprivation (OGD) (e.g., ischemia) can produce considerably longer or sustained changes in ionic concentrations characterized by enhanced K+ efflux and Na+, Ca2+, and Cl− influx (Sick et al, 1982; Hansen, 1985; Jiang and Haddad, 1991; Müller and Somjen, 2000a, b; Galeffi et al, 2004; Martinez-Sanchez et al, 2004). The loss of ionic gradients across the membrane is believed to be central to anoxia-induced depolarization, which may lead to neuronal death. In particular, the steep increase in extracellular K+ has been shown to be closely associated with anoxia-induced depolarization (Hansen, 1985). Several lines of evidence have suggested that K+ fluxes play an important role in the neuronal death that occurs in various conditions (Yu et al, 1997; Wei et al, 2003; Liu et al, 2003). Cultured mouse cortical neurons undergoing apoptotic death because of serum deprivation or staurosporine treatment exhibited an increase in extracellular K+ (Yu et al, 1997). Blockade of K+ efflux has been shown to attenuate hypoxia- and ischemia-induced neuronal death (Huang et al, 2001; Wei et al, 2003; Liu et al, 2003), suggesting that inhibition of K+ fluxes and maintaining cellular K+ homeostasis may be of therapeutic benefit in the treatment of stroke and related neurodegenerative conditions (Wei et al, 2003; Liu et al, 2003). This suggests that delaying/reducing the magnitude of the disruption of K+ homeostasis during anoxiainduced depolarization may protect neurons from anoxic injury. In support of this is the observation that some species like turtles have a remarkably higher endurance for anoxia than mammals, and exhibit very little increase in neuronal K+ efflux under anoxia as compared with mammalian neurons (Sick et al, 1982). Interestingly, we recently observed that the turtle brain has a much higher DOR density than rat brain (Xia and Haddad, 2001). This unique property may play a role in, at least partially, the regulation of K+ homeostasis in the turtle brain under hypoxic/excitotoxic stress.
Based on our previous work, we hypothesize that DOR may play its neuroprotective role by modulating anoxia-induced K+ derangement during the early stages of hypoxia. In the present study, we aimed to determine (1) whether activation of DOR inhibits anoxia-induced increase in extracellular potassium; (2) if so, whether DOR's effect can be blocked by its antagonist; and (3) what are the main mechanisms of the intracellular signaling.
Materials and Methods
Animals
Male C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA, USA). All animal procedures were performed in accordance with the guidelines of the Animal Care and Use Committee of Yale University School of Medicine, which is accredited by the American Association for Accreditation for Laboratory Animal Care.
Slice Preparation
Cortical slices were prepared from 24- to 33-day-old mice. The mice were decapitated under inhalational anesthesia with halothane and the brain was rapidly removed from the skull and placed in chilled artificial cerebrospinal fluid (ACSF) that had been saturated with carbogen (95% O2, 5% CO2) for 1 to 2 mins. Transverse cortical slices (400 μm) were cut on a vibrotome containing carbogen-saturated ice-cold ACSF. Slices were then transferred to an incubation holder placed in a beaker containing 150 mL ACSF vigorously bubbled with carbogen at ~ 35°C. Artificial cerebrospinal fluid consisted of (in mmol/L) NaCl 125, KCl 3.1, NaHCO3 26, CaCl2 2.4, MgSO4 1.3, NaH2PO4 1.25, and D-glucose 10 at pH 7.4. After an equilibration period of at least 90 mins in carbogen-saturated ACSF at ~ 35°C, slices were used for recording.
Induction of Anoxia or Oxygen–Glucose Deprivation in Cortical Slices
A slice was transferred to the recording chamber (Model RC-22C, Warner Instrument Co, Hamden, CT, USA) after equilibration with carbogen-saturated ACSF. The chamber was perfused with carbogen-saturated ACSF (35.5°C ± 0.5°C) with a flow rate of 2 to 3 mL/min. Slices were completely submerged 0.5 to 1 mm below the ACSF surface in the tissue chamber and kept under normoxic conditions for at least 15 mins at ~35.5°C before experimental measurements were taken.
Anoxia was induced by switching from the control superfusate (95% O2, 5% CO2) to one continuously aerated with 95% N2 and 5% CO2. Each slice was subjected to a single period of anoxia that continued for approximately 1.5 mins after the onset of anoxic depolarization (as assessed by a rapid increase in extracellular [K+] and negative shift of extracellular DC potential), or for a period of 20 mins if anoxic depolarization did not occur.
Oxygen–glucose deprivation was produced by exposing slices to D-glucose-deficient ACSF (D-glucose was substituted by equimolar mannitol) continuously aerated with 95% N2 and 5% CO2, as performed in anoxic induction.
Measurements of Extracellular Potassium and DC Potential
Extracellular K+ concentrations ([K+]e) were measured using K+-sensitive microelectrodes. K+-sensitive microelectrodes were prepared according to Ammann et al (1987). In brief, electrodes were pulled from standard wall borosilicate glass capillaries without filament. The electrodes were silanized by exposure to N-(trimethysilyl)dimethylamine (Fluka 41420) and baked at approximately 180°C for at least 2 h. The microelectrode tips were then broken back to approximately 5 μm. Afterwards, the internal filling solution (10 mmol/L KCl) was injected from the back into the electrode using a fine hand-drawn plastic capillary attached to a syringe. A column of optimized membrane phase (with height approximately 2 mm) was sucked into the microelectrode tips. The membrane phase contained 5% wt valinomycin (Fluka 94675), 2% potassium terrakis(p-chlorophenyl)borate (Fluka 60591), and 93% 2,3-dimethylnitrobenzene (Fluka 40870). The reference electrode was a Ag/AgCl bridge electrode embedded in 2% agar in 3 mol/L KCl. Calibrations were performed by detecting the responses generated in KCl solutions (1, 3.1, 5, 10, 20, 40, 80, 100, 160 mmol/L) in triplicate. For each concentration, the average of voltage changes in three separate tests was used as the final voltage change. Above this range, electrode response was near ideal, showing a logarithmic relationship to [K+]. Average slope of K+-sensitive electrodes was 55.08±0.21 mV per log 10 unit increase in [K+] at 25°C (n = 81).
Electrical signals were monitored on an oscilloscope and recorded by a DC amplifier (Model IE-210, LPF 200, Warner Instrument Co., Hamden, CT, USA) and digitized by an Axon minidigitizer acquisition system (Model miniDigi 1A, Axon Instruments, Union City, CA, USA) at a sampling rate of 100 Hz. The electrical signal of a K+ sensitive electrode and extracellular DC potential were recorded either separately or simultaneously with two electrodes with the tips positioned very closely. Because most of our experiments were either K+ potential or extracellular DC potential records, the data were not corrected by subtracting DC potential from K+ potential.
The following parameters were derived to assess K+ homeostasis: (1) the latency of anoxia- or OGD-induced [K+]e increase (Latency), which was defined as a time period from the beginning of anoxia/OGD to the time point when anoxia/OGD induced a K+ electrode voltage change greater than 1 mV; (2) maximal [K+]e ([K+]max), which was the peak change in extracellular potassium concentration induced by anoxia/OGD; (3) the rate of rise of [K+]e from latency level to peak (noted as latency–max rate of [K+]e), which was the ratio of the [K+]max and the time interval from the latency to the time point of anoxia/OGD-induced maximal [K+]e change; (4) the undershooting of [K+]e (undershoot), which referred to the minimal value of [K+]e during reoxygenation; and (5) anoxia-induced negative shift of extracellular DC potential (ΔDC).
After recording of a stable baseline for at least 5 mins, the slices were subject to experimental treatments (anoxia or OGD with/without drug administration). The electro-physiological recordings were continuously performed from the baseline to the time when a steady recovery was seen during reoxygenation after the experimental treatments. Because there were major differences in different groups in terms of experimental treatments (e.g., larger changes in [K+]e in OGD than those of anoxia alone; DOR agonist versus antagonist), the duration of each experiment and the subsequent recovery periods were different among various groups.
Because K+ homeostasis, including the resting and hypoxia-altered levels of extracellular K+ in the brain slices, has been well described previously (Jiang and Haddad, 1991; Roberts, 1993; Müller and Somjen, 2000a, b ), we focused, in this work, on relative changes in extracellular K+ in cortical slices under stress with/without DOR activation with all recordings being performed in the exact same conditions.
Chemicals and Reagents
N-(trimethysilyl)dimethylamine (Fluka 41420), valinomycin (Fluka 94675), potassium terrakis(p-chlorophenyl) borate (Fluka 60591), 2,3-dimethylnitrobenzene (Fluka 40870), [D-Ala2,D-Leu5]-enkephalinamide (DADLE), a selective DOR agonist, naltrindole hydrochloride (NTI), a highly selective DOR antagonist, phorbol 12-myristate 13-acetate (PMA, PKC activator), N-(2-[p-bromocinnamylamino]-ethyl)-5-isoquinolinesulfonamide dihydrochloride (H89, protein kinase A (PKA) inhibitor), and mannitol were purchased from Sigma Chemicals Co. (St Louis, MO, USA). Chelerythrine chloride (CHEL, a PKC inhibitor) was purchased from Alexis Co. (San Diego, CA, USA). DADLE, NTI, and CHEL were prepared in high concentrations in ACSF as stock solutions, and diluted with ACSF to final concentration before experiments. Phorbol 12-myristate 13-acetate and H89 were first dissolved in dimethyl sulfoxide at a high concentration as stock solutions, and diluted with ACSF to final concentration before experiments to a concentration of no more than 0.1% dimethyl sulfoxide in the final solution. For the latter, control solution also contained 0.1% dimethyl sulfoxide.
Drug Administration
Drugs were applied to cortical slices by switching from control superfusate to one containing drugs, which was controlled by a six-channel valve-controlled solution perfusion system (Model VC-6, Warner Instrument Co, Hamden, CT, USA). All drugs were perfused for 20 mins before induction of anoxia or OGD, and continued to the end of anoxic induction/OGD. In NTI and DADLE co-perfusion experiments, NTI was perfused for 10 mins before DADLE, and then continued with DADLE to the end of anoxia induction.
Statistics
All data are expressed as mean ± s.e.m. and the number of experiments (n) referred to the number of slices investigated. To ensure the independence of data, no more than three slices from the same mouse were used in the same experiments. To assess the significance, two-tailed, unpaired Student's t-test was used for comparison of two experimental groups, and one-way analysis of variance test followed by Newman-Keuls test was used for multiple pairwise tests. Changes were identified as significant if probability value was < 0.05.
Results
Anoxia or OGD Induced a Major Change in [K+]e and DC Potential
The baseline [K+]e was found to be around 3.1 mmol/L at rest in the mouse cortical slices and stable for at least 90 mins when equilibrated in ACSF. Anoxia caused a slow increase in [K+]e in the first 5 to 6 mins, which was first described as phase 1 by Hansen (1978) during ischemia in the rat cortex. When [K+]e reached a threshold level of 5.13±0.21 mmol/L (n = 17), a rapid and large increase in [K+]e accompanied by a negative shift in extracellular DC potential was observed (Figure 1A). This change corresponded to the anoxic depolarization (phase 2) (Hansen, 1978) (Figure 1A). Within 20 secs, [K+]e reached a peak value (40.88±3.22 mmol/L, n = 17), then gradually decreased. On return to normoxia, [K+]e fell rapidly, and often undershot to below the basal levels by 1.54±0.13 mmol/L (n = 17) before returning to the preanoxia level. Oxygen-glucose deprivation evoked a similar change in [K+]e with larger amplitude than anoxia alone (Figure 1B). Compared with anoxia alone, OGD induced a larger increase in [K+]max (34.85%), rate of rise of [K+]e from latency to peak (20.95%), and undershoot (55.84%), indicating that OGD induced a greater derangement of potassium homeostasis in cortical slices. Our data are consistent with those in other reports (Hansen, 1978; Jiang and Haddad, 1991).
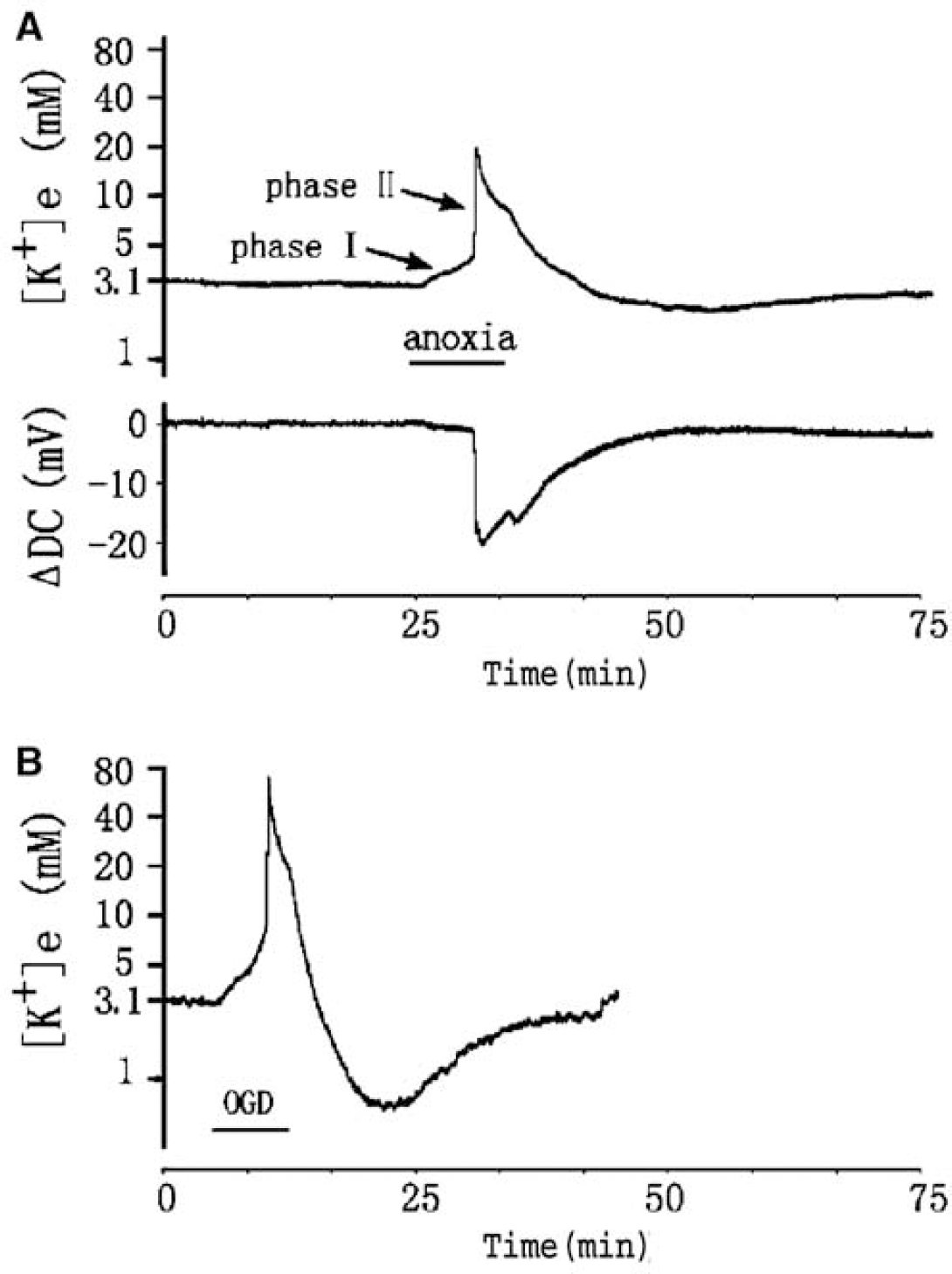
Anoxia- and OGD-induced changes in extracellular potassium concentration ([K+]e) and extracellular DC potential (ΔDC) in mouse cortical slice. (
DOR Activation Attenuated Anoxia/OGD-Induced Change in [K+]e and DC Potential
To determine whether DOR activation would prevent the K+ derangement during acute hypoxic stress, DOR agonist DADLE (Moore et al, 1994; Yao et al, 2003; Zhang et al, 2000, 2002; Ma et al, 2005) was applied 20 mins before anoxia and maintained during the stress. DADLE in 1 μmol/L (DADLE1) (n = 9) significantly delayed the response of cortical slices to anoxia, but did not reduce the peak increase in [K+]e and the rate of rise of [K+]e to peak in comparison with control (Figure 2). At a concentration of 10 μmol/L DADLE (DADLE10) (n = 17), the latency of the response to anoxia was greatly prolonged, similar to treatment with 1 μmol/L DADLE. The maximal increase in [K+]e was largely attenuated by 41.7% (n = 17, P < 0.01) and the rate of rise of [K+]e from latency to peak was also significantly reduced (0.060±0.010 mmol/L/sec versus control 0.148±0.026 mmol/L/sec, n = 17, P < 0.05). During reoxygenation, the maximum undershoot of [K+]e was not affected by DADLE at both 1 and 10 μmol/L (Figure 2). We also tested the effect of DADLE on the anoxia-induced change in extracellular DC potential in cortical slices. Anoxia induced a negative shift of DC potential to −18.38±0.98 mV (n = 22), whereas application of DADLE (10 μmol/L) attenuated this negative shift to −12.36±1.63 mV (n =14, P =0.0019) (Figure 3).
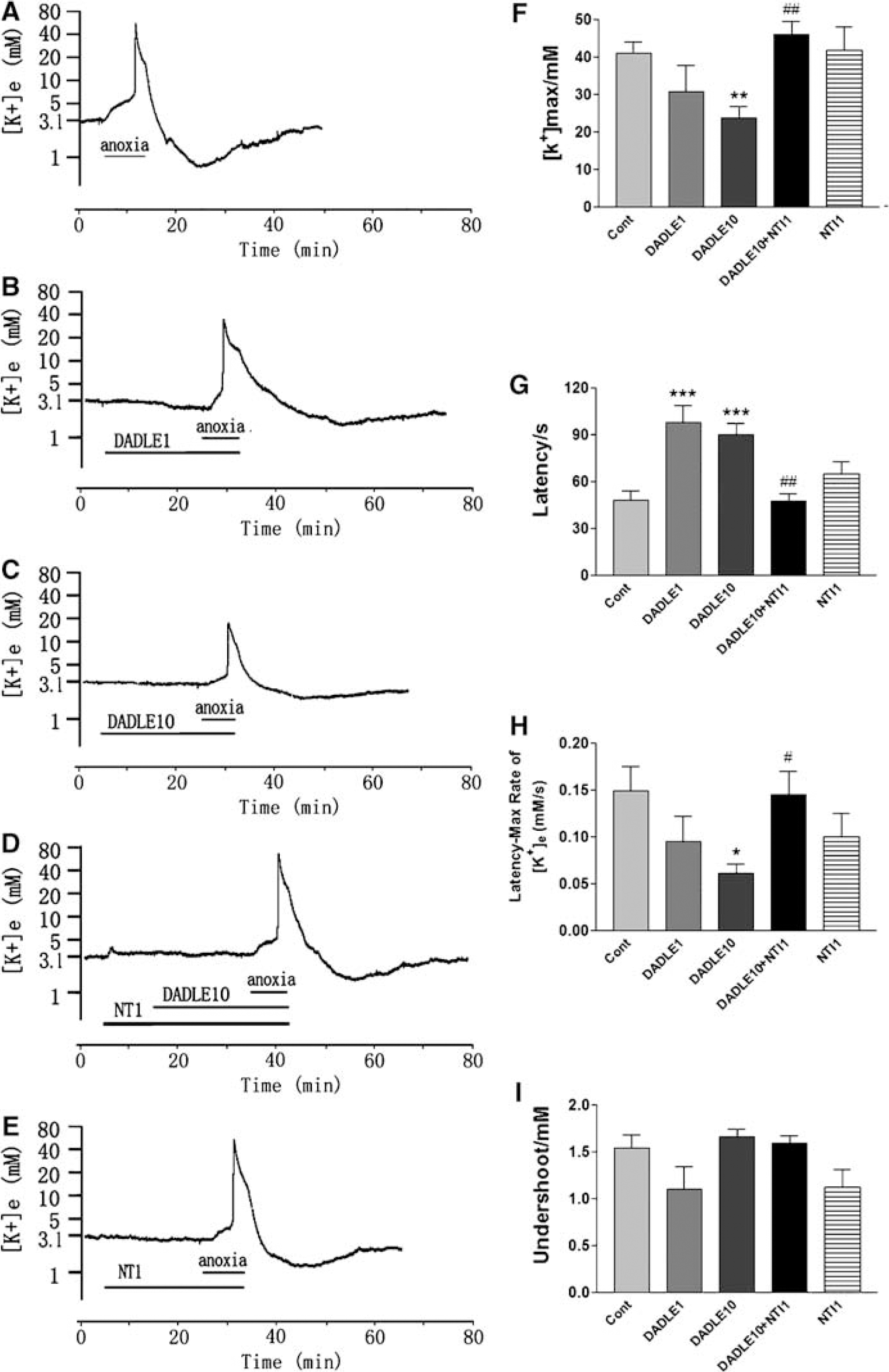
δ-Opioid receptor activation attenuates anoxia-induced increase in extracellular potassium. Trace recordings of (
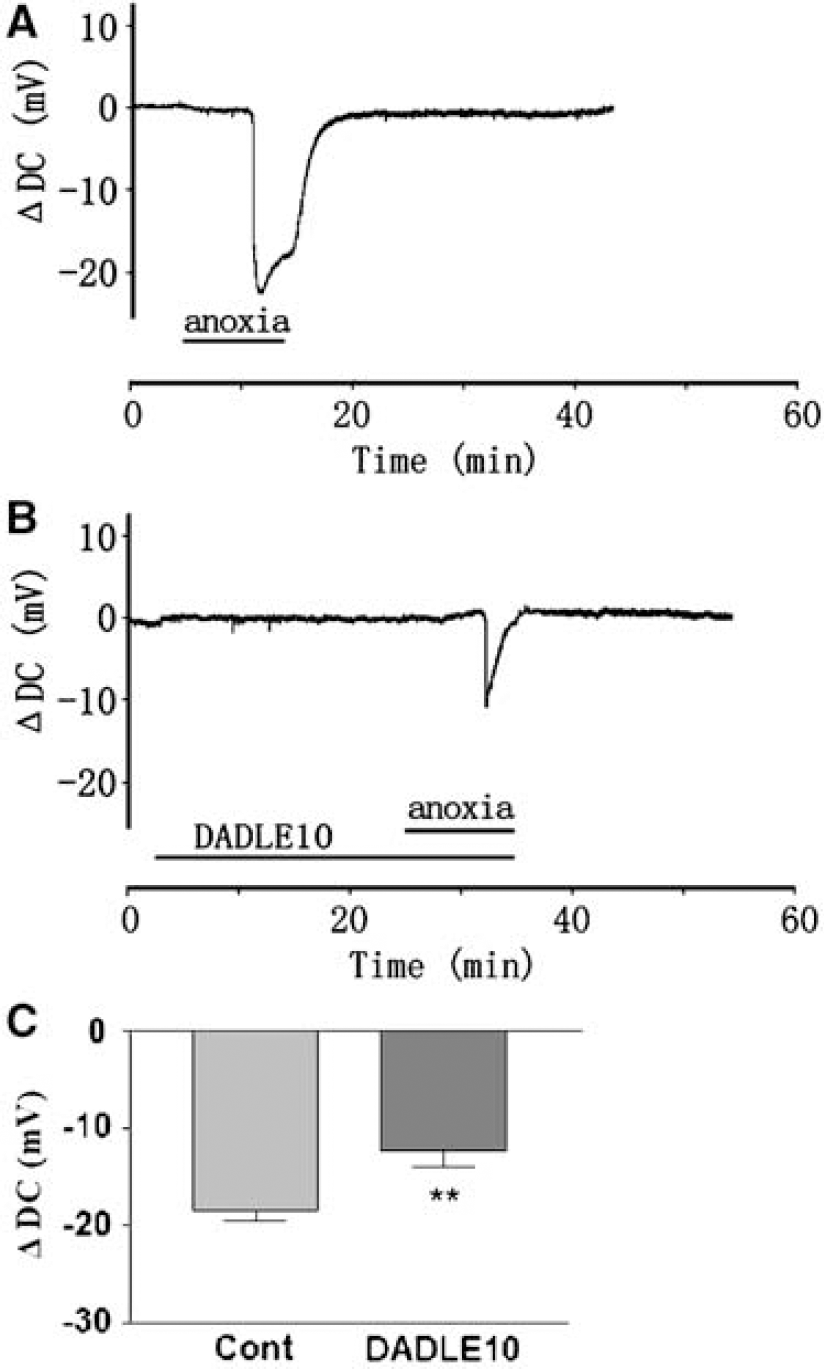
Effect of DOR activation on anoxia-induced decrease in extracellular DC potential (ΔDC). (
All the above results suggest that DOR activation had a preventive effect on anoxia-induced change in [K+]e and DC potential in cortical slices.
Because glucose levels have been shown to alter the rate of K+ efflux during ischemia in the rat brain (Hansen, 1978; Roberts, 1993), and because in clinical situations such as stroke concurrent lack of both oxygen and glucose often occurs, we extended our studies from oxygen deprivation alone to examining the effect of DOR activation on cortical slices deprived of both oxygen and glucose (OGD), to mimic ischemia. Oxygen-glucose deprivation induced a maximal increase in [K+]e to 55.10±6.75 mmol/L (n = 14) with a latency of approximately 66 secs, and a rate of rise of 0.179±0.033 mmol/L/sec from latency to peak; the undershoot of [K+]e reached 0.68±0.12 mmol/L on returning from anoxia to normoxia. Applying DADLE (10 μmol/L) to the OGD slices significantly delayed the OGD-induced response. The maximal [K+]e was decreased to 35.56±3.04 mmol/L (P =0.0058, n = 21), the rate of rise of [K+]e to peak was also significantly reduced (0.122±0.018 mmol/L/sec, P =0.0157), and the undershoot of [K+]e was also attenuated (1.03±0.11 mmol/L, P =0.039) (Figure 4). These results indicate that DADLE had a similar effect on both anoxia- and OGD-induced K+ derangement in mouse cortical slices.
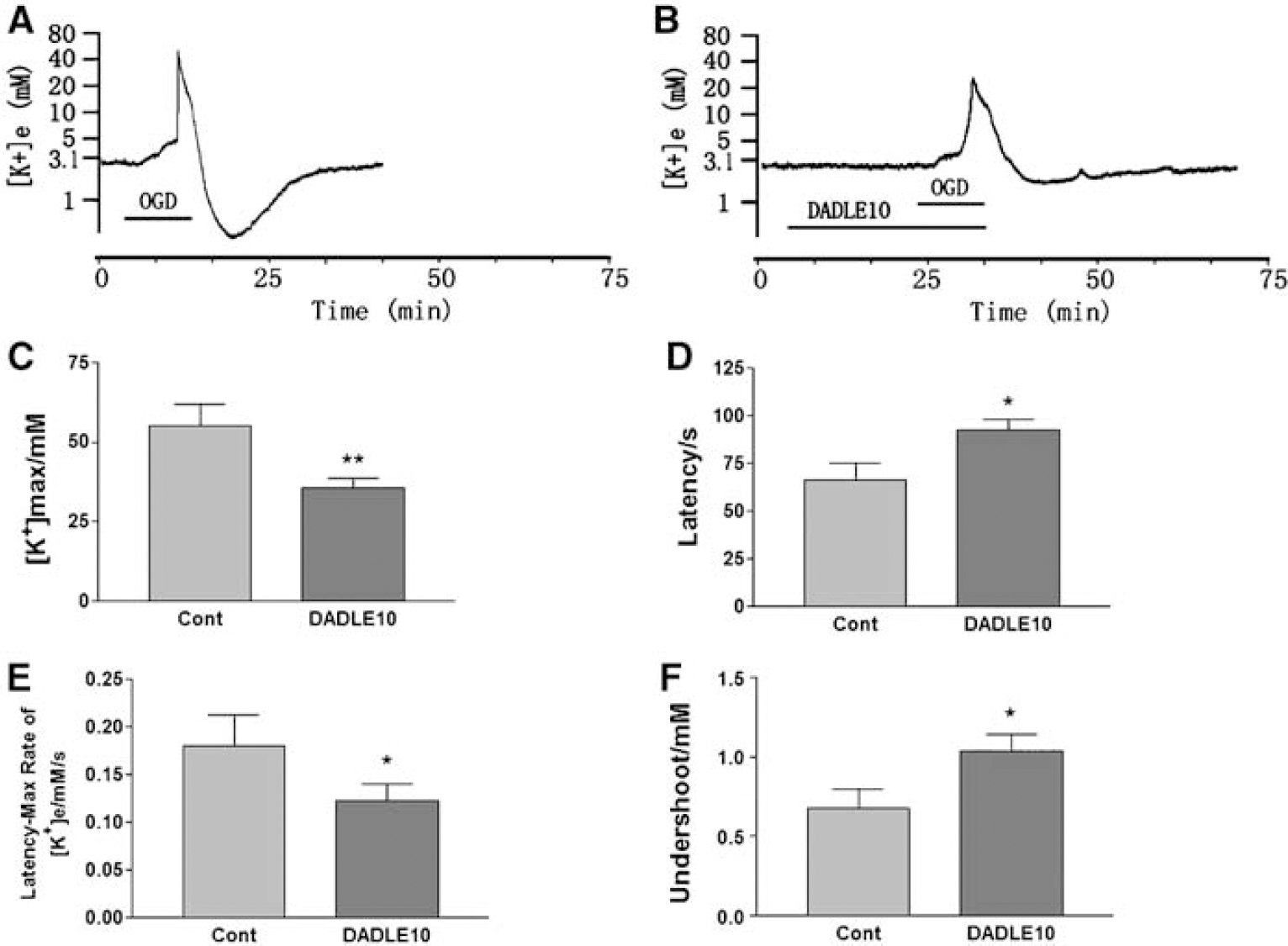
Inhibitory effect of DOR activation on OGD-induced derangement of potassium. Trace recordings of (
DOR Antagonist Blocked the Effect of DOR Activation on the Cortical Slices
To further ascertain the protective role of DOR on K+ derangement, we asked whether the effect of DOR activation could be reversed by DOR antagonists. Because activation of DOR induced a similar effect on both anoxia- and OGD-induced disruption of K+ homeostasis, and the anoxic cortical slice is a simpler model as compared with the OGD model, we chose anoxic cortical slices to test the effect of NTI, a DOR antagonist (Yao et al, 2003; Ammon-Treiber et al, 2005; Lim et al, 2004; Zhang et al, 2000, 2002, 2006; Ma et al, 2005). Because our previous data have shown that 1 to 10 μmol/L of NTI blocked DOR protection (Zhang et al, 2000; Ma et al, 2005), we tested NTI at 1 and 10 μmol/L in this work, respectively. NTI at 1 μmol/L had no appreciable effect on [K+]e during anoxia, and the changes in [K+]e were comparable to those seen in anoxia alone (41.64±6.30 versus 40.88±3.22 mmol/L, n = 10, P>0.05). In the presence of 1 μmol/L of NTI, however, DOR activation with DADLE no longer attenuated the changes in [K+]e. In 11 out of 14 slices from seven animals, NTI completely blocked the protective effect of DADLE (10 μmol/L) on anoxia-induced changes in [K+]e. The latency of response to anoxia significantly decreased from 90±7 secs (10 μmol/L DADLE) to 47±5 secs (10 μmol/L DADLE plus 1 μmol/L NTI) (P < 0.001). The DADLE-attenuated change in peak [K+]e was reversed by NTI (from 23.83±2.95 mmol/L in DADLE only to 45.88±3.66 mmol/L in DADLE plus NTI, P < 0.01). The DADLE-decreased rate of rise of [K+]e from latency level to peak was also reversed (from 0.060±0.010 mmol/L/sec in DADLE only to 0.145±0.026 mmol/L/sec in DADLE plus NTI, P < 0.05) (Figure 2). All the parameters of latency, peak, rate of rise of [K+]e, and undershoot were no different from anoxia alone (Figure 2). Next, we increased the concentration of NTI to 10 μmol/L. This dose of NTI itself evoked an extremely large increase in [K+]e during anoxic induction (n = 6). The voltage changes in the NTI-treated slices were so large that they were beyond the range of ideal electrode response of a logarithmic relationship to [K+] we had tested in the range of 1 to 160 mmol/L KCl.
DOR-Mediated Protection Against Anoxic K+ Derangement was Dependent on the PKC, but not PKA, Pathway
Our previous studies and those of others have shown that DOR regulates the activities of PKA and PKC (Lou and Pei, 1997; Yao et al, 2003; Ma et al, 2005), suggesting that they are involved in DOR signaling. To determine whether PKA and PKC play a role in DOR-mediated attenuation of anoxia-induced K+ derangement, we applied inhibitors of PKA and PKC to cortical slices and tested their role in DOR effect on K+ homeostasis.
To examine the role of PKA, a PKA inhibitor H89 (0.5 μmol/L, ~10 times its IC50) was applied simultaneously with DADLE (10 μmol/L) to cortical slices. H89 (0.5 μmol/L) (n = 8) did not cause any significant change in anoxia-induced [K+]e, nor did it alter the effect of DADLE on the anoxia-induced changes in [K+]e (n =18; Figure 5). These findings suggest that DOR-mediated protection against anoxia-induced disruption of K+ homeostasis is independent of the PKA pathway in the cortical slices.
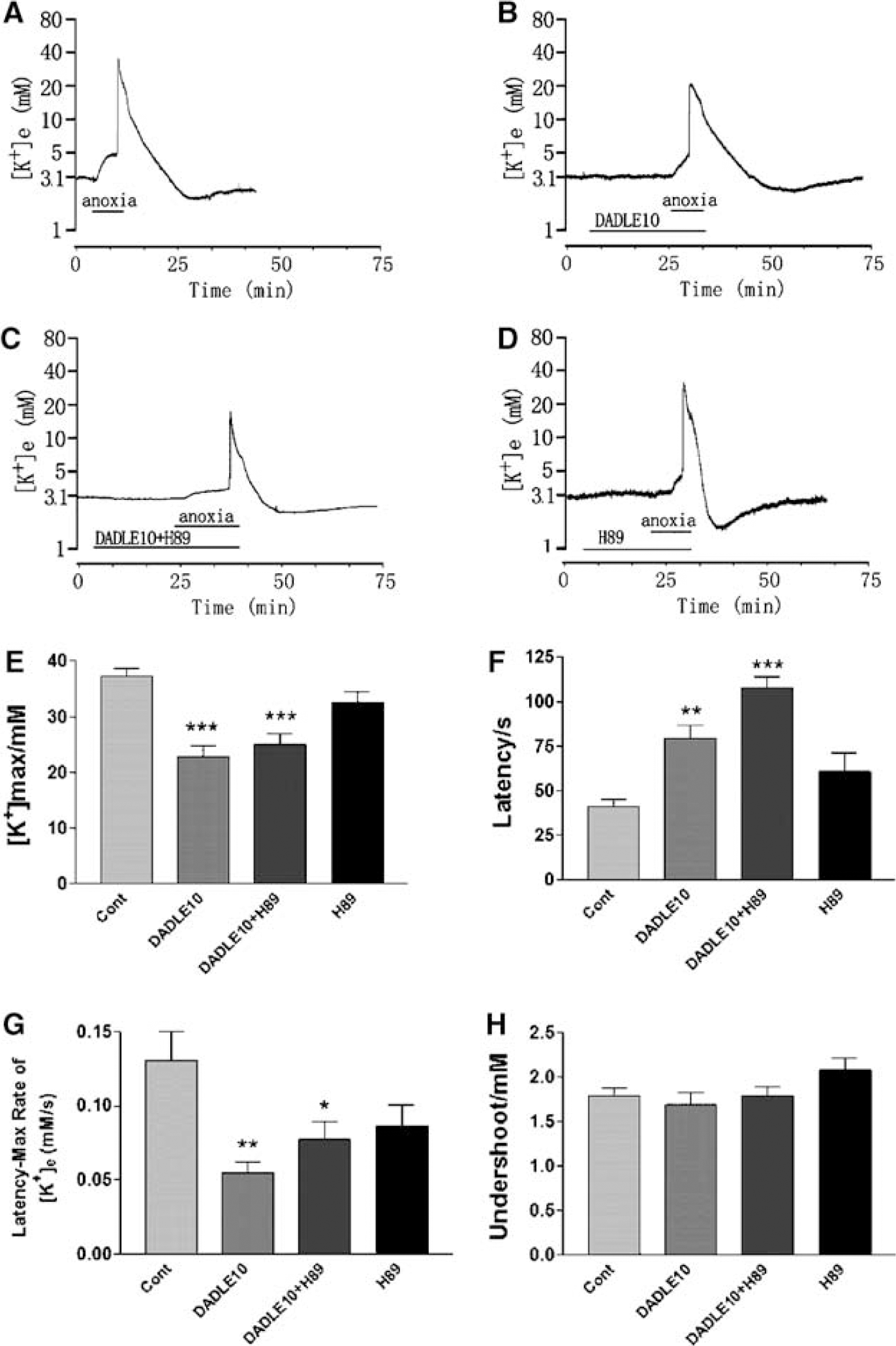
Effect of PKA inhibitor on DOR inhibition of anoxia-induced derangement of potassium. Trace recordings of (
We then tested the effect of CHEL (10 μmol/L), a PKC inhibitor, on the DOR effect using the same experimental procedures as described above. Chelerythrine chloride (10 μmol/L) itself had no effect on anoxia induced changes in [K+]e (n = 6) (Figure 6). In sharp contrast to PKA inhibitor, CHEL reversed the effect of DADLE (10 μmol/L) on [K+]e, resulting in a significantly shortened latency from 80±8 secs (DADLE group, n = 8) to 56±5 secs (DADLE plus CHEL group, n = 8), an increase in peak [K+]e from 22.90±1.87 mmol/L in the DADLE group to 34.81±1.95 mmol/L (P < 0.05), and an increase in the rate of rise of [K+]e from 0.055±0.008 to 0.114±0.021 mmol/L/sec (P < 0.05). These results suggested that PKC is involved in the DOR-mediated attenuation of anoxia-induced disruption of potassium homeostasis in cortical slices.
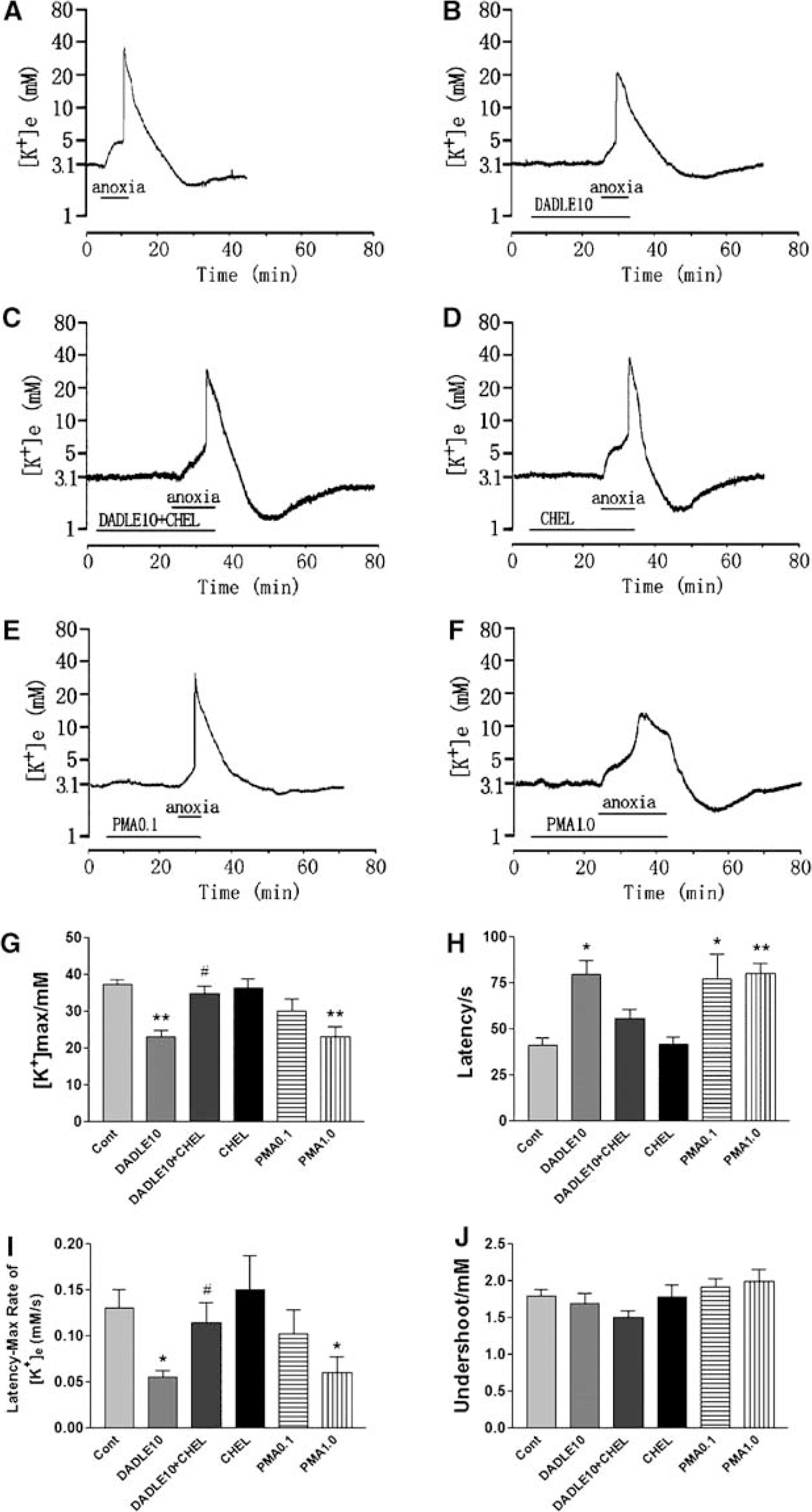
δ-Opioid receptor-mediated inhibition of anoxia-induced derangement of potassium involves a PKC pathway. Trace recordings of (
We further asked whether a PKC activator could mimic the DOR effect. PMA (a PKC activator) was applied to cortical slices. At 0.1 μmol/L (PMA0.1), PMA significantly increased the latency period (n = 9, P < 0.05 as compared with control) and had a tendency to decrease the anoxia-induced increase in [K+]e, and slow the rate of rise to peak. When the concentration was increased to 1 μmol/L (PMA1.0) (n = 8), an effect similar to that of DADLE was seen (Figure 6), that is, increased latency (from 41±4 to 80±6 secs, n = 11, P < 0.01), decreased maximal [K+]e (from 37.24±1.36 to 23.03±2.84 mmol/L, n = 11, P < 0.001), and rate of rise of [K+]e (from 0.130±0.020 to 0.090±0.034 mmol/L/sec, n = 11, P < 0.05). These results further confirm that PKC plays a role in the cellular regulation of K+ homeostasis in the cortical slices.
Discussion
The main findings in this study are the following: (1) DOR activation attenuates anoxia- and OGD-induced K+ derangement and/or a fast negative deflection in extracellular DC potential at the time of the steep increase in the [K+]e in cortical slices; (2) a DOR antagonist abolishes the DOR-mediated protection on K+ homeostasis; and (3) the observed effects of DOR activation are mediated via an intracellular PKC, but not PKA, pathway.
Much light has recently been shed on what appear to be benefits of activation of DORs, especially regarding the protective role of DORs in hypoxia/ischemia-induced cellular injury. Activation of DORs has been shown to increase ischemic/hypoxic tolerance and decrease ischemic/hypoxic injury in several organs and cells including neurons (Ma et al, 2005; Zhang et al, 2002, 2006; Lim et al, 2004; Borlongan et al, 2004). However, the mechanisms underlying the DOR-mediated neuroprotection are largely unknown. No information is available as to whether DOR-mediated neuroprotection occurs via regulation of ionic homeostasis although it is well known that disruption of K+ homeostasis is a major early event in hypoxic cells. Because K+ efflux with resultant depolarization triggers pathways that lead to neuronal death, there is great interest in finding strategies to reduce the rate of K+ efflux. In recent reports, it has been shown that using K+ channel blockers to prevent excessive K+ efflux through K+ channels and from the intracellular compartment attenuates hypoxia/ischemia-induced neuronal death (Huang et al, 2001; Wei et al, 2003; Liu et al, 2003). In the present study, we show that activation of δ-opioid receptor effectively attenuates anoxia/OGD-induced K+ derangement (as indicated by a decreased change in [K+]e and increased latency to the change) as well as the anoxia-induced negative shift in DC potential in cortical slices. We also show that the protection is reversed by a highly selective, non-peptide DOR antagonist naltrindole (Yao et al, 2003; Ammon-Treiber et al, 2005; Zhang et al, 2006), suggesting that DOR protects cortical neurons, at least partially, through regulation of K+ homeostasis during anoxia. To our knowledge, this work represents the first demonstration that DOR modulates K+ homeostasis in neurons during hypoxic stress.
We previously observed that naltrindole, if administrated at 10 μmol/L in neuronal culture, increased hypoxic injury in cortical neurons (Zhang et al, 2002). In this work, we found that at the same concentration in the cortical slice, naltrindole induced an extremely larger increase in [K+]e as compared with that of anoxia alone. It is very likely that excessive inhibition of DOR may cause neuronal injury through severe disruption of K+ homeostasis. Indeed, there is evidence showing that disruption of K+ homeostasis plays an important role in neuronal death (Yu et al, 1997; Wei et al, 2003; Liu et al, 2003). DADLE is a preferential δ-agonist that is widely used for DOR activation in cultured neurons and brain slices in the micromolar range (Moore et al, 1994; Zhang et al, 2000, 2002; Yao et al, 2003; Ma et al, 2005). In previous studies on neuronal cultures (Zhang et al, 2000, 2002), we observed that 10 μmol/L of DADLE was effective in inducing neuroprotection. This has been confirmed in the present work. Conversely, a lower concentration of DADLE, that is, 1 μmol/L, did not reduce the peak increase in [K+]e and the rate of rise of [K+]e to peak in anoxia, although it delayed the response of cortical slices to anoxia. These results suggest that 10 μmol/L is a suitable concentration for DADLE to induce protection from cortical stress. We did not use a higher concentration of DADLE, as we were concerned that this may cause a complex effect on cortical neurons because more μ-opioid receptors (MOR) and other functional protections could become activated. Because 1 to 10 μmol/L of DADLE may still bind to MOR, we performed experiments to determine whether MOR plays a role in the protection shown in this work. We have repeated the experiments with a more specific and potent DOR agonist, H-Dmt-Tic-NH-CH(CH2-COOH)-Bid (Balboni et al, 2002). We observed that 1 μmol/L of H-Dmt-Tic-NH-CH(CH2-COOH)-Bid mimics the protection induced by 10 μmol/L of DADLE, but a 10-fold increase in the concentration did not further enhance the effect of H-Dmt-Tic-NH-CH(CH2-COOH)-Bid on extracellular K+ (Chao et al, manuscript submitted). This suggests that MOR plays little, if any, role in the DOR agonist-induced effect. If MOR were critically involved in the effect seen in our first set of experiments, we would have expected to see a major increase in the protection induced by increased H-Dmt-Tic-NH-CH(CH2-COOH)-Bid, as it has a very low affinity for MOR (Balboni et al, 2002). In fact, there is evidence showing that MOR activation aggravates neurotoxic effects of hypoxia/hypoglycemia, induces apoptosis of human neurons and microglia, and inhibits neuronal survival in the mouse (Ammon-Treiber et al, 2005; Hu et al, 2002; Hauser et al, 1994). In addition, the blockade of MOR activation has been shown to be beneficial to cerebral ischemia/reperfusion insult (Liao et al, 2003). Our previous work has shown that DOR, but not MOR, plays a role in protection against neuronal injury in cortical neurons (Zhang et al, 2000). Taken together, these data support the conclusion that DOR, and not MOR, plays a critical role in attenuating the extracellular increase in potassium during hypoxic stress.
Our previous data have shown that stimulation of DOR reduces neuronal injury in culture exposed for a prolonged hypoxia (Zhang et al, 2002) and this protection is because of an upregulation of the intracellular activity of the G protein-PKC-pERK-Bcl2 pathway and downregulation of the phos-phorylated p38 and cytochrome c death signals (Ma et al, 2005). The present data indicate that DOR reduces the disruption of K+ homeostasis during short-term hypoxia. Taken together, our data suggest that DOR may protect neurons from hypoxic injury in two major ways: (1) in the initial stage of hypoxia, DOR signals maintain ionic homeostasis, and (2) during prolonged hypoxia, DOR signals upregulate benefit-signaling molecules and downregulate those involved in cell injury or death.
There was a major [K+]e undershooting during reoxygenation after anoxia or OGD as shown in this work. Corresponding to the change in [K+]e, neuronal membrane potential (DC potential) usually becomes more negative as compared with prehypoxic conditions (Müller and Somjen, 2000b). The mechanisms underlying the [K+]e undershooting are unclear at present and may be related to compensatory overtransportation of K+ from outside to inside of the membrane by a sodium-potassium pump, which we did not measure in this study. Because we did not observe any significant effect of DOR activation/inhibition on [K+]e undershooting in this work, DOR is less likely involved in the process of K+ transportation after anoxic or OGD stress.
Our present data have shown that protein kinases play an important role in signal transduction of the DOR-mediated regulation of potassium homeostasis. Protein kinase A and PKC are important signaling molecules in a variety of cellular functions, including modulation of neurotransmitter release, regulation of ion channels and enzymes, control of growth and differentiation, and modification of neuronal plasticity (Majewski and Iannazzo, 1998; Leenders and Sheng, 2005). In addition to involvement in normal physiologic events, these two kinases have also been shown to play an important role in pathophysiologic events such as response to hypoxia/ischemia (Tanaka, 2001; Selvatici et al, 2002; Raval et al, 2003; Libien et al, 2005). Our previous studies and those of others have shown that DOR regulates PKA and PKC activities under certain conditions (Lou and Pei, 1997; Yao et al, 2003; Ma et al, 2005), suggesting an involvement of these protein kinases in DOR signaling. In this work, we could not show the involvement of PKA in the DOR protection against increase in extracellular K+, because blocking PKA with H89 did not result in any change in the DOR effect. In sharp contrast, PKC inhibition with chelerythrine reversed, and PKC activation by PMA mimicked, the protective effect of DOR activation. Indeed, we have previously observed that DOR-mediated neuroprotection by hypoxia preconditioning could be blocked by a PKC inhibitor, but not by PKA inhibition (Ma et al, 2005). Collectively, our data suggest that the effect of DOR activation on anoxia-induced K+ derangement is mainly via a PKC-dependent, but PKA-independent, pathway. The interaction between DOR and PKA may be important in signaling other cellular events and requires further investigation.
Protein kinase C signaling may play a role in multiple processes in the neurons. Phosphorylation of K+ channel by PKC has been reported to reduce potassium conductance of mouse neurons (Grega et al, 1987), which may be attributed to the blockade of voltage-dependent, rapidly inactivating K+ efflux from brain synaptosomes (Chaki et al, 1994). Our observations regarding the interaction of DOR and PKC are consistent with this mechanism. A second mechanism may be related to glutamate release, based on work showing that blockade of PKC significantly potentiated OGD-induced glutamate release (Selvatici et al, 2002) that may consequently cause neuronal K+ loss and neurotoxicity (Lopachin et al, 2001; Yu et al, 1999; Xiao et al, 2001). It is therefore possible that in cortical slices, PKC signaling resulting from DOR activation may decrease anoxia-induced glutamate release and in turn reduce potassium efflux.
The precise mechanisms underlying the DOR regulation of potassium homeostasis could be very complex. During anoxia, the increase in extracellular K+ could be caused by increased K+ efflux, decreased K+ influx, or both. In addition, neurons along with glial cells could contribute to this net [K+]e increase. The increase in [K+]e seen in adult brain tissue during anoxia is, to a great extent, secondary to neuronal loss of K+ (Jiang and Haddad, 1991). Many mechanisms have been proposed for this K+ loss, including inhibition of Na+-K+ ATPase, increased leakage of K+ because of an increase in excitability and action potential generation in neurons, activation of ATP-sensitive K+ channels, Ca2+-activated K+ channels, and Na+-activated K+ channels (Jiang and Haddad, 1991; Reid and Paterson, 1996; Erdemli et al, 1998; Yuan et al, 2000). In addition, the ionotropic glutamate receptor (NMDA (N-methyl-D-aspartate), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid), and kainate receptors)-mediated neuronal K+ loss may be another reason for anoxia-induced [K+]e increase (Lopachin et al, 2001) because blockade of ionotropic glutamate receptors with NMDA receptor blocker 3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid or AMPA receptor blocker 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) partially decreased OGD-induced intraneuronal K+ loss in hippocampus (Lopachin et al, 2001). Activation of NMDA, AMPA, and kainate receptors of mouse cortical neurons, even in a condition of decreased inward cation influx by lowering extracellular Na+ and Ca2+, induced a large outward K+ current, which caused loss of approximately 50% to 80% intraneuronal K+ and great shrinkage of cell body and consequently neuronal apoptosis (Yu et al, 1999; Xiao et al, 2001). Therefore, it is possible that activation of DOR attenuates anoxic potassium derangement by directly or indirectly decreasing neuronal K+ loss and/or increasing its influx. Indeed, electrophysiological studies in vivo and in vitro have shown that activation of DOR depresses spontaneous and stimulus-evoked action potential discharge as well as the amplitudes of stimulus-evoked excitatory postsynaptic potentials/currents of neocortical neurons (Stanzione et al, 1989; Tanaka and North, 1994; Ostermeier et al, 2000), which at least reduces increased leakage of K+ because of an increase in excitability and action potential generation in cortical neurons. In fact, it has been shown that in hippocampal slices, when neuronal excitability was decreased and spontaneous and evoked impulse firing were blocked by TTX or lidocaine, hypoxia-induced sharp [K+]e increase was significantly dampened (Raley-Susman et al, 2001; Müller and Somjen, 2000a, b). Studies at the cellular level have indicated that DOR activation inhibits diverse calcium currents that link to Ca2+ influx through different high-voltage-activated channels types in neurons as well as neuroblastoma cell lines (Toselli et al, 1999; Acosta and Lopez, 1999), and decreases [Ca2+]i level in resting state as well as depolarization-induced Ca2+ uptake in rat cerebral cortex (Vlaskovska et al, 1997; Thorlin et al, 1998). Because hypoxia/ischemia causes a great increase in [Ca2+]i (Martinez-Sanchez et al, 2004), a high cytosolic concentration of Ca2+ would be expected to activate the Ca2+-activated K+ channels and induce K+ efflux. Therefore, it is possible that activation of DOR attenuates anoxic K+ derangement by restricting Ca2+ entry into neurons. An alternative mechanis underlying DOR attenuation of anoxic K+ derangement may involve DOR regulation of glutamate signaling. Immunocytochemical studies revealed that DOR is localized at presynaptic terminals in a variety of neurons, including those of mammalian cortex (Bausch et al, 1995; Svingos et al, 1995). δ-Opioid receptor activation has been shown to prevent the release of glutamate from presynaptic vesicles, thereby reducing glutamate excitability (Tanaka and North, 1994; Ostermeier et al, 2000), which would decrease hyperexcitability-induced K+ leakage through ionotropic glutamate receptors in neurons. It is very likely that DOR attenuation of anoxic K+ derangement might involve multiple strategies involved in ionic homeostasis in the neurons although the precise mechanisms are not yet clear.
It is interesting to note that anoxia-induced changes in extracellular potassium and DC potential are similar to those that occurred in spreading depression, in which K+ release by a group of neurons raised extracellular K+ and subsequently depolarized nearby cells, providing the feedback responsible for the propagation of spreading depression (Müller and Somjen, 2000a). Because DOR activation attenuates anoxic [K+]e increase and the fast negative deflection in DC potential in our work, we speculate that DOR activation may be preventive against spreading depression.
In summary, we present the first data to show that activation of DOR protects cortical neurons from anoxia-induced potassium derangement, and this protection is dependent on a PKC-dependent and PKA-independent, pathway. We conclude that an important aspect of DOR-mediated neuroprotection is its early action against ionic disruption during anoxia or ischemia.