
Abstract
Astrocytic energy demand is stimulated by K+ and glutamate uptake, signaling processes, responses to neurotransmitters, Ca2+ fluxes, and filopodial motility. Astrocytes derive energy from glycolytic and oxidative pathways, but respiration, with its high-energy yield, provides most adenosine 5' triphosphate (ATP). The proportion of cortical oxidative metabolism attributed to astrocytes (~30%) in in vivo nuclear magnetic resonance (NMR) spectroscopic and autoradiographic studies corresponds to their volume fraction, indicating similar oxidation rates in astrocytes and neurons. Astrocyte-selective expression of pyruvate carboxylase (PC) enables synthesis of glutamate from glucose, accounting for two-thirds of astrocytic glucose degradation via combined pyruvate carboxylation and dehydrogenation. Together, glutamate synthesis and oxidation, including neurotransmitter turnover, generate almost as much energy as direct glucose oxidation. Glycolysis and glycogenolysis are essential for astrocytic responses to increasing energy demand because astrocytic filopodial and lamellipodial extensions, which account for 80% of their surface area, are too narrow to accommodate mitochondria; these processes depend on glycolysis, glycogenolysis, and probably diffusion of ATP and phosphocreatine formed via mitochondrial metabolism to satisfy their energy demands. High glycogen turnover in astrocytic processes may stimulate glucose demand and lactate production because less ATP is generated when glucose is metabolized via glycogen, thereby contributing to the decreased oxygen to glucose utilization ratio during brain activation. Generated lactate can spread from activated astrocytes via low-affinity monocarboxylate transporters and gap junctions, but its subsequent fate is unknown. Astrocytic metabolic compartmentation arises from their complex ultrastructure; astrocytes have high oxidative rates plus dependence on glycolysis and glycogenolysis, and their energetics is underestimated if based solely on glutamate cycling.
Introduction
Astrocytes are More Than Housekeepers
Recent studies in many different fields have shown much more active roles of astrocytes in brain function than previously portrayed by their traditionally ascribed ‘bystander or housekeeping’ functions. Emerging roles of astrocytes include their interactions with the vasculature, neurons, and other astrocytes via signaling, biosynthetic, and transport processes to regulate blood flow, modulate impulse transmission, and synthesize and degrade glucose-derived neurotransmitters, for example, glutamate and γ-aminobutyric acid (GABA) (Takano et al, 2006; Hertz and Zielke, 2004; Volterra and Meldolesi, 2005). All of these processes are energyrequiring or dependent on energy-related metabolic pathways, thereby directly linking astrocyte functions, energetics, and metabolite fluxes.
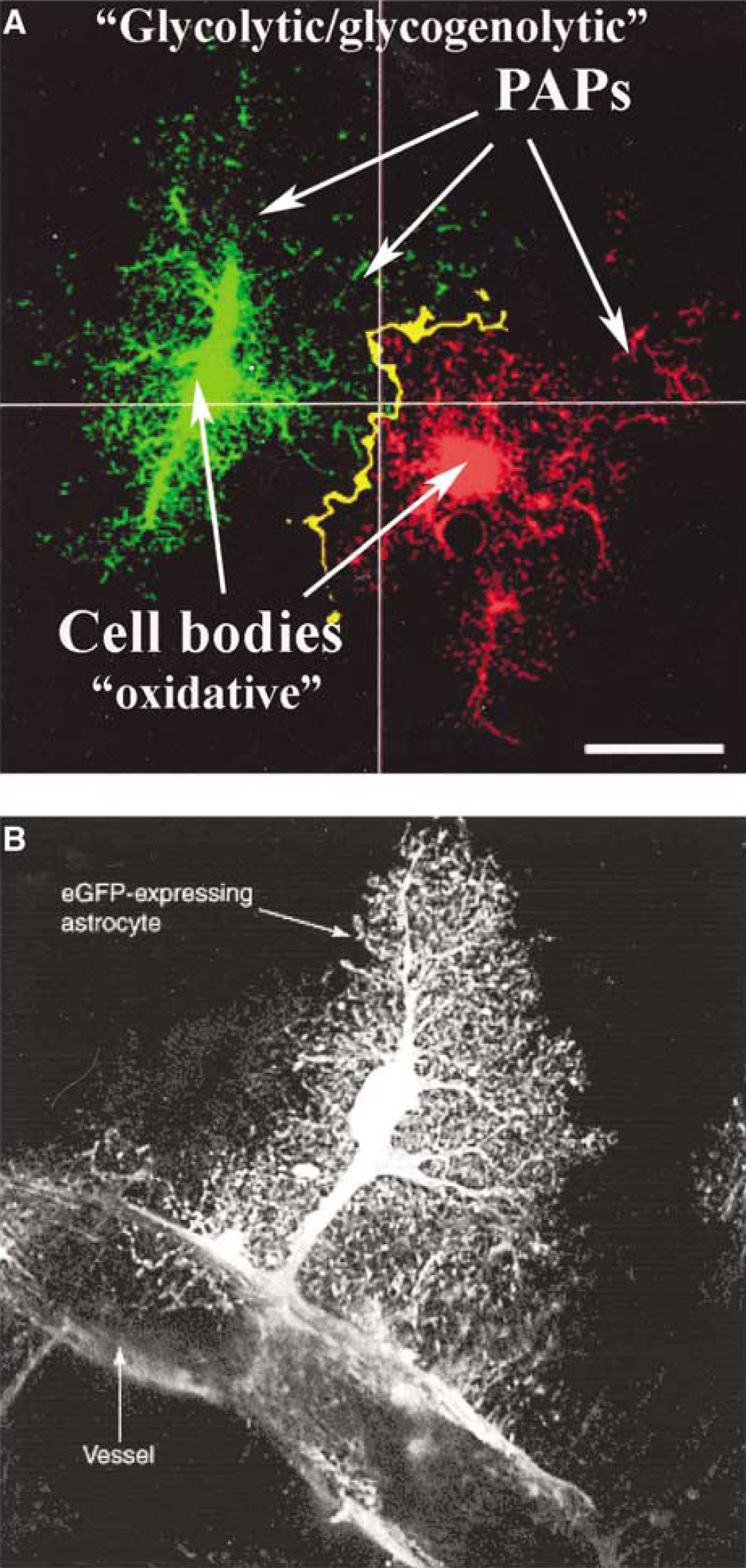
The narrow astrocytic surface extensions (lamellae and filopodia, also called peripheral astrocytic processes or PAPs), which are in close apposition with neuronal elements (including synapses), the larger processes (including those associated with arterioles and venules as endfeet), and the cell bodies all have different and specialized activities. Therefore, subcellular regions of astrocytes differ in their spatial and temporal dependence on the glycolytic and oxidative pathways (Figure 1). Although oxidative metabolism provides by far the majority of the energy needed, glycolysis and glycogenolysis in the PAPs appear temporarily to provide most of the energy required during an abrupt energy demand, for example, that created by fluctuations in the extracellular contents of ions or transmitters triggering uptake into the peripheral astrocytic processes.
Astrocyte structure, interactions between astrocytes, and interactions with the vasculature. (
Cellular Basis of Brain Energetics
Functionally activated energy production in brain cells contributes to the generation of metabolic, magnetic resonance, and redox signals used to monitor and evaluate brain function in situ using various technologies (e.g., positron emission tomography (PET), nuclear magnetic resonance (NMR), fluorescence microscopy, autoradiography). Elucidation of the cellular basis of metabolic responses to functional activity in brain has been an important goal of neuroscientists for decades, but its realization has been elusive, because technical difficulties are formidable due to anatomic complexity and metabolic heterogeneity. Astrocytes are a major but understudied cell type in brain, and this review emphasizes functional energetics of astrocytes with a brief overview below and in-depth description later (see in Sections Glucose metabolism in astrocytes and Brain activation and neuromodulators increase astrocytic energy generation).
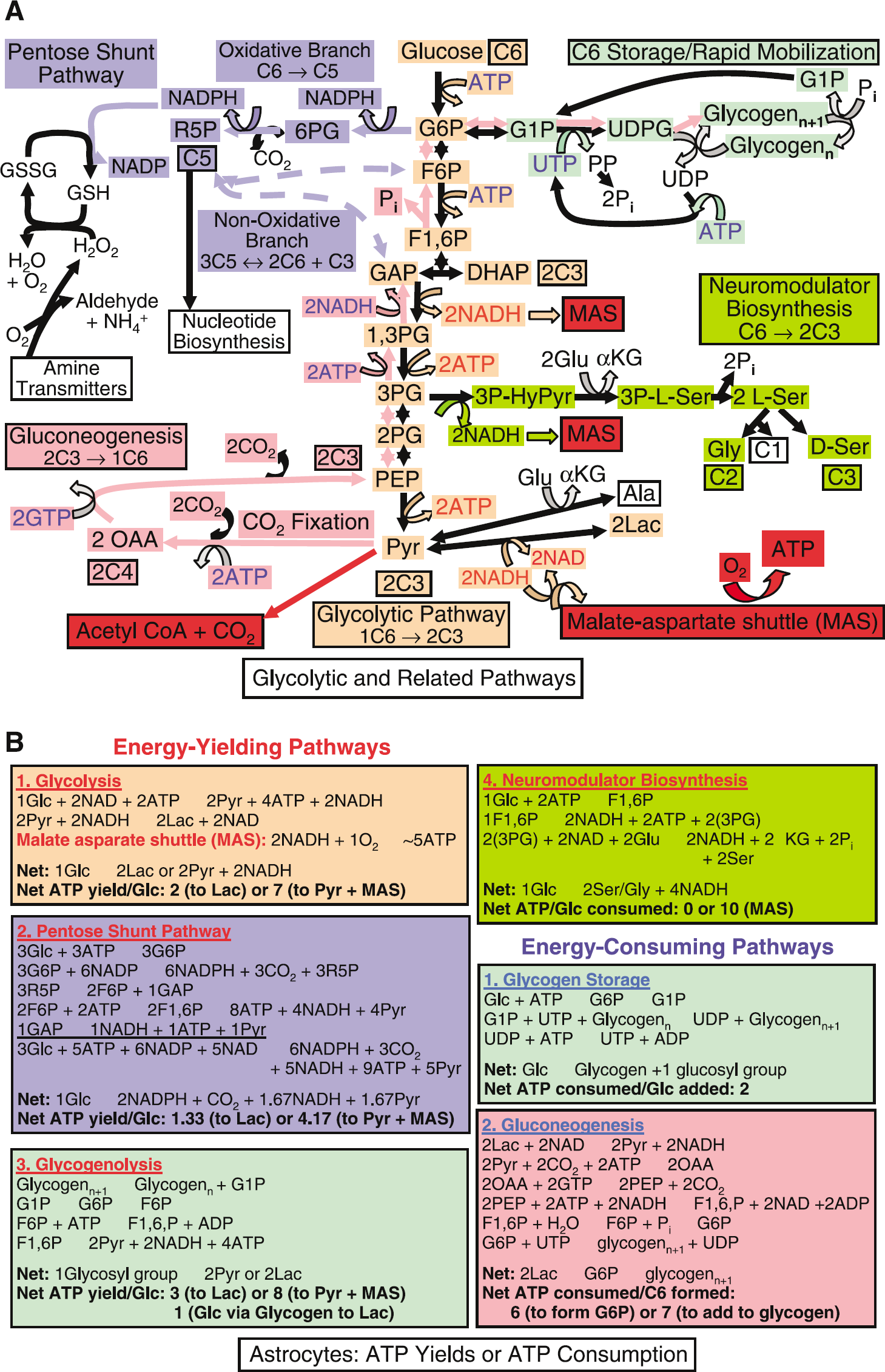
High Oxidative Rates in Astrocytes in the Brain In Vivo
Within the last 5 years and for the first time, NMR spectroscopic studies of brain metabolism in situ under resting conditions have led to the conclusion drawn by several different groups that oxidative metabolism in cortical glial cells (Blüml et al, 2002) or astrocytes (Gruetter et al, 2001; Lebon et al, 2002; Öz et al, 2004) accounts for ~30% of total tissue oxygen consumption in brain cortex. This fraction is similar to, or perhaps even higher than, the portion of the average total tissue volume occupied by astrocytes, estimated to be at most 20% to 25% (Williams et al, 1980; Wolff and Chao, 2004), although the astrocytic volume fraction does vary from region to region and from one cortical layer to another. An astrocytic oxidative capacity comparable to that in neurons is also suggested by the similar mitochondrial volume fraction in neuronal and astrocytic cell bodies (Pysh and Khan, 1972). The notion that astrocytes and neurons have equivalent oxidative capabilities on a cellular basis under resting conditions is a paradigm-shifting concept with broad implications for functional roles of astrocytes.
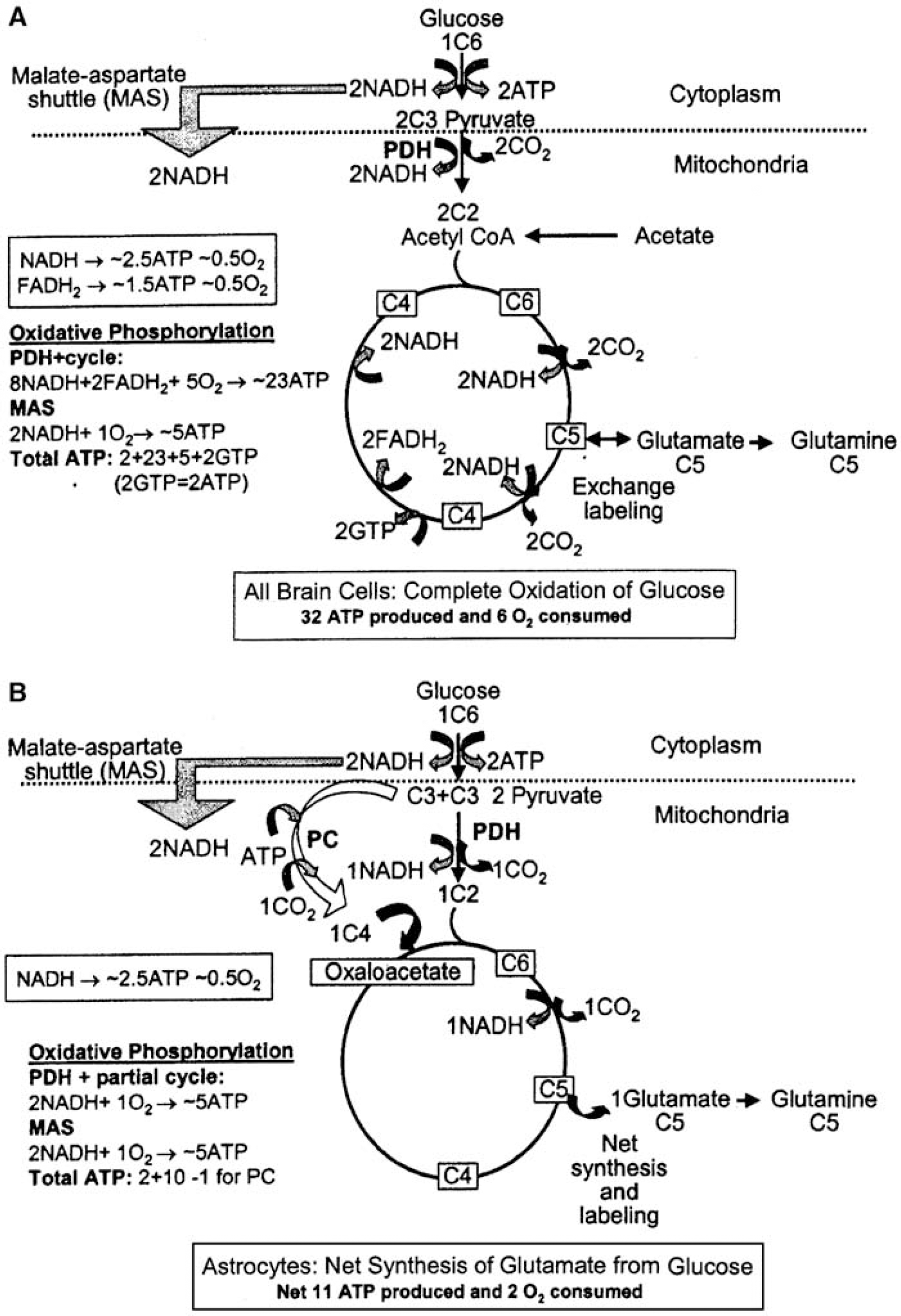
Interpretation of the in situ NMR measurements of oxidative metabolism in astrocytes is based on two major findings made by histochemistry and in cultured cells. First, the ability of astrocytes but not of neurons (Muir et al, 1986; Waniewski and Martin, 1998) and oligodendrocytes (Edmond, 1992) to accumulate acetate and other short- or medium-length fatty acids and metabolize them via the tricarboxylic acid (TCA) cycle (Figure 2A) is the basis for interpretation of measurement of rates of acetate oxidation in terms of oxidative metabolism in astrocytes. Second, the ability of astrocytes to carry out carbon oxide (CO2) fixation via pyruvate carboxylation enables de novo synthesis of TCA cycle intermediates and their amino-acid derivatives (Figure 2B), and astrocytic expression of pyruvate carboxylase (PC) is the basis for interpretation of pyruvate carboxylation rates as astrocytic. This enzyme is absent in neurons (Yu et al, 1983; Shank et al, 1985) and probably in oligodendrocytes (see Section Pyruvate carboxylation and oxidative degradation of TCA cycle intermediates and their derivatives). Any contribution by microglia is quantitatively insignificant due to their low volume fraction (Wolff and Chao, 2004). The specialization of transport and oxidative pathways in astrocytes not only enables their assay in situ, but it also generates energy for astrocytes and fulfills critical functions for neurons, as described in more detail in Section Glucose metabolism in astrocytes.
Glucose metabolism in astrocytes and energetics of glucose oxidation and of glutamate formation. (
Astrocytes Metabolize Half the Glucose Taken Up Into Brain and Store Glycogen
Glucose is the major, obligatory fuel for all brain cells because it is the only substrate that is normally both present in the systemic circulation in a high, hormonally regulated concentration and rapidly transported across the blood–brain barrier. Astrocytes and neurons metabolize glucose via glycolytic, pentose shunt, and oxidative pathways, whereas only astrocytes have the ability to store glucose as glycogen, a high molecular-weight glucose polymer, in an energy-requiring process (Hamprecht et al, 2005). In brain, the glycogen level is stable under resting conditions and its turnover is slow (Watanabe and Passonneau, 1973; Choi et al, 2003), but glycogenolysis can increase substantially during brain activation (Swanson et al, 1992; Cruz and Dienel, 2002).
Glucose enters the glycolytic pathway after phosphorylation by hexokinase to glucose-6-phosphate (G6P), and under resting conditions, about half of the phosphorylation of blood-borne glucose in brain of conscious rats in vivo occurs in astrocytes (Nehlig et al, 2004). Glucose-6-phosphate is also generated from glycogen after initial conversion of glucosyl units to glucose-1-phosphate (G1P) by glycogen phosphorylase. Nearly all of the hexose-phosphate is further metabolized via the glycolytic pathway to produce 2 molecules of pyruvate. Pyruvate can be converted to alanine, lactate, or oxaloacetate, or be oxidatively metabolized in the TCA cycle (Figure 2A), which is its major overall fate in brain tissue. Pyruvate equilibrates with a ~10 times higher concentration of lactate via the lactate dehydrogenase (LDH) reaction because of the LDH equilibrium constant and the redox state and pH in the tissue; the normal resting rat brain lactate level is ~ 1 μmol/g wet wt (e.g., Siesjö, 1978; Dienel et al, 2002).
Activating Conditions Stimulate Oxidative Metabolism and Glycogenolysis in Astrocytes
During a specific activation paradigm, such as sensory stimulation, glycogen breakdown (glycogenolysis), and oxidative metabolism in astrocytes increase in brain in situ (Swanson et al, 1992; Cruz et al, 2005; R Gruetter, presented at the Third Wierzba Conference, 2005). The response to activation is greater and more complex than that calculated on the basis of only the two adenosine 5' triphosphate (ATP)-dependent reactions involved in neurotransmitter glutamate-glutamine cycling (e.g., Attwell and Laughlin, 2001). The increase in cerebral rate of oxidative metabolism (CMRo2) usually coincides with an even larger activation-dependent stimulation of cerebral glucose utilization (CMRglc) (reviewed by Dienel and Cruz, 2004). The CMRo2/CMRglc ratio accordingly falls from its theoretical maximal value of 6.0 (because oxidation of one glucose molecule requires six molecules of oxygen (C6H12O6+ 6O2→6CO2 + 6H2O)), reflecting nonoxidative metabolism of glucose. A concomitant rise in whole-brain lactate content from ~1 to ~2 μmol/g wet wt during activation only partly explains the reduction in CMRo2/CMRglc ratio. Some of the elevated lactate content is probably astrocytic in origin (Elekes et al, 1996; Kasischke et al, 2004), arising from both glucose and glycogen. In many situations, glycogenolysis increases from a negligible rate in resting brain to values that approach or exceed rates of glucose degradation. When a net glycogenolysis is taken into account, the CMRÖ2/CMRglc ratio in brain falls much further (Dienel and Cruz, 2004).
Summary
Recent findings indicate that astrocytic energy metabolism is much more intense in magnitude than commonly believed. Under resting conditions (i.e., no specific stimulus) astrocytes account for ~ 30% of oxidative metabolism in brain and perhaps 50% of glycolysis, whereas very little glycogenolysis occurs. During activation, flux through all of these astrocytic pathways increases, but overall glucose utilization often rises disproportionately more than oxidative metabolism causing the CMRo2/CMRglc ratio in the brain in vivo to fall, often well below 6, especially when the rate of glycogenolysis also is taken into account. The much higher rates of oxidative metabolism and larger functional increase in metabolism in astrocytes than commonly acknowledged suggest the presence of functional activities that are not commonly recognized or that are supplied with energy via specific pathways due to their unique location or characteristics.
Astrocytic Localization, Ultrastructure, and Functional Activities
Astrocytic Ultrastructure Influences Metabolic Changes During Brain Activation
Major anatomic regions of astrocytes: The dependence of astrocytes on stimulation of glycolysis and/or glycogenolysis during activation, even though oxidative metabolism provides nearly all of the energy they need, arises from their ultrastructure and its relation to function. Astrocytes are located at strategic positions around neurons and blood vessels in the brain. They virtually enclose the synaptic cleft, giving rise to the expression ‘the tripartite synapse’ (Araque et al, 1999). Equally important is that their peripheral processes also ensheathe the larger part of many dendrites (e.g., those of Purkinje cells), cover the circumference of many neuronal somata, surround most, but not all, of the surface of brain capillaries, and attach as endfeet to pre- and postcapillary vessels (Peters et al, 1991; Hirrlinger et al, 2004; Wolff and Chao, 2004). Although astrocyte shape can differ with localization, a typical astrocyte has four major anatomic components, the soma (cell body), the larger processes, the very fine PAPs (Figure 1A), and the large endfeet (Figure 1B). Endfeet surround precapillary arterioles (Wolff and Chao, 2004) and participate in Ca2+ -mediated blood flow control by synthesis and release of compounds that regulate vascular diameter (Takano et al, 2006). Contrary to general belief, these relatively large endfoot processes are not associated with capillaries but with slightly larger vessels (Wolff and Chao, 2004). The common misconcept is due to the inability of the classical Cajal studies to distinguish between capillaries and small arterioles.
The cell body of a typical astrocyte accounts for only ~ 2% of its total volume, with larger, branching organelle-containing processes constituting ~ 60%. Astrocytic oxidative capability resides in the soma and larger cell processes (Figure 1) that contain the mitochondria (Wolff and Chao, 2004). Threadlike filopodia (~3 μm long by ~0.2 μm wide) and the equally thin, sheet-like lamellipodia account for ~40% of the cell volume, and are highly mobile (Hirrlinger et al, 2004). These peripheral astrocyte processes (Figure 1A) constitute a specific compartment (Derouiche and Frotscher, 2001) that cannot accommodate mitochondria, which are approximately twice as wide. These small extensions can, however, rapidly respond to energy demand with glycolytically-derived ATP (Figure 1A) because they have access to glucose and contain glycogen deposits and express glycogen phosphorylase activity (Peters et al, 1991; Hamprecht et al, 2005); diffusion of ATP and phosphocreatine generated in mitochondria may subsequently help supply their energy and underwrite re-synthesis of glycogen. The PAPs account for 80% of the cell's surface area (Wolff and Chao, 2004) and interact with other astrocytes (Figure 1), extracellular fluid, and capillaries; most of the cell's energy-requiring active transport occurs in these thin proceses.
Energy-dependent functions involving extracellular glutamate and K+: It is generally accepted that astrocytes are responsible for the major part of glutamate uptake after its release from neurons (Danbolt, 2001), but subtypes of astrocytes differ with respect to their ion channels, glutamate transporters, and glutamate receptors (Matthias et al, 2003; Kimelberg, 2004; Volterra and Meldolesi, 2005). Glutamate is accumulated by cotransport with sodium ions (Na+) along the inwardly directed electrochemical gradient of Na+. Therefore, glutamate uptake is not immediately dependent on energy, whereas the subsequent Na+ extrusion, catalyzed by the Na+,K+-ATPase, consumes ATP (Figure 3). A pathway for glutamate return to neurons is called the glutamate-glutamine cycle, and involves its ATP-requiring conversion to glutamine by the astrocyte-specific glutamine synthetase (Norenberg and Martinez-Hernandez, 1979; Derouiche, 2004); this process is well established, as is oxidative degradation of part of the accumulated glutamate (Section Neurotransmitter glutamate-glutamine cycling).
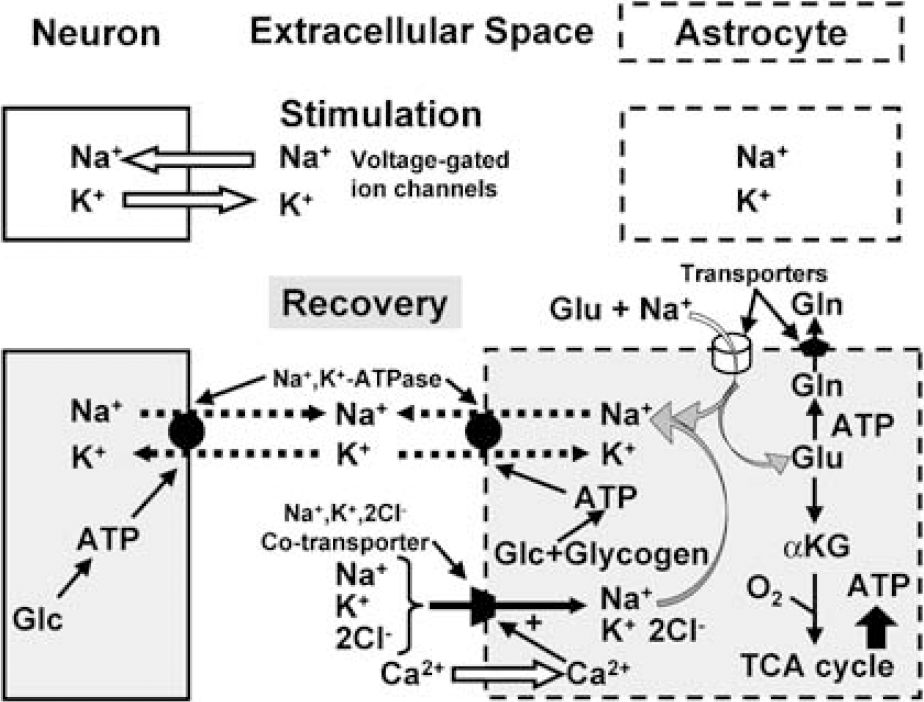
Uptake of potassium, sodium, and glutamate into astrocytes after neuronal excitation. During neuronal stimulation, sodium ions (Na+) enter and potassium (K+) ions exit passively from neurons via voltage-gated channels (denoted by open arrows), whereas there are no ion movements in astrocytes. During recovery, K+ clearance from extracellular space occurs by two active transport mechanisms, the Na+, K+ -ATPase, exchanging extracellular K+ with intracellular Na+ (in both astrocytes and neurons), and a cotransport system, transporting in conjunction 1 Na +, 1 K+ and 2 Cl− ions into the cell. In astrocytes, the Na+, K+ -ATPase is stimulated at its extracellular K+ -sensitive site by the increase in extracellular K+ concentration resulting from neuronal excitation and in neurons at its intracellular Na+-sensitive site by the stimulus-induced increase in intracellular Na+ concentration. Uptake of Na+, K+ and Ch by the cotransport system is energetically driven by the Na+ gradient. The cotransport system is active in astrocytes, and its operation stimulated by entry of calcium ions (Ca2+) through voltage-sensitive Ca2+ channels (open arrow) by K+ - mediated depolarization. The combined operation of the Na +, K+ -ATPase and the cotransport system leads to a reaccumulation of K+, together with Cl−, but without any concomitant net uptake of Na+, because accumulated Na+ is extruded by stimulation of the intracellular, Na+ -sensitive site of the astrocytic Na+, K+ -ATPase. The astrocytic Na+, K + -ATPase is also activated by a rise in the level of extracellular glutamate, stimulating glutamate uptake into astrocytes along with Na+, which subsequently activates the intracellular, Na+ -sensitive site of the Na+, K+ -ATPase, leading to extrusion of Na+ and additional uptake of K+. Stimulation of Na+, K+ -ATPase activity, regardless of triggering mechanism, leads to conversion of ATP to ADP, and the locally altered ATP/ADP ratio stimulates both mitochondrial oxidative metabolism and cytosolic glycolysis. The accumulated glutamate is partly converted to glutamine, which is released, and partly converted to α-ketoglutarate and oxidized in the TCA cycle, stimulating the rate of oxygen consumption and contributing energy for its own uptake and glutamine formation.
Astrocytes play a similar active role in the clearance of extracellular potassium ions (K+) (Walz, 2000, 2004), although this is less-generally acknowledged. During neuronal excitation, extracellular K+ level rises from 3 mmol/L to a maximum of 12 mmol/L, and may remain elevated for at least 15 to 20 secs (Figure 4A). The importance of glial cell function for K+ clearance in cerebral cortex of adult anesthetized rats has been demonstrated by Lian and Stringer (2004), and the same group found pronounced inhibition of K+ removal from the extracellular fluid of hippocampal slices by ouabain, an inhibitor of the Na+, K+ -ATPase (Xiong and Stringer, 2000). Passive re-distribution of a local increase in extracellular K+ through a glial syncytium to different extracellular locations by a current-mediated redistribution of K+ (‘spatial buffering’) can take place, but its importance in the mammalian central nervous system has been disputed (Walz, 2000, 2004). It now seems clear that spatial buffering does not contribute to clearance of extracellular K+ when it rises above the resting level, but buffering has a small effect on the magnitude of the resting extracellular K+ concentration and reduces the poststimulation undershoot by a ‘reverse’ spatial buffer mechanism (D'Ambrosio et al, 2002). Another mechanism, Donnan type uptake of KCl, does not occur under physiological conditions because the normal astrocyte has a very limited Cl− conductance, but such an uptake does occur in reactive astrocytes (Walz, 2004).
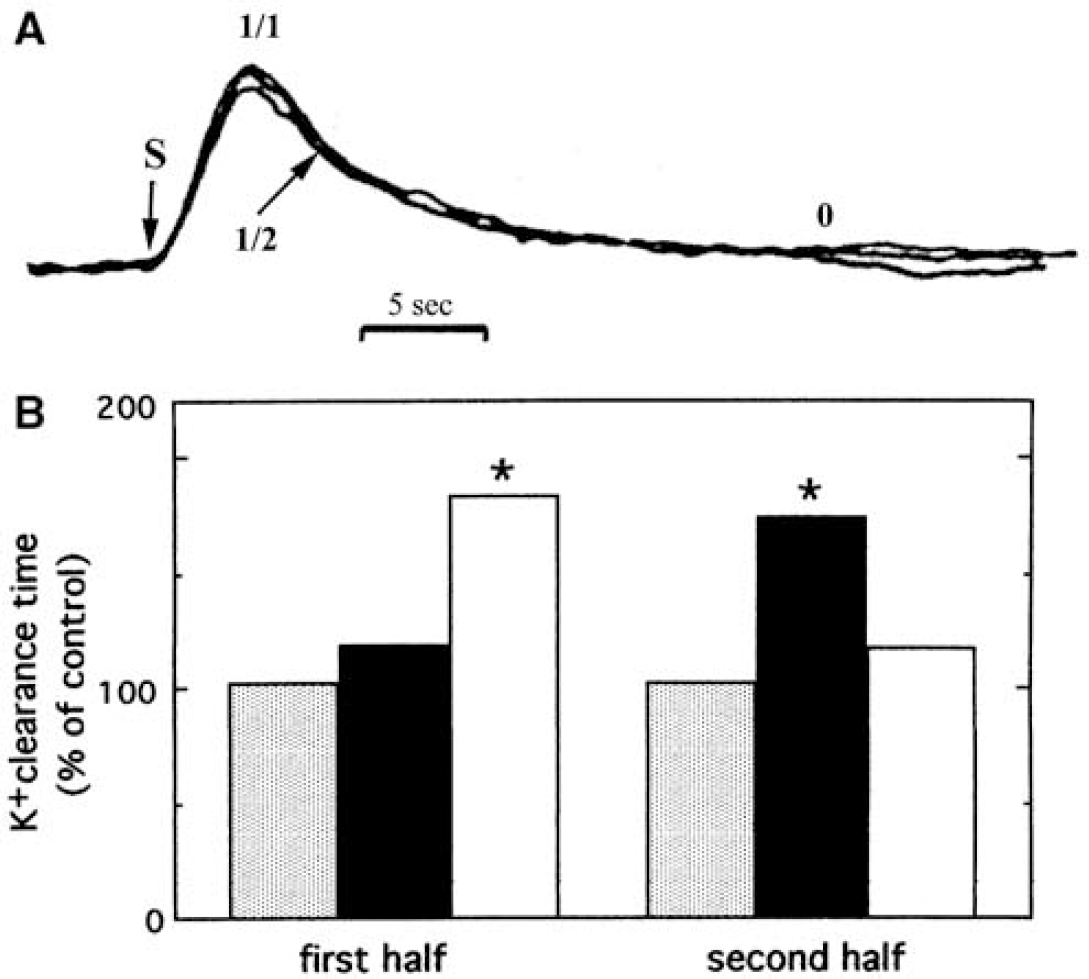
Temporal profile of extracellular K+ concentration ([K+]e) and energy pathways linked to [K+]e clearance after neuronal excitation. (
The K+ uptake into astrocytes occurs by two different mechanisms: (i) activation of the extracellular K+ -sensitive site of the Na+, K+ -ATPase and (ii) stimulation of the Na +, K+, 2Cl− cotransporter (Figure 3). Uptake of K+ by the astrocytic Na+, K+ -ATPase occurs by direct stimulation at its extracellular K+ -sensitive site by any increase in extracellular K+ concentration and requires an immediate supply of ATP. In contrast, uptake by the cotransporter is driven by the Na+ gradient; this process may be associated with a slightly larger increase in extracellular K+ concentration because it requires entry of calcium ions (Ca2+) through depolarization-dependent L-channels (Su et al, 2000). Like glutamate uptake, cotransporter-mediated uptake is initially independent of energy supply, but subsequently dependent on ATP-mediated extrusion of accumulated Na+ in exchange with K+ to maintain a transmembrane Na+ gradient (Walz and Hinks, 1986). The combined actions of the cotransporter and Na+-K+-ATPase mediate uptake of 2K+ and 2Cl− (Figure 3).
The energy for the initial and later phases of K+ clearance appears to be generated by different pathways because they can be differentially delayed. Glycolysis and/or glycogenolysis must provide energy during the initial phase of K+ uptake in brain in situ, because the first half of extracellular K+ clearance is delayed by iodoacetate, an inhibitor of glycolysis (and glycogenolysis), but not by hypoxia (Figure 4B). In contrast, the later part of K+ uptake (beginning at ~ 3 secs after the peak rise in extracellular K+, denoted by 1/2 in Figure 4B) is fueled mainly by oxidatively derived energy, reflected by its inhibition by severe hypoxia (Raffin et al, 1992). It is likely that most of the initial K+ uptake occurs into astrocytes (Walz, 2000, 2004), particularly into peripheral processes that are devoid of mitochondria. This notion is consistent with the initial dependence of K+ clearance on glycolysis or glyogenolysis and subsequent dependence on oxidative metabolism. Little is known about diffusion of ATP and phosphocreatine generated by mitochondria into PAPs, but these thin processes are readily filled by diffusion and visualized in fluorescent dye microinjection studies (Figure 1A), and the gap junction literature provides evidence for long-distance diffusion of dyes and metabolites among astrocytes, including hexoses, adenine nucleotides, and IP3 (Scemes and Spray, 2004). Diffusion coefficients of ~1 μm2/sec for phosphocreatine and ATP have been determined in myofibrils in vivo (de Graaf et al, 2000), and if diffusion rates are equivalent in astrocytes, a few seconds may suffice for ATP and phosphocreatine to reach peripheral astrocyte extensions. Also, diffusion of accumulated neurotransmitter glutamate from PAPs to mitochondria would provide a timely source of oxidative fuel (Figure 3) and diffusion of pyruvate would allow pyruvate carboxylation (Figure 2B).
Glycogenolysis is especially well suited to rapidly provide large amounts of glycolytically derived energy because, in contrast to glycolysis, it requires no initial ATP expenditure to phosphorylate glucose, and breakdown of glycogen to pyruvate produces more energy than pyruvate formation from glucose (Section Glycogen: an endogenous astrocytic source of glucose-6-Phosphate). Consistent with the concept that the initial part of K+ uptake into astrocytes is energized by glycolysis/glycogenolysis is the finding that elevated K+ stimulates glycogenolysis in brain slices (Hof et al, 1988) and in cultured astrocytes (Subbarao et al, 1996). In contrast, glutamate does not activate glycogenolysis (Magistretti et al, 1981), perhaps reflecting the following: (i) glutamate uptake itself is not directly dependent on energy (whereas the subsequent Na+ extrusion is ATP dependent); (ii) some glutamate might be oxidized to support sodium extrusion (Figure 3); (iii) glutamine synthetase, the enzyme catalyzing the subsequent energy-requiring glutamine formation, is expressed throughout the entire cytoplasm of the astrocyte, not restricted to the filopodia and lamellipodia (Derouiche, 2004); and (iv) the amount of extracellular glutamate may be smaller than that of K+ released from neurons during the entire propagation of the action potential.
In addition to responding to and acting on neuronally released compounds, astrocytes can influence the activities of neurons by releasing signaling agents. Astrocytes cause neuronal excitation by exocytotic and cytosolic release of glutamate (Evanko et al, 2004; Volterra and Meldolesi, 2005), or they induce metabolic inhibition by release of ATP, which is metabolized extracellularly to form adenosine (Newman, 2003). Stimulation of astrocytes by neuronally released glutamate generates an increase in free cytosolic Ca2+ concentration ([Ca2+]i) in the astrocytes, which either can remain locally and lead to exocytotic glutamate release from astrocytes, or travel through an astrocytic syncytium as a Ca2+ wave, mediated by transport of IP3 through gap junctions and/or by ATP release and subsequent stimulation of purinergic receptors (Cornell-Bell et al, 2004; Haas et al, 2006). Exocytotic glutamate release and propagation of Ca2+ waves (including reestablishment of a low resting [Ca2+]i) require energy, and release of ATP as a transmitter represents utilization of an energy-rich molecule, which subsequently has to be re-constituted by active uptake of adenosine and the sequential re-synthesis of adenosine monophosphate (AMP), adenosine diphosphate (ADP), and ATP (Peng et al, 2005). No details or quantitative estimates of energy demand for these functions and filopodial motility are available.
Summary: Regulation of the extracellular fluid composition by uptake of K+ and extrusion of Na+ after its uptake together with glutamate are major energy-requiring processes, especially in the peripheral astrocytic processes. These structures are devoid of mitochondria but contain glycogen deposits and express glycogen phosphorylase activity. K+ uptake stimulates glycogenolysis in a concentration-dependent manner, and the glycolytic pathway fuels the initial part of this energy-requiring process. Respiration supports the later phase of the K+ uptake, presumably by diffusion of phosphocreatine and/or ATP generated in centrally located mitochondria. Filopodial motility, astrocytic signaling to neurons, and to other astrocytes by release of glutamate and of ATP and increases in [Ca2+]i place additional, quantitatively-unknown demands for energy on astrocytes.
Glucose Metabolism in Astrocytes
Pathways for Glucose Metabolism and their Energetic Yield
Introduction: Astrocytes are sometimes considered to have low energy requirements and correspondingly minor ATP production rates. An extremely low rate of oxidative metabolism in primary cultures of rat astrocytes was reported by Itoh et al (2003) and may be due, in part, to the use of a very high glucose concentration in the culturing medium, which reduces oxidative capacity by about 50% (Abe et al, 2006). In contrast, high respiratory rates in freshly isolated and cultured astrocytes with 6 to 7 mmol/L glucose as the metabolic fuel have been consistently observed by Hertz and co-workers (Hertz and Schousboe, 1975; Hertz and Hertz, 1979). Accordingly Hertz and Schousboe (1975) concluded that the relative contribution of astrocytes to cerebrocortical oxygen consumption corresponded to the relative volume of astrocytes in brain cortex, a conclusion in agreement with the recent NMR observations in brain in vivo described above.
In the brain in vivo different subcellular regions of astrocytes have different functions and capacities for glycolytic and oxidative energy generation, thereby preferentially influencing the fluxes of different pathways to satisfy local energy demands. Astrocytic ‘work’ consumes ATP and generates ADP, an obligatory substrate for and regulator of energygenerating reactions (see Hertz and Dienel, 2002). Overall glucose utilization is measured at the hexokinase step that generates G6P from glucose (Figure 5) by the deoxyglucose method (Sokoloff et al, 1977) using autoradiography or PET. Oxidation rates are evaluated by NMR spectroscopy and modeling of rates of incorporation of label from labeled precursors (glucose, acetate) into TCA cyclederived amino acids (mainly glutamate and glutamine, Figure 2), and redox shifts are detected by changes in the concentrations of endogenous fluorescent metabolites (e.g., cytoplasmic and mitochondrial nicotinamide adenine diucleotide (NADH) and mitochondrial flavin adenosine dinucleotide). Interpretation of signals derived from metabolic activity requires understanding of the major pathways, their interrelationships, and regulation, and their energy yields.
Glucose utilization pathways that provide or consume ATP (
Glycolysis generates adenosine 5' triphosphate, redox substrates, and precursors for biosynthesis and oxidative metabolism: The first step for glycolytic metabolism of glucose (Figure 5A; see legend for more details) is formation of G6P by hexokinase. Hexokinase is feedback inhibited by G6P, and in the presence of mitochondria, it preferentially uses oxidatively derived ATP (see Hertz and Dienel, 2002). The major regulatory enzyme governing glycolytic flux is phosphofructokinase, which converts fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6P), and is activated by ADP and AMP and inhibited by ATP and phosphocreatine. As illustrated in Figure 5B, production of 2 pyruvate from glucose requires 2 ATP and generates 4 ATP, for a net yield of 2 ATP.
The glycolytic process leading to the formation of pyruvate involves an oxidative, NADH-generating step. This creates a potential problem for the cell because the NAD must be rapidly regenerated by coupling to an oxidative reaction for glycolysis to continue, and NADH cannot cross the mitochondrial membrane. When the glycolytic rate exceeds that of pyruvate oxidation or during hypoxic–anoxic conditions, NADH is reoxidized by conversion of pyruvate to lactate (Figure 5A). In normoxic brain, cytoplasmic reoxidation of NADH occurs in the malate-aspartate shuttle (MAS) (Figure 2A). Cytoplasmic NAD+ is regenerated by conversion of oxaloacetate to malate and subsequent transfer of malate into the mitochondria, where it is oxidized. Completion of the shuttle process requires transfer of the oxaloacetate precursor, aspartate, in the opposite direction and linkage to mitochondrial conversion of malate to oxaloacetate to generate mitochondrial NADH that is oxidized via the electron transport chain, generating ~2.5 ATP (~5 ATP per molecule of glucose, Figures 2 and 5B). If pyruvate is reduced to lactate, the 5 ATP generated via MAS are not produced.
An alternative pathway for transfer of reducing equivalents, the glycerol phosphate shuttle, has lower activity in brain (Siesjö, 1978; Nguyen et al, 2003). Evidence that MAS (or another mechanism for transfer of reducing equivalents) functions in astrocytes is provided by the ability of both cultured and noncultured astrocytes to efficiently oxidize glucose (Dienel and Cruz, 2006).
The glycolytic pathway in astrocytes has several important branch points (Figure 5A) that increase the fraction of glucose metabolized by nonoxidative routes, lower ATP yield per glucose consumed Figure 5B, and reduce the CMRo2/CMRglc ratio below 6. Carbon is diverted for synthesis of L-serine and glycine, as well as for the astrocyte-specific synthesis of glycogen and D-serine, a neuromodulator; these biosynthetic steps consume ATP (Figure 5A). Glucose-6-phosphate also enters the pentose shunt pathway, which is present in all brain cells and produces substrate for synthesis for nucleic acids, as well as nicotinamide adenine dinucleotide phosphate (NADPH) for reductive biosynthesis (e.g., of lipids) and for hydrogen peroxide detoxification by glutathione peroxidase. Pyruvate can be converted to alanine, lactate, oxaloacetate, or acetyl coenzyme A (CoA).
Glycogen: an endogenous astrocytic source of glucose-6-phosphate: Glycogen and its degrading enzyme glycogen phosphorylase are located throughout the astrocyte including in the PAPs (Peters et al, 1991; Hamprecht et al, 2005), and glycogen is synthesized from G6P (Figure 5A) at a cost of 2 ATP per glucose moiety incorporated (Figure 5B). Glucose is the major source of G6P, but it can also be formed via gluconeogenesis from pyruvate/lactate and TCA cycle constituents and their derivatives, including glutamate. This multistep process (see Figure 5A and legend for details) consumes much more energy than glycogen synthesis from glucose (Figure 5B) and requires two astrocyte-specific enzymes, fructose-1,6-bisphosphatase (Schmoll et al, 1995) and PC (Yu et al, 1983; Shank et al, 1985). Pyruvate carboxylation is required to produce the TCA cycle intermediate oxaloacetate (Figure 1B), from which phosphoenolpyruvate, then fructose1,6-phosphate and, ultimately, G6P are generated (Figure 5A).
Glycolytic breakdown of preformed glycogen to produce 2 pyruvate from one glucosyl unit has a net yield of 3 ATP (Figure 5B). Glycogenolysis has a 50% higher yield compared with glycolytic metabolism of glucose because glycogen phosphorylase forms G1P from glycogen plus inorganic phosphate; G1P and G6P are in equilibrium via a mutase reaction, and no ATP is required to generate G6P (Figure 5A). Many signaling molecules and neurotransmitters stimulate glycogenolysis; the glycogen phosphorylase isozymes are highly regulated, and stimulated by protein kinase A and elevated [Ca2+]i (Hamprecht et al, 2005). The capability for rapid response and higher yield of ATP by utilization of glycogen rather than of glucose may represent a major advantage during acute energy demand, particularly in the astrocytic filopodia and lamellipodia.
In contrast to net degradation of stored glycogen, stimulation of glycogen turnover, such that blood-borne glucose is, in effect, rapidly metabolized to pyruvate via glycogen (a ‘glucose–glycogen shunt’; Shulman et al, 2001), yields a net gain of only 1 ATP. Near-simultaneous synthesis and degradation of glycogen consumes 2 ATP and produces 3 ATP per glucose moiety, for a net ATP yield half that of glucose processed strictly via glycolysis (Figure 5B). The resulting increase in ADP in peripheral astrocytic processes would stimulate glycolysis, and twice as much glucose would be required to produce the same amount of glycolytically derived ATP. Glucose–glycogen shunting during brain activation would therefore stimulate glycolysis and lead to a disproportionately larger utilization of glucose compared with oxygen and reduce the CMRo2/CMRglc ratio even during normal, normoxic conditions. This shunting may occur in brain in vivo as suggested by the observation by Schmalbruch et al (2002) that the β-adrenergic antagonist propranolol, which impairs transmitter-induced glycogenolysis, abolished the CMRo2–CMRglc mismatch in stimulated rats.
Lactate: a metabolite at the crossroad of glycolysis and oxidation: Lactate concentration is the net difference between its rates of formation and clearance, that is, the summation of rates of glycolysis, glycogenolysis, oxidative metabolism, diffusion, and facilitated transport. Simultaneous changes of different parameters complicate interpretation of lactate level as changes in rates of metabolic pathways because metabolite level does not provide flux information, only net formation versus disappearance. All brain cell types can produce and oxidize lactate, and changes in any or all of the above processes, as well as local pH and oxygen availability, can affect the lactate level. Lactate has received a large amount of attention as a possible endogenous fuel, but this role is particularly difficult to evaluate in living brain because of many technical analytical difficulties and the complexity of the factors that can influence lactate level.
A doubling of its concentration to ~ 2 mmol/g wet wt is often observed during brain activation (Prichard et al, 1987; Dienel et al, 2002), but the net gain corresponds to a very small fraction of total glucose plus glycogen consumed during the assay interval (Dienel and Cruz, 2004). A small (~10%), brief initial dip in lactate level after stimulation has been reported and modeled to represent cellular uptake (Aubert et al, 2005). However, the lactate responses used for this modeling, a brief fall and subsequent, more prolonged rise, were generated only by a 5 secs electrical stimulus, whereas shorter stimuli (1, 2, 3, or 4 secs) caused no change in glucose, lactate, or oxygen levels even though cellular activation is likely, suggesting a threshold response. Modeling has also been performed based on changes in NADH/NAD+ ratio, which may be increased during enhanced glycolysis or glycogenolysis and decreased by an ADP-mediated stimulation of oxidative phosphorylation (e.g., Kasischke et al, 2004). Like lactate content, this ratio is, however, not an unequivocal indicator of metabolic fluxes.
After metabolic activation, it is essential that the intracellular concentration of lactate does not rise to reach levels that would impair continued aerobic glycolysis by its effect on the redox state of the cytosol (Siesjö, 1978; Dienel and Hertz, 2001). It is not known where or if extracellular lactate is metabolized, or whether it is released from the tissue, or both. Local lactate level in astrocytes can be rapidly reduced by its transport through the astrocytic syncytium by barrier-free diffusion (Medina and Tabernero, 2005) and by release to extracellular space, facilitated by the low-affinity, highcapacity monocarboxylate transporters (MCTs) in astrocytes (Hertz and Dienel, 2005; Aubert et al, 2005). If elevated extracellular lactate re-enters cells, it is more likely to enter other astrocytes than neurons because neurons express the high-affinity MCT2 that is 60% saturated at the resting lactate level of 1 μmol/g wet wt (Hertz and Dienel, 2005). However, the most important parameter governing sustained net cellular lactate uptake is intracellular metabolism, which is necessary to maintain a low intracellular concentration of lactate needed for concentration gradient-driven net uptake (Dienel and Hertz, 2001; Hertz and Dienel, 2005).
Pyruvate dehydrogenase-mediated pyruvate entry into the oxidative pathway. As the CMRo2/CMRglc ratio is close to 6 in resting brain, most pyruvate formed from blood-borne glucose must be oxidatively decarboxylated via the highly regulated mitochondrial pyruvate dehydrogenase (PDH) step to form acetyl CoA. This process generates 2.5 adenosine 5' triphosphate (Figure 2A and its legend) per pyruvate and consumes oxygen via the electron transport chain when NADH is oxidized to regenerate NAD+. Acetyl CoA condenses with oxaloacetate to form the six-carbon intermediate, citrate, in a reaction common to neurons and astrocytes. During each turn of the TCA cycle, 2 CO2 are produced, oxygen is consumed, 10 ATP are produced and the four-carbon catalytic intermediate, oxaloacetate, is regenerated. Oxidation of the 2 cytosolic NADH via the MAS adds 5 ATP, for a total of 30 ATP from oxidation of one molecule of glucose; this is lower than earlier estimates of 36 because some proton leakage occurs across the mitochondrial membrane. Inclusion of the 2 ATP from glycolysis yields 32 ATP for each molecule of glucose metabolized (Figure 2A).
Pyruvate carboxylation and oxidative degradation of tricarboxylic acid cycle intermediates and their derivatives: The energy balance described above does not account for biosynthesis of any TCA cycle intermediate, a process that requires net, not catalytic, formation of oxaloacetate (Figure 2B). In brain, this reaction is predominantly or exclusively catalyzed by PC (see Hertz and Dienel, 2002), which can be regarded as astrocyte-specific based on several lines of evidence: (i) the only existing immunohistochemical study (Shank et al, 1985) shows astrocytic expression of PC, as in the cerebellar Bergmann fibers, but no neuronal expression (Figure 6A); (ii) in cultured cells, PC activity is higher in mature astrocytes than in adult brain cortex, where the astrocytes are ‘diluted’ by neurons, but it is absent in neurons (Figure 6B); (iii) pyruvate carboxylation occurs in cultured astrocytes, but not in cultured neurons (Figure 6C). The possibility of pyruvate carboxylation in oligodendrocytes has not been directly investigated, but a histochemical study in an ‘astrocyte-enriched’ culture (versus a highly purified astrocyte culture) by Cesar and Hamprecht (1995) revealed no cells with apparent oligodendrocytic morphology that were immunostained for this enzyme. Thus, astrocytes carry out net synthesis of TCA cycle intermediates and their derivatives, including glutamate, GABA, aspartate, and glutamine, whereas neurons on their own cannot produce these amino acid transmitter precursors and transmitters.
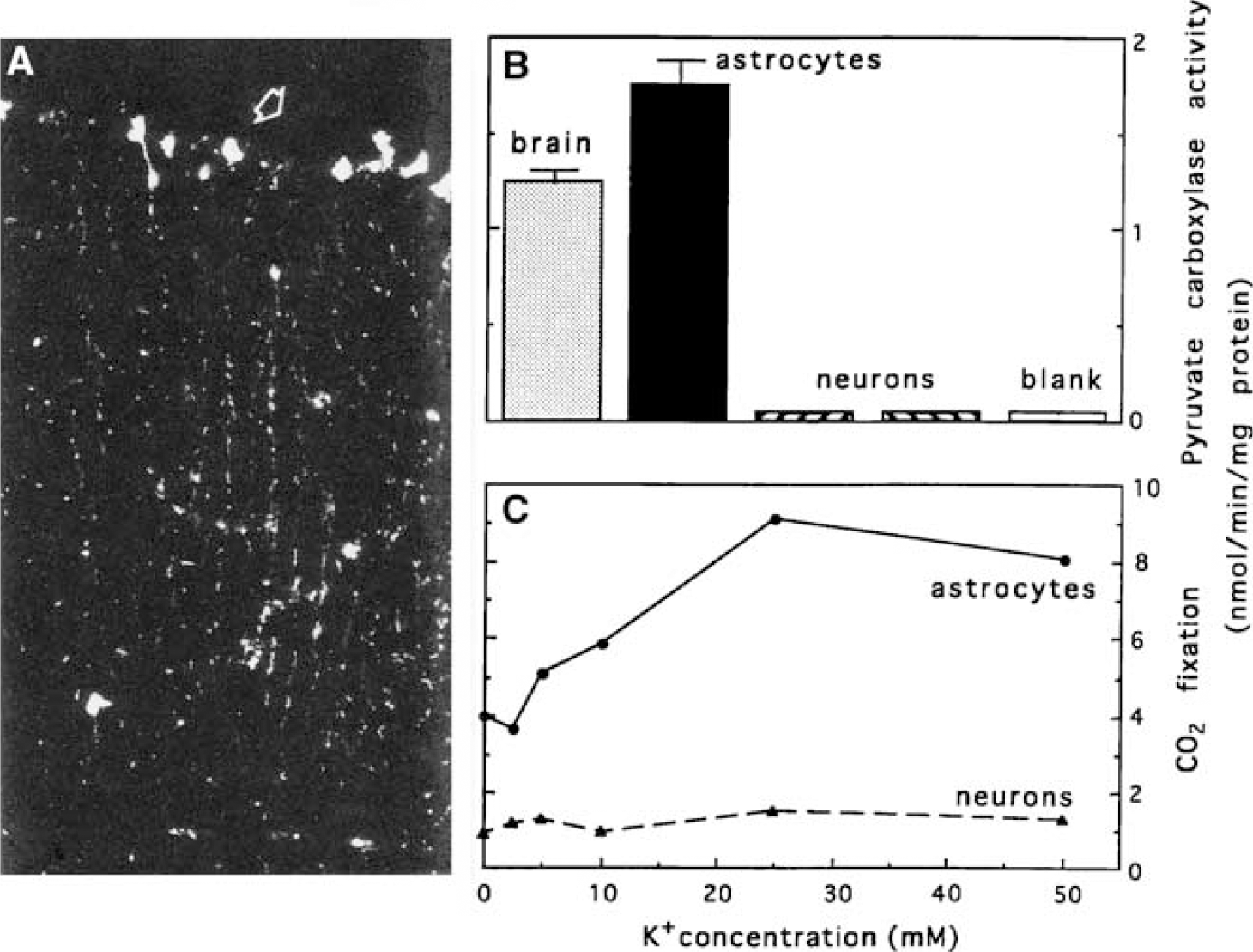
Pyruvate carboxylase expression, PC activity, and pyruvate carboxylation are astrocyte-specific. (
The astrocytic TCA cycle generates ATP during net synthesis of glutamate and glutamine, but not of aspartate, which is formed by transamination of oxaloacetate. Glutamate synthesis requires two pyruvate molecules, one for oxaloacetate formation, the other for synthesis of acetyl CoA; together they generate a ‘new’ 6-carbon compound that is decarboxylated to form α-ketoglutarate, and transaminated to form glutamate. This biosynthetic process involves partial oxidation of glucose and requires oxygen consumption, with net yield of 11 ATP, derived as sketched in Figure 2B. Pyruvate carboxylation occurs continuously in brain of the awake, resting subject (Section Fluxes of glucose oxidation pathways at the cellular level in brain in situ), and under steady-state conditions, when amino-acid levels are constant, there is a stoichiometrically equivalent rate of degradation of glutamate, glutamine, aspartate, and GABA.
Complete oxidation of the amino-acid transmitters and glutamine requires exit of a 4-carbon compound from the cycle, mainly or exclusively in astrocytes (Sonnewald et al, 2005) and re-entry as acetyl CoA (Figure 7; see its legend for details). The first step in glutamate degradation is formation of α-ketoglutarate, in mouse astrocytes mainly by oxidative deamination (Yu et al, 1982), and malate formation from glutamate via α-ketoglutarate yields a minimum of 4 ATP plus one GTP. When malate exits from the cycle and is converted to pyruvate more ATP is generated. After its exit to the cytosol, malate is decarboxylated to pyruvate by malic enzyme (Figure 7), which is expressed in both astrocytes and neurons. Neurons have only the mitochondrial isozyme, whereas astrocytes have mitochondrial and cytosolic isozymes (reviewed by Hertz and Dienel, 2002). For thermodynamic reasons, this enzyme operates in the direction of oxaloacetate decarboxylation, and a reported carboxylation of pyruvate to malate in neurons by reversal of its direction is not quantitatively significant (see Hertz and Zielke, 2004). Astrocytes also accumulate glutamate much more avidly than neurons (Danbolt, 2001), and most, if not all, oxidative decarboxylation of malate occurs in astrocytes, as verified in mouse brain by comparison between labeling of the C2 carbon in lactate (formed from pyruvate) by [2-13C]acetate and [1-13C]glucose via TCA cycle intermediates (Hassel et al, 1995; Sonnewald et al, 2005). Re-entry of pyruvate into the TCA cycle as acetyl CoA completes the overall process involving 5 oxidative decarboxylation steps, generating a total of 20 ATP and consuming 4 molecules of oxygen (Figure 7).
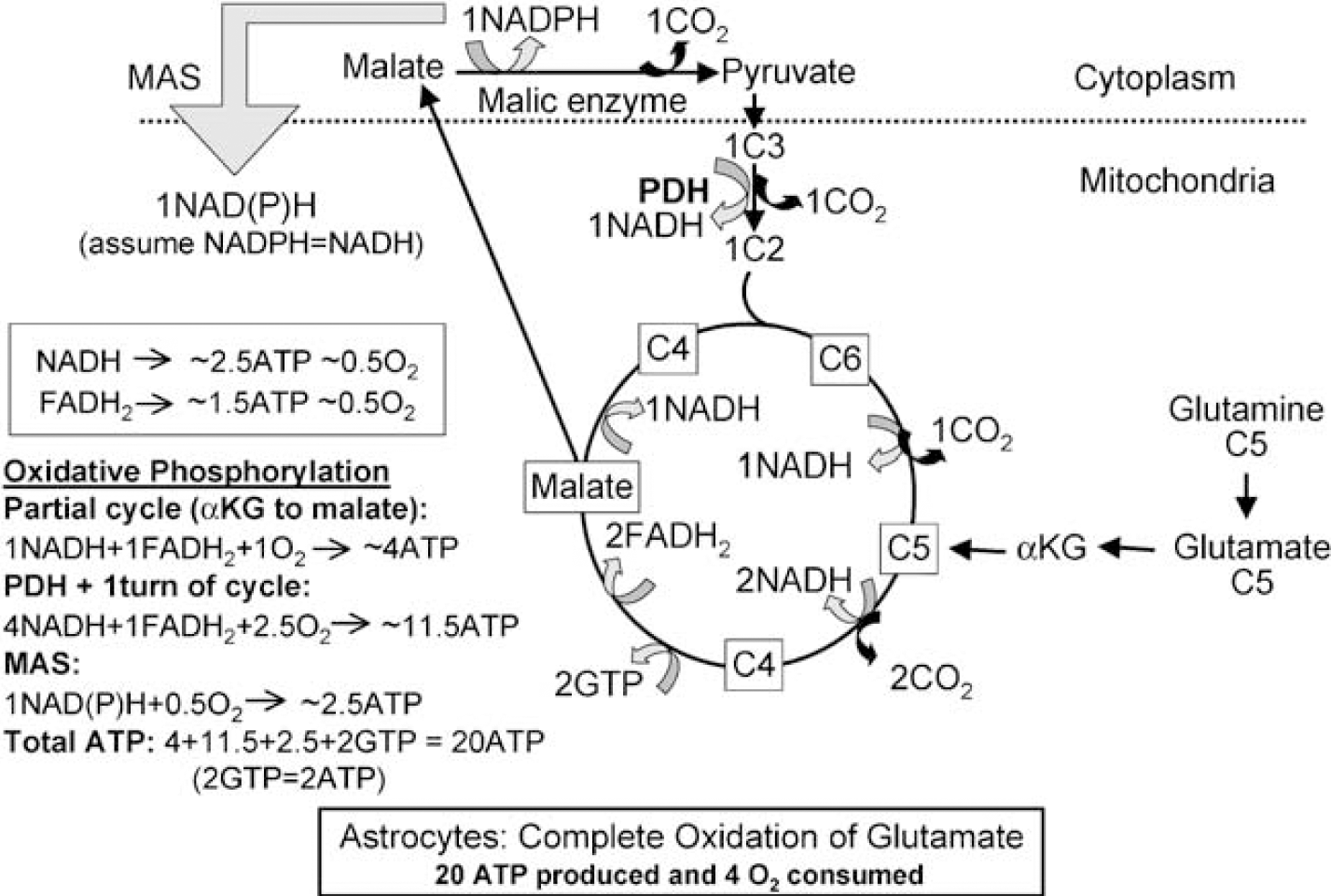
Metabolism and energetics of glutamate and glutamine oxidation in astrocytes. Astrocytic glutamate can be converted to α-ketoglutarate, metabolized in the TCA cycle to malate, which exits the TCA cycle to the cytoplasm, and decarboxylated by cytosolic malic enzyme to pyruvate, which subsequently can be converted to acetyl CoA, catalyzed by PDH, and metabolized in the TCA cycle. Glutamine can be metabolized in the same manner after initial conversion to glutamate. The partial turn of the TCA cycle from α-ketoglutarate (C5) to malate (C4), oxidative decarboxylation of malate to pyruvate, oxidative degradation of pyruvate by PDH mediated oxidative decarboxylation, and one turn of the TCA cycle altogether produce 20 ATP, provided the one NADPH which is formed in the cytosol is oxidized via MAS. Accordingly, complete oxidative oxidation of one molecule of glucose after conversion to glutamate (yielding 11 ATP (Figure 1B)) and subsequent oxidation of glutamate consumes the same amount of O2 as oxidation without any detour via glutamate and generates 31 ATP, that is, only one molecule less ATP than direct oxidation of glucose exclusively via PDH-mediated formation of acetyl CoA. If the newly synthesized glutamate is transferred to neurons in the glutamate-glutamine cycle after conversion to glutamine, requiring one ATP, net formation of ATP will be reduced by one. If the one pyruvate exiting the TCA cycle during complete oxidation of glutamate is not oxidized in astrocytes, the net production of astrocytic ATP will be reduced by 12.5 molecules and that of O2 consumption by 2.5 molecules.
The sum of the 31 ATP generated from glutamate turnover (11 from synthesis (Figure 2B) plus 20 from oxidation (Figure 7)) essentially equals the 32 derived from glucose oxidation (Figure 2A). Owing to the glutamate-glutamine neurotransmitter cycling process, more ATP is consumed for synthesis of glutamine, packaging of neuronal glutamate into synaptic vesicles, and the costs of astrocytic glutamate signaling (Volterra and Meldolesi, 2005). Also, much less energy would be recovered from glutamate degradation if the cytosolic pyruvate derived from malate were converted to lactate and released from the cell. Finally, if there is net glutamate synthesis during brain activation (Section Pyruvate carboxylation and glutamate content respond to activation), the yield of ATP will be greatly reduced, and the CMRO2/CMRglc ratio will decrease.
Summary: Net formation of ATP from glycolytic flux amounts only to 2 ATP per glucose molecule, whereas transfer of cytosolic reducing equivalents to mitochondria plus oxidation of pyruvate raises the total to 32. Recent studies suggest an important role for glycogen as a metabolic fuel in normal working astrocytes, probably especially in their thin peripheral processes. Net pyruvate formation from glycogen gives an 50% higher ATP yield than glycolysis, but rapid shunting of glucose via glycogen cuts the ATP yield in half and may stimulate glycolysis as a result of increased ADP accumulation in the peripheral processes. The lactate level is relatively low in resting brain and its rise during activation represents only a small fraction of the glucose and glycogen consumed. Simultaneous changes in lactate production and degradation (or transport) complicate interpretation of changes in lactate level, because metabolite level does not provide flux information, only net formation versus disappearance. Glutamate turnover is essentially an astrocytic process, and combined biosynthesis and oxidative degradation of glutamate produces almost as much ATP as glucose oxidation. Enhanced glycolysis, glycogenolysis, pentose shunt flux, and biosynthesis can contribute to lowering the CMRo2/CMRglc ratio during activation.
Determination of Oxidative Activity in Astrocytes Via Three Routes into the Tricarboxylic Acid Cycle
Introduction: Carbon flows into the astrocytic TCA cycle via PDH, via PC, and via the acetyl CoA formed from acetate (Figure 2), as well as from ketone bodies and medium chain-length fatty acids, including octanoate (Edmond, 1992). All substrates entering the cycle via these routes are at least partially oxidized and generate much more ATP per mole than glycolysis. Fluxes of these pathways in astrocytes in vivo can be quantified by use of specific labeled precursors and assay of labeled products, as described below.
Acetate utilization in brain of normal conscious rats is 15% to 25% of CMRgIc: Acetate is a normal constituent of plasma at a low concentration (< 1 mmol/L) that is readily transported into astrocytes by facilitated diffusion and quickly oxidized via the TCA cycle. Acetate serves as a ‘reporter molecule’ for astrocytic oxidative metabolism due to its cell-type specificity, as shown in a histochemical study by Muir et al (1986); acetate-derived label was highly localized in astrocytes compared with neurons in cerebellar cortex, cerebral cortex, and retinal Müller cells. Also, astrocytes in primary cultures rapidly produce 14CO2 from [1-14C]acetate, whereas virtually no 14CO2 production or acetate uptake occur in synaptosomes (Figure 8A) or cultured neurons (Figure 8B). In cultured astrocytes, net accumulation of labeled acetate metabolites within the cell rises within 10 mins to levels exceeding by 20- to 30-fold the content of intracellular acetate that would be attained by its passive equilibration with that in the extracellular medium (Figure 8B); this time-dependent increase in labeled products of a compound taken up by ‘facilitated’ diffusion is a ‘metabolism-driven’ uptake (Hertz and Dienel, 2005). Label accumulation is due mainly to isotope ‘trapping’ by dilution in larger glutamate and glutamine pools after net synthesis in astrocytes (Section Fluxes of glucose oxidation pathways at the cellular level in brain in situ), followed by labeling of other amino-acid pools by intercellular ‘metabolic trafficking’ and exchange reactions (Figure 8C).
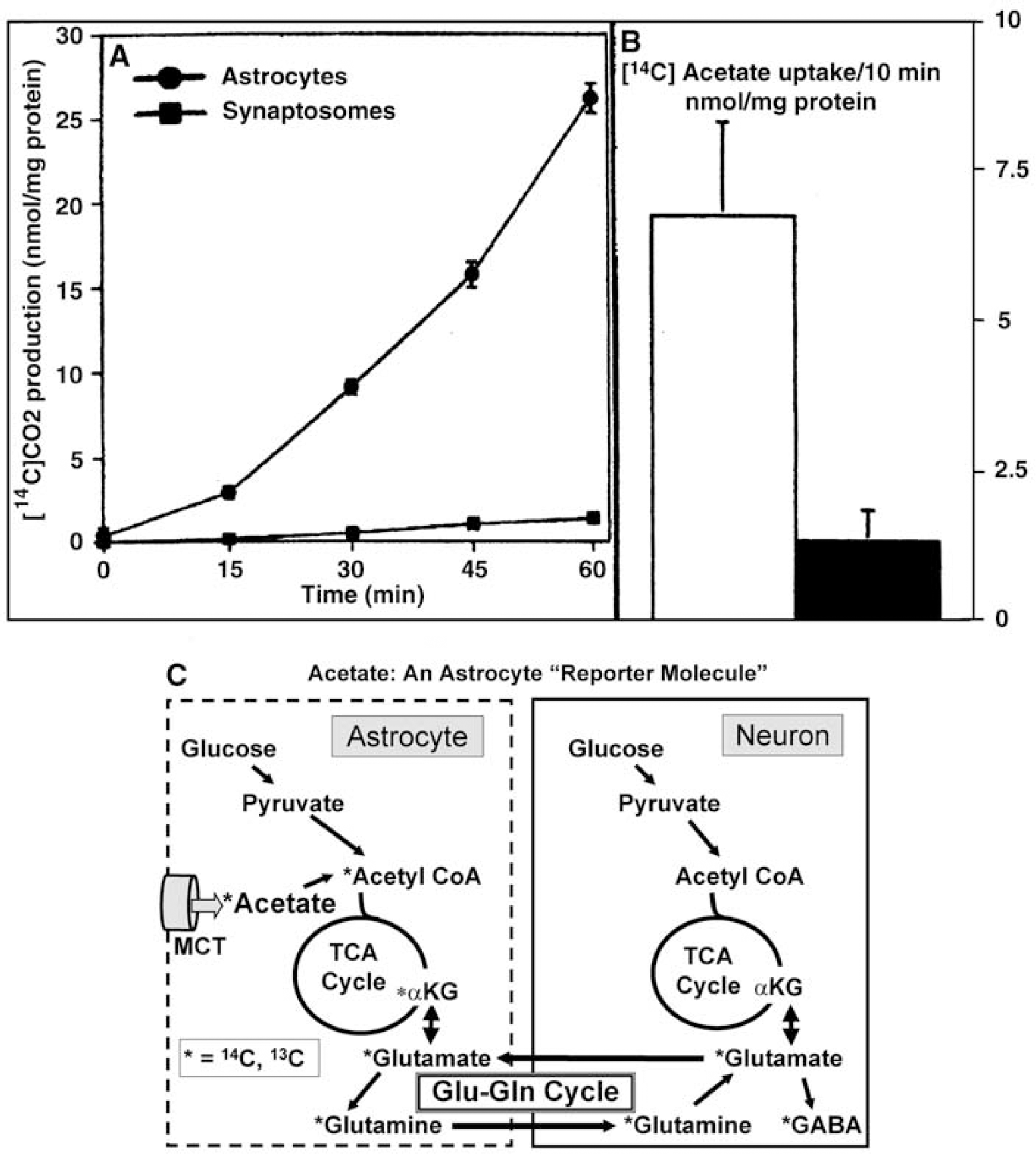
Acetate is an ‘astrocyte reporter molecule’. (
When normal, conscious rats were administered an intravenous pulse injection of tracer amounts of [2-14C]acetate, and local rates of acetate utilization in brain were calculated, the values in representative gray matter structures ranged from 0.11 to 0.16 μmol/g min, which are 15% to 25% of the corresponding total glucose utilization rates in all cells, measured in parallel with [14C]deoxyglucose (Cruz et al, 2005). Thus, oxidation of acetate, a minor blood-borne fuel, can generate a substantial amount of energy for astrocytes, and the rate of acetate utilization is similar to the 20% of total glucose oxidized via acetyl CoA in astrocytes (Table 1 and Section Fluxes of glucose oxidation pathways at the cellular level in brain in situ).
Metabolic fluxes in human and rat brain measured by nuclear magnetic resonance (NMR) spectroscopy
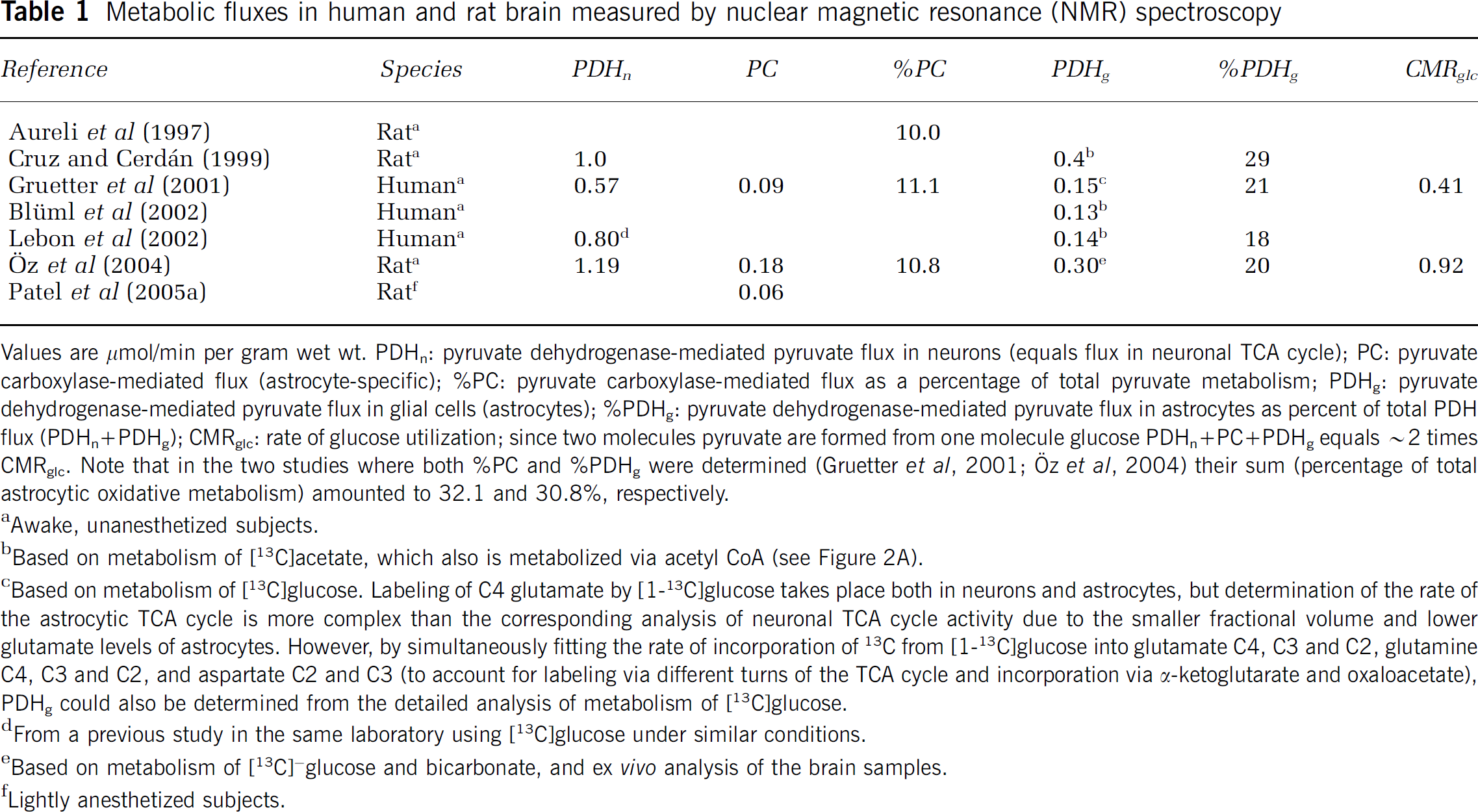
Values are μmol/min per gram wet wt. PDHn: pyruvate dehydrogenase-mediated pyruvate flux in neurons (equals flux in neuronal TCA cycle); PC: pyruvate carboxylase-mediated flux (astrocyte-specific); %PC: pyruvate carboxylase-mediated flux as a percentage of total pyruvate metabolism; PDHg: pyruvate dehydrogenase-mediated pyruvate flux in glial cells (astrocytes); %PDHg: pyruvate dehydrogenase-mediated pyruvate flux in astrocytes as percent of total PDH flux (PDHn+PDHg); CMRglc: rate of glucose utilization; since two molecules pyruvate are formed from one molecule glucose PDHn+PC+PDHg equals ~2 times CMRglc. Note that in the two studies where both %PC and %PDHg were determined (Gruetter et al, 2001; Öz et al, 2004) their sum (percentage of total astrocytic oxidative metabolism) amounted to 32.1 and 30.8%, respectively.
Awake, unanesthetized subjects.
Based on metabolism of [13C]acetate, which also is metabolized via acetyl CoA (see Figure 2A).
Based on metabolism of [13C]glucose. Labeling of C4 glutamate by [1-13C]glucose takes place both in neurons and astrocytes, but determination of the rate of the astrocytic TCA cycle is more complex than the corresponding analysis of neuronal TCA cycle activity due to the smaller fractional volume and lower glutamate levels of astrocytes. However, by simultaneously fitting the rate of incorporation of 13C from [1-13C]glucose into glutamate C4, C3 and C2, glutamine C4, C3 and C2, and aspartate C2 and C3 (to account for labeling via different turns of the TCA cycle and incorporation via α-ketoglutarate and oxaloacetate), PDHg could also be determined from the detailed analysis of metabolism of [13C]glucose.
From a previous study in the same laboratory using [13C]glucose under similar conditions.
Based on metabolism of [13C]~glucose and bicarbonate, and ex vivo analysis of the brain samples.
Lightly anesthetized subjects.
Fluxes of glucose oxidation pathways at the cellular level in brain in situ: Nuclear magnetic resonance methodology can identify labeled atoms in specific compounds, and modeling facilitates evaluation of in vivo rates of metabolism via different pathways. For example, metabolism of [1-13C]glucose leads to the formation of [4-13C]glutamate (and glutamine) in the first turn of the TCA cycle due to the bidirectional exchange reaction between α-ketogluta-rate and glutamate (Figure 2A), with an efficiency that depends on the rate of exchange compared with the cycling rate. Accordingly, label incorporation into glutamate is a measure of the rate of TCA cycle turnover (Sibson et al, 1998; Gruetter et al, 2001). Most glutamine labeling by [1-13C]glucose occurs after astrocytic metabolism of the glutamate that is labeled by the exchange reaction, mainly in neurons, because neurons occupy a larger volume than astrocytes and have a much higher glutamate content than astrocytes (Ottersen et al, 1992), facilitating isotope trapping. Therefore, glutamine labeling from [1-13C]glucose lags behind that of glutamate (Figure 9A) even though some ‘direct’ glutamine labeling must occur in astrocytes.
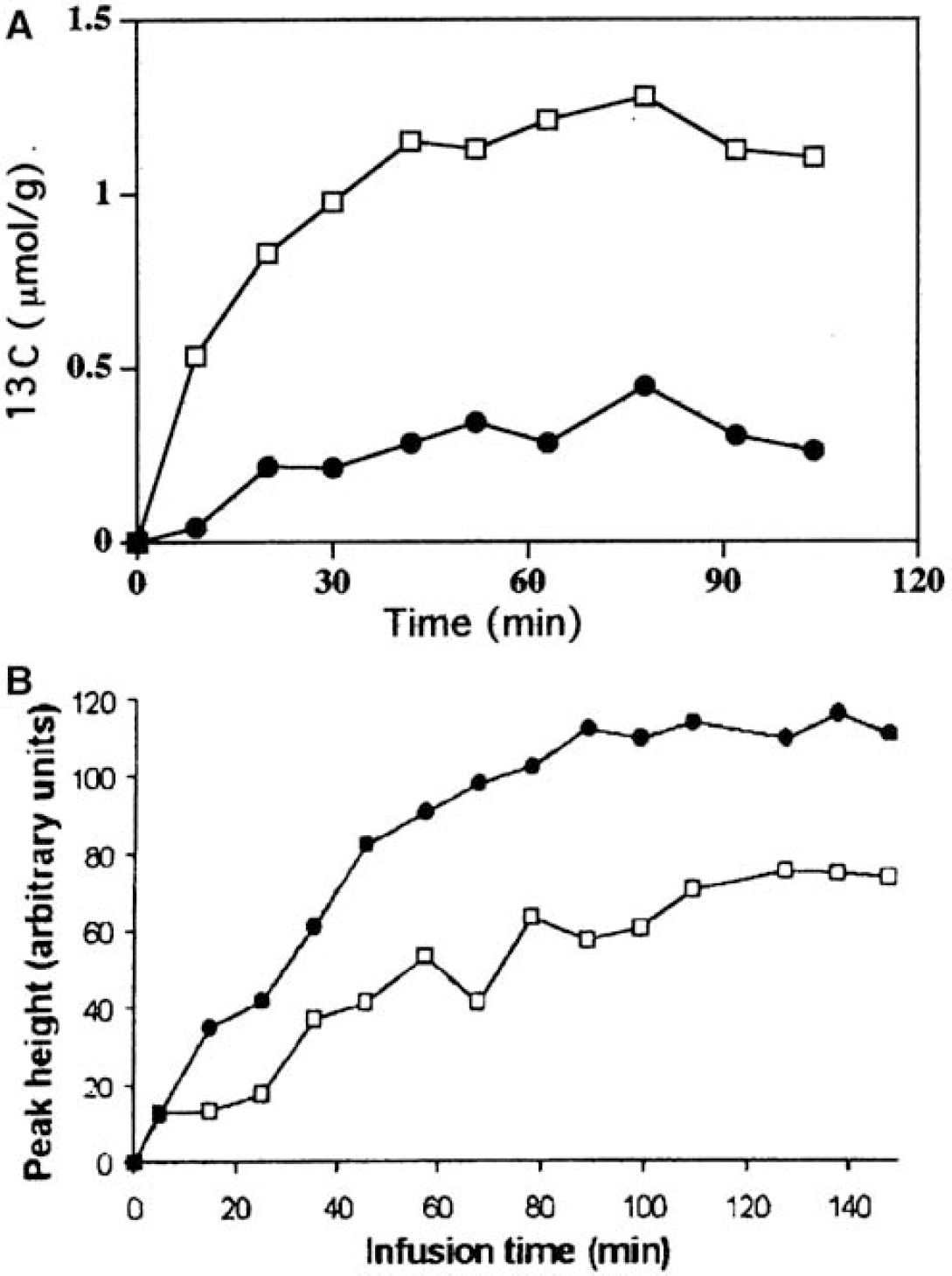
Labeling of [4-13C]glutamate and [4-13C]glutamine after in vivo administration of [1-13C]glucose or [2-13C]acetate. (
When the astrocyte-specific substrate, [2-13C]acetate, is the precursor, carbon 4 in glutamate and glutamine also becomes labeled, but in this case astrocytic [4-13C]glutamine is the precursor for neuronal [4-13C]glutamate, and the increase in labeling of [4-13C]glutamine precedes that of [4-13C]glutamate (Figure 9B). In contrast to exchange-labeling of glutamate in neurons (Figure 2A), glutamine labeling from acetate is due to net synthesis (Figure 2B). This conclusion is drawn from the fact that virtually no label from [1-14C]acetate was incorporated into glutamine in brain slices when acetate was the only substrate, whereas there was a pronounced incorporation when the incubation medium also contained unlabeled glucose (Gonda and Quastel, 1966). Inclusion of glucose enables pyruvate carboxylation, consistent with the requirement for net synthesis of glutamate/glutamine to take place for acetate to label glutamine. This finding also suggests that the α-ketoglutarate-glutamate exchange rate in astrocytes is slow compared with the TCA cycle rate, so that label from [1-14C]acetate is lost by the TCA cycle decarboxylation reactions faster than it can be incorporated via the exchange into glutamate/glutamine and trapped (Gonda and Quastel, 1966; Dienel et al, 2001); label loss would cause underestimation of astrocytic TCA cycle rates unless corrections are made.
Based on metabolism of [13C]acetate both Lebon et al (2002) and Blüml et al (2002) calculated the rate of the astrocytic TCA cycle to be ~ 20% of total oxidative metabolism in human brain, and Cruz and Cerdan (1999) calculated flux through the glial and neuronal TCA cycles in rat brain as 0.4 and 1 μmol/g mins, respectively (Table 1). Using [13C]glucose and a complex modeling approach based on label incorporation into additional metabolites (see legend of Table 1), Gruetter et al (2001) obtained rates for PDH-mediated TCA cycle activity in astrocytes in human brain that are similar to that obtained based on metabolism of [13C]acetate (Table 1).
When [1-13C]glucose is converted into pyruvate and introduced into the TCA cycle by pyruvate carboxylation, C2 of glutamate and glutamine becomes labeled, rather than the C4 that is labeled by the PDH-mediated reaction. Using this labeling pattern for modeling, rates of pyruvate carboxylation corresponding to ~10% of total pyruvate metabolism in brain (Table 1) have consistently been demonstrated in both animal and human brain (Aureli et al, 1997; Gruetter et al, 2001; Oz et al, 2004). Sizeable rates of pyruvate carboxylation have also been determined in rabbit brain, using a different NMR-based approach (Lapidot and Gopher, 1994).
Taken together, in vivo NMR spectroscopic studies show that the fraction of total oxidative metabolism ascribed to astrocytes by PDH-mediated formation of acetyl CoA and by pyruvate carboxylation is ~ 30%. These results are in good agreement with the high respiration rates in freshly isolated and cultured astrocytes found by Hertz and Schousboe (1975), and they form the basis for the conclusion that the relative oxidative contribution of astrocytes in the brain in vivo corresponds to their volume fraction (Section High oxidative rates in astrocytes in the brain in vivo). The high rate of oxidative metabolism in astrocytes is important for modeling studies, and the conclusions of a recent study by Aubert and Costalat (2005) hinges critically on the incorrect assumption that oxidative metabolism in astrocytes is four to five times lower than in neurons.
Astrocytic utilization of lactate: Nuclear magnetic resonance spectroscopy has also been used to try to evaluate the cellular metabolic fate of extracellular lactate in brain in situ. Bouzier et al (2000) and Tyson et al (2003) postulated that exogenous lactate is primarily oxidized in neurons in vivo, based on their observations that intravenous injection of large amounts of [3-13C]lactate gives rise to identical incorporation of label into the C2 and C3 positions of glutamate and glutamine, rather than to selective incorporation into the C2 position, indicative of pyruvate carboxylation. However, oxaloacetate produced by pyruvate carboxylation readily equilibrates with the symmetrical fumarate, thereby giving rise to labeling of both C2 and C3 in glutamate and glutamine. Moreover, pyruvate carboxylation from exogenous lactate in intact brain has been demonstrated by other investigators, although it was less pronounced than when glucose was the substrate (Qu et al, 2000).
Astrocytic utilization of exogenous lactate as an oxidative fuel has been conclusively demonstrated in cultured cells. However, direct comparison of relative rates of glucose and lactate oxidation are difficult to interpret because of the metabolic consequences of administering ‘loading’ (versus tracer) doses of labeled lactate and the many metabolic steps and metabolite pools between the labeled precursor and metabolites that must be analyzed to draw firm conclusions. Waagepetersen et al (1998) concluded that lactate is less efficient than glucose as a metabolic fuel for astrocytes, based on assays of trapping 13C in glutamate after incubation of cultured astrocytes with [13C]lactate or [13C]glucose. However, equivalent rates of oxidative release of 14CO2 from labeled glucose and lactate were observed in similar cultures, after correction for the correspondence of one glucose to two lactate (Peng et al, 1994; Abe et al, 2006). Rapid metabolism-driven accumulation of labeled products of [14C]lactate is also evident in cultured astrocytes (Dienel and Hertz, 2001).
In most ‘competition’ assays using mixtures of labeled plus unlabeled substrates (e.g., labeled glucose and unlabeled lactate, or vice versa), the extent of isotopic dilution of the pyruvate pool is not determined by direct measurement of pyruvate specific activity or isotopic enrichment. After determination of this value, it must be used in oxidation rate calculations as the true specific activity of the glucose-derived pyruvate instead of the initial value for glucose, which has been reduced by downstream dilution. Thus, an apparent change in rate of utilization of labeled glucose because of addition of unlabeled lactate could simply arise from isotope exchange between lactate and the small (~100 μmol/L) labeled pyruvate pool, rather than net conversion (reviewed by Hertz, 2004b). Recycling of extracellular lactate after isotope exchange with intracellular pyruvate is substantial in C6 glioma cells (Rodrigues et al, 2005) and in primary cultures of astrocytes, but less so in primary cultures of neurons (Fonseca et al, 2004). However, when large exogenous amounts of lactate are provided to C6 glioma cells, mass action-driven lactate transport and dehydrogenase-mediated conversion of lactate to pyruvate may lead to a high rate of lactate oxidation and concomitant reduction of glucose utilization (Rodrigues et al, 2005). Thus, here is no doubt that lactate can be a substrate for cultured astrocytes and glioma cells.
Blood-borne lactate is a fuel for human brain during exhaustive exercise: Severe muscular activity activates the brain by impulses to and from working muscles as well as by mental effort and causes plasma lactate levels to rise substantially. Lactate therefore enters the brain down its concentration gradient and is oxidized at a rate that equals glucose oxidation. Simultaneously, the metabolic ratio between (CMRo2) and the glucose plus lactate metabolized falls as low as ~3 (Dalsgaard, 2006). The cells that oxidize the lactate taken up into brain are not known, but this ratio reduction may occur for at least two reasons: (i) CSF, and presumably brain, levels of noradrenaline are greatly increased (Dalsgaard, 2006), suggesting a strong, widespread noradrenergic stimulation of glycogenolysis in astrocytes (Section Biogenic amines activate astrocytic metabolism). A large rise in glycogen utilization involving glucose degradation via glycogen (‘shunting’ of glucose via glycogen) would yield only 1 ATP per glucose, creating a deficit in glycolytically produced ATP and triggering a disproportionate increase in glycolysis compared with oxidative metabolism (Section Glycogen: an endogenous astrocytic source of glucose-6-phosphate); (ii) demand for glycolytically derived ATP or carbon (e.g., for filopodial transport processes or pyruvate carboxylation) would be expected to rise during activation, and contrary to glucose oxidation, no glycolytically derived energy is produced during lactate oxidation.
Summary: Oxidative metabolism in astrocytes via PDH-mediated formation of acetyl CoA accounts for 20% of total oxidative metabolism in brain cortex. Both pyruvate dehydrogenation and pyruvate carboxylation are required for net synthesis of TCA cycle constituents (Figure 2) and the rate of pyruvate carboxylation is about one-half of the PDH-mediated oxidative rate in astrocytes (see Table 1). Accordingly one half of the PDH flux in astrocytes in brain in vivo is used for biosynthesis. In other words, two out of every three pyruvate molecules metabolized in astrocytes are used for de novo synthesis of TCA cycle constituents and their derivatives, especially glutamate; a similar conclusion was reached by Hassel et al (1995). This has very little impact on the amount of energy generated, since glutamate formation plus its subsequent oxidation generates essentially the same ATP yield as direct PDH-mediated glucose oxidation.
Neurotransmitter Glutamate–Glutamine Cycling
Astrocyte-neuron interactions and excitatory neuro-transmission: Astrocytic and neuronal energy metabolism is closely linked via excitatory neurotransmission, glucose oxidation, and glutamate turnover. Most pyruvate carboxylation serves the purpose of generating glutamine, GABA, aspartate and especially glutamate (Hertz and Zielke, 2004; Oz et al, 2004). Under steady-state conditions, when glutamate synthesis and degradation are equal, glutamate formation accounts for most of the two thirds of the oxidative metabolism of glucose performed in astrocytes and 20% of glucose oxidation in brain (see Section Neurotransmitter glutamate-glutamine cycling). Evidence will be presented below (Section Pyruvate carboxylation and glutamate content respond to activation) that during excitation contents of glutamate and/or glutamine can increase, suggesting that rate of glutamate formation may have exceeded its rate of degradation.
Astrocytes also accumulate most of neuronally released transmitter glutamate (Danbolt, 2001) and convert part, but not all (Section Glutamate turnover in vivo), of it to glutamine by glutamine synthetase, an astrocyte-specific enzyme (Norenberg and Martinez-Hernandez, 1979; Derouiche, 2004). Glutamine then returns to neurons and is converted back to glutamate in a process called the glutamate–glutamine cycle (reviewed by Hertz and Zielke, 2004). Most glutamate newly synthesized in astrocytes from glucose also appears to be transported to neurons via this cycle (Lebon et al, 2002), although some intercellular trafficking of α-ketoglutarate cannot be excluded. Owing to the concept of cycling, it might be inferred that the same molecules continuously travel back and forth between neurons and astrocytes during the neurotransmission process, but this is not the case. Continuous oxidative degradation of a portion of the glutamate taken up from extracellular fluid (Yu et al, 1982; McKenna et al, 1996) requires astrocytic pyruvate carboxylation to compensate for this loss (Hertz, 2004a; Oz et al, 2004). Also, 20% to 25% of the glutamine carried in the cycle originates from neuronally-released GABA, not from transmitter glutamate (Patel et al, 2005b), so metabolite trafficking associated with the glutamate-glutamine cycle involves interchange among excitatory and inhibitory neurons.
Glutamate turnover in vivo: A high degradation rate of any biologic molecule confers the ability to rapidly change level according to prevailing conditions, because the rate of approach to a new concentration is governed by its half-life; the shorter the half-life, the faster the rise or fall. Glutamate appears to be such an ‘adaptive molecule’ because the turnover of at least a portion of the large glutamate pool corresponds to a high fraction of the in vivo cycle rate. From calculated rates of the glutamate-glutamine cycle (Sibson et al, 1998; Gruetter et al, 2001) and pyruvate carboxylation (Table 1), it can be concluded that de novo synthesis of glutamate in vivo equals 30% of the glutamate-glutamine cycle rate (Lapidot and Gopher, 1994; Gruetter et al, 2001; (Öz et al, 2004; Xu et al, 2004). Thus, at steady state when its level is constant, transmitter glutamate is, on average, only recycled a couple of times before being degraded. This conclusion consistent with the finding that interruption of the astrocytic TCA cycle by the astrocyte-specific toxin fluoroacetate abolishes glutamatergic transmission within minutes; continuous operation of the biosynthetic portion of the astrocytic TCA cycle is necessary for de novo synthesis of glutamate and neuronal function (reviewed by Hertz and Zielke, 2004).
Astrocytic oxidation of extracellular glutamate in astrocytes rises with its level: The more glutamate released during excitatory transmission, the higher the energy demand on astrocytes surrounding glutamatergic neurons. Glutamate and glutamine are oxidized in rat brain in vivo (Zielke et al, 1998), but cellular adjustments to intense demand are difficult to assess in vivo. This knowledge gap is filled by studies in cultured astrocytes that show that glutamate stimulates both respiration and its own oxidation in a graded manner. Exposure of astrocytes to 100 mmol/L glutamate enhances oxygen consumption by 50% (Eriksson et al, 1995). As the extracellular glutamate concentration is increased from 100 to 500 μmol/L (i.e., levels below the 1 to 3 mmol/L peak in the synaptic cleft), the fraction oxidized compared with that converted to glutamine rises from 10% to 50% (McKenna et al, 1996), and an even larger fraction of oxidation was reported by Yu et al (1982). When either glutamate or glutamine is the sole substrate for cultured astrocytes, a high rate of oxidative metabolism is maintained, sufficient to completely oxidize glutamate (Yu and Hertz, 1983; Hertz and Hertz, 2003). Amino-acid oxidation by astrocytes is directly demonstrable by NMR, and such studies prove that (i) metabolism of [13C]glutamate involves label re-entry into the TCA cycle after conversion to pyruvate (Waagepetersen et al, 2002), and (ii) there is formation of a lactate isotope that can only be formed when 13C-labeled aspartate, glutamate or glutamine exit the TCA cycle (Sonne-wald et al, 2005). These findings provide conclusive evidence that oxidative degradation of glutamate, aspartate, and glutamine is quantitatively significant and occurs mainly in astrocytes.
Summary: Glutamate turnover in astrocytes is an integral and dynamic component of excitatory neurotransmission and of energy metabolism. On average, a glutamate molecule is re-cycled only a few times in the glutamate-glutamine cycle before it is oxidatively degraded in astrocytes; de novo synthesis of glutamate from glucose in astrocytes compensates for oxidation. Under steady state conditions, when glutamate synthesis and degradation are equal, glutamate formation accounts for about 2/3 of the oxidative metabolism of glucose performed in astrocytes and 20% of glucose oxidation in brain.
Brain Activation and Neuromodulators Increase Astrocytic Energy Generation
Astrocytic Glycolysis and Glycogenolysis In Vivo Respond to Sensory Stimulation
Disproportionate stimulation of glycolysis: Working brain cells consume ATP and produce ADP, which stimulates phosphofructokinase and PDH activities thereby increasing flux through the glycolytic and oxidative pathways. Unfortunately, there are no cell type-specific tracers for glycolysis, contrasting the labeled tracers that are useful for dissecting out the fluxes of oxidative pathways in astrocytes and neurons. However, as discussed elsewhere, glycogenolysis (Section Glycogen: an endogenous astrocytic source of glucose-6-phosphate) and formation of glutamate (Section Pyruvate carboxylation and glutamate content respond to activation) are enhanced during many types of sensory stimulation along with a concomitant decrease in the CMRo2 /CMRglc ratio, so part of the glycolytic activation can be ascribed to astrocytes. A substantial activation of glycolysis is also consistent with results obtained during visual and acoustic stimulation of conscious rats. The stimulus-induced rise in glucose utilization is greatly underestimated when assayed with [14C]glucose compared with [14C]deoxyglucose, indicating very rapid formation and clearance from activated tissue of labeled metabolites of [1 or 6-14C]glucose (reviewed by Dienel and Cruz, 2004). As lactate has a high specific activity (half that of glucose) shortly after administration of [14C]glucose, more [14C]lactate is probably released compared with labeled CO2 and amino acids (Cruz et al, 1999). The precise cellular locus of glycolysis remains, however, to be identified, and novel experimental approaches will be required to evaluate lactate formation and release from activated astrocytic processes, which are likely to be highly glycolytic.
Stimulation of glycogenolysis in working astrocytes: When Swanson et al (1992) reported that glycogen is consumed during whisker stimulation, they changed the historical concept of the role of glycogen in the brain from being an energy reservoir used in emergency situations to that of an energy store that is rapidly consumed in response to enhanced neuronal activity. Since glycogen is located almost exclusively in astrocytes, there is no doubt that glycogenolysis is an astrocytic response. Even when blood and brain glucose levels are normal, glycogenolysis occurs during and after brain activation with an apparent rate of 0.5 mmol glucose equiva-lents/min per gram wet wt during activation (Dienel et al, 2002). This value approaches the entire glucose utilization rate in rat brain (0.7 mmol/min per gram wet wt; Sokoloff et al, 1977) under nonstimulated condition. Immediately after onetrial aversive training glycogen level in forebrain of day-old chickens falls by 0.8 mmol glucose equivalent min per gram wet wt (Hertz et al, 2003), and subsequently glycogen levels are restored to pretraining values at a rate of at least 0.2 to 0.3 μmol/min per gram. During spreading depression (Section TCA cycle activity responds to activation), the glycogen content in brain decreases by one quarter to one-third (Krivanek 1958; Lauritzen et al, 1990), and it can recover within 10 mins, indicating capacity for rapid resynthesis in vivo.
Glycogenolysis during brain activation raises intriguing, but currently unresolved questions as to the location and fate of generated lactate. Rates of glycogenolysis can approximate or exceed the net increase in CMRglc during stimulation, suggesting that during activation pyruvate/lactate production in astrocytes is probably higher than in neurons, consistent with the part of the hypothesis by Pellerin and Magistretti (1994) suggesting that brain activation increases glycolytic metabolism in astrocytes and with the demonstration by Kasischke et al (2004) of an increase in astrocytic NADH level. If lactate accumulates to high levels in the activated astrocytes in which it was generated, it would impair aerobic glycolysis (Siesjö, 1978; Dienel and Hertz, 2001), so lactate is probably released through gap junctions and by facilitated diffusion across the cell membranes, mediated by low-affinity monocar-boxylate transporters MCT1 and 4 (Hertz and Dienel, 2005). A decrease in the CMRo2/CMRglc ratio suggests that the pyruvate/lactate is not oxidized within the activated region (i.e., not by stimulated neurons or astrocytes) during activation because CMRo2 would then increase markedly (reviewed by Dienel and Cruz, 2004).
Different metabolic fates of glucose and glycogen metabolites: Pyruvate/lactate formed from glycogen may be segregated from that produced from blood-borne glucose in brain (Dienel and Cruz, 2004) and from extracellular glucose in cultured astrocytes (Sickmann et al, 2005); a small decrease in labeling of extracellular glutamine in the cultured astrocytes when glycogenolysis is inhibited may suggest that glycogen is preferred glutamate precursor. This notion is supported by the observation that glycogenolysis coincides temporally with increased forebrain contents of both glutamate and glutamine during memory formation in day-old chicks (Hertz et al, 2003). Moreover, after intracerebral injection of a glycogenolysis inhibitor in the chick brain, both a precursor for acetyl CoA (the astrocyte-specific acetate) and a precursor for oxaloacetate (aspartate) are required to overcome the lack of glycogenolysis and restore establishment of memory. In contrast, acetate alone can rescue memory formation after administration of unlabeled 2-deoxyglucose in sufficiently large amounts to inhibit transport and metabolism of glucose (Gibbs et al, 2006a, b). These observations show that pyruvate generated from glycogen in the non-inhibited brain is needed for pyruvate carboxylation and metabolism via acetyl CoA (as required for glutamate formation) whereas that generated from glucose is required exclusively for metabolism via acetyl CoA (i.e., for provision of energy). This does not mean that glycogen metabolism does not yield energy, and it should be kept in mind that glutamate formation creates ATP. Glycogen may also supply glucose-6-P for the NADPH-generating pentose phosphate pathway in astrocytes, even in the presence of normal levels of glucose, because rapid glycogen depletion occurs during peroxide treatment and activation of NAD(P)H-dependent glutathione redox cycling (Figure 5A) (Rahman et al, 2000).
Summary: On the basis of a limited number of in vivo studies, higher rates of astrocytic glycolysis can be inferred from stimulation of overall glucose utilization along with astrocytic oxidative activity and CO2 fixation during brain activation. Both glycogen synthesis and degradation increase during brain activation in normoglycemic animals, and can occur very rapidly. When glycogenolysis is taken into account, formation of pyruvate/lactate must in many cases of brain activation greatly exceed that in neurons, and lactate must be released from astrocytes. The generated lactate is in most cases not oxidized locally, and its fate is an important, but presently unanswered question.
Astrocytic Tricarboxylic Acid Cycle Activity and Pyruvate Carboxylation in Brain In Vivo Respond to Sensory Stimulation
Tricarboxylic Acid cycle activity responds to activation: Direct evidence that TCA cycle activity increases in activated astrocytes comes from assays of [14C]acetate incorporation into amino acids via the oxidative pathway in vivo. Unilateral acoustic stimulation by a broadband click stimulus significantly increases (by 15% to 18%) acetate utilization in two structures in the auditory pathway, inferior colliculus and lateral lemniscus, compared with contralateral structures (Figure 10A). The stimulus-induced percent rise in astrocytic oxidative activity is about 20% or 40%, respectively, of the increase in glucose utilized by all cells in these two structures during stimulation, as determined by the [14C]deoxyglucose method (Cruz et al, 2005). Photic stimulation of the retina also stimulates oxidative metabolism in astrocytes in activated pathways. During 16 Hz on-off photic stimulation [14C]acetate uptake increased ~ 20% (Figure 10B), a rise that was about one-third of that measured with [14C]deoxyglucose; in contrast, removal of retinal input to the superior colliculus reduced [14C]acetate uptake when assayed in the dark (Figure 10B). These autoradiographic images reflect acetate oxidative metabolism because 90% to 95% of the acetatederived label in brain extracts is recovered in metabolites, with only 5% to 10% in unmetabolized acetate; most label was recovered in purified glutamate and glutamine and the Gln/Glu specific activity ratio is high, consistent with in vivo acetate oxidative metabolism mainly via the ‘small’ astrocytic glutamate pool (Dienel et al, 2001; Cruz et al, 2005). Acetate utilization also increased in specific brain regions by 15% to 24% after behavioral training (Dienel et al, 2003).
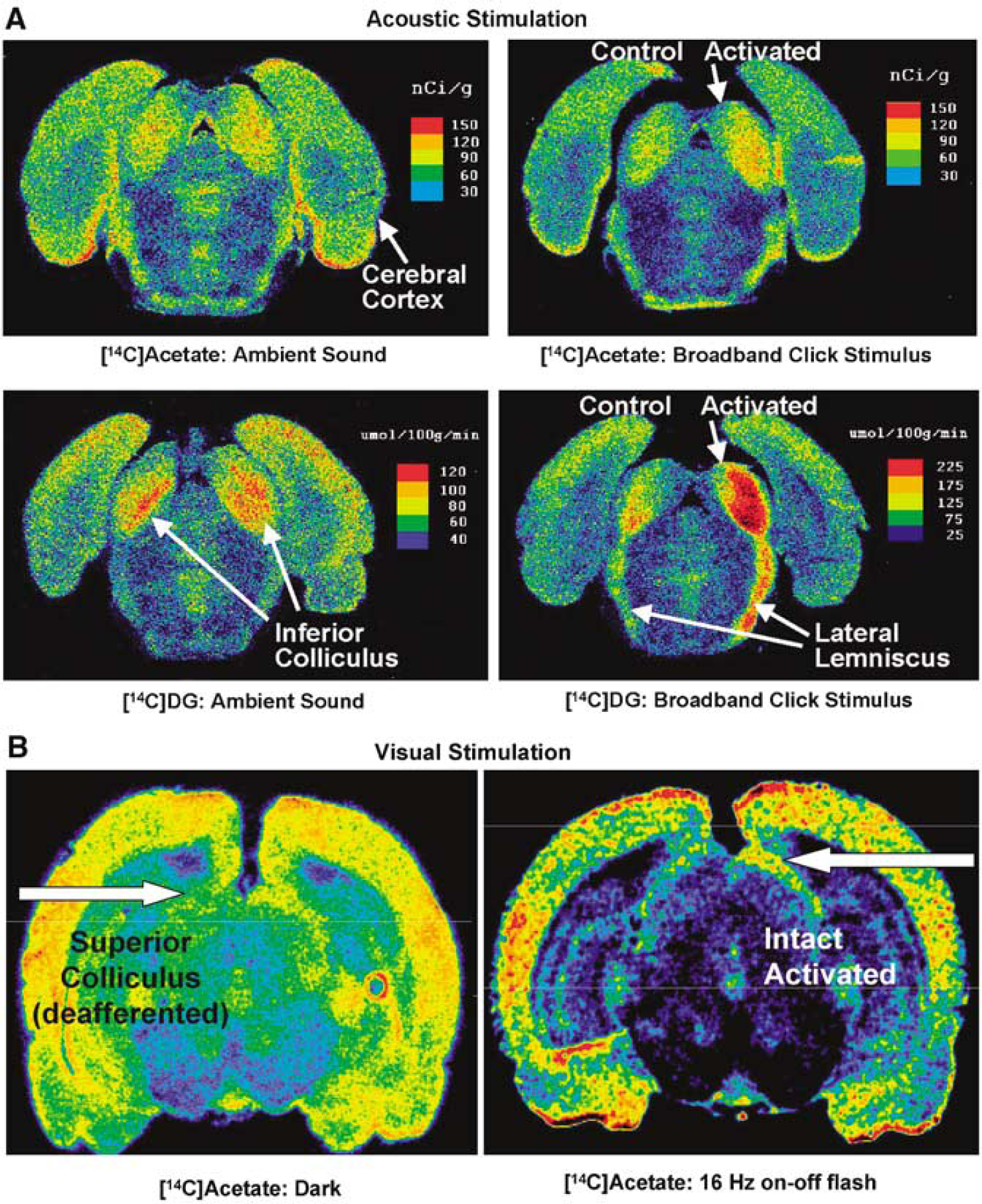
Functional metabolic activation of astrocytes by acoustic and visual stimulation. (
Pathophysiological conditions also alter metabolic demand in astrocytes and activate their oxidative pathway. For example, spreading depression is a peculiar electrophysiological phenomenon, during which a wave of suppression of electrical activity slowly spreads from its point of origin across the cortex. The wave is preceded by brief electrical hyperactivity, is accompanied by a substantial release and subsequent active re-accumulation of K+, mainly into astrocytes (Lian and Stringer, 2004), and causes a 40% increase in [1-14C]acetate utilization in tissue surrounding its origin, with an average increase of 23% throughout cerebral cortex (Dienel et al, 2001).
Pyruvate carboxylation and glutamate content respond to activation: CO2 fixation rates can also change under various experimental conditions, but not necessarily in the same direction as oxidative activity. In contrast to the higher rates of utilization of acetate during spreading depression, incorporation of labeled bicarbonate into the acid soluble metabolite pool fell by 14% (Dienel et al, 2001), and no significant increase in the rate of pyruvate carboxylation was found in the brains of lightly anaesthetized rats during bicuculline-induced convulsions (Patel et al, 2005a). However, pyruvate carboxylation increases 30% in the intact rat retina during dark adaptation, when both glucose metabolism and glutamatergic activity increase (KF LaNoue, personal communication), and it is several fold higher in awake compared with anaesthetized rats (Oz et al, 2004). Changes in amino-acid levels do not necessarily reflect increased synthesis, but the following constellation of changes, along with increased CO2 fixation, makes increased synthesis a likely interpretation: (i) glutamate content in visual cortex increases during visual stimulation (R Gruetter, Presented at the Third Wierzba Conference, 2005); (ii) glutamate and glutamine levels rise significantly in day-old chick forebrain during specific phases of one-trial passive avoidance training that is associated with increased glutamatergic activity and memory consolidation (Hertz et al, 2003); (iii) hippocampal glutamine content is increased in amygdaloid-kindled rats (Shirayama et al, 2005); and (iv) glutamate, aspartate, glutamine and GABA levels are elevated in a chronic phase of epilepsy after pilocarpine admistration to rats (Cavalheiro et al, 1994). Even when amino-acid levels do not change during activation (Cruz et al, 2005), synthesis rates may change in parallel with degradation rates.
Summary. During sensory stimulation acetate uptake and metabolism is increased as an indication of increased oxidative metabolism in astrocytes. Pyruvate carboxylation increases during some, but not all, types of stimulation and contents of glutamate and glutamine may increase, suggesting enhanced net synthesis. Besides functioning as fuel, glycogen may serve as preferred substrate for biosynthesis of glutamate/glutamine and NADPH. During these processes energy is also produced, but no ‘energy budget’ for astrocytes has been created that includes stimulation of astrocytic TCA cycle activity and biosynthesis in response to cerebral activation.
Sources of Astrocytic Energy to Support Glutamate Cycling are Debated
Glutamate turnover is a major component of astrocytic oxidative metabolism, and glutamate-glutamine cycling links brain function to astrocyteneuron metabolic rates. However, the source of energy to support the astrocytic components of this process cannot be readily determined in vivo, and results of in vitro studies are discrepant. There is consensus that glutamate uptake occurs in cotransport with Na+, which enters the cells along its electrochemical gradient and exits the cells by stimulation of the Na+, K+ -ATPase at its intracellular Na+ -sensitive site (Figure 3). However, the source of energy for ATPase activity and subsequent glutamine synthesis from part of the accumulated glutamate is debated because conflicting results have been obtained in different culture preparations.
Two groups of investigators, Pellerin and Magistretti (1994) and Sokoloff et al (1996), reported increases in glucose phosphorylation and lactate release to the medium, and concluded that glycolytically derived energy fuels glutamate uptake. However, these studies did not assess oxidative metabolism, and Eriksson et al (1995) demonstrated a large, ouabain-inhibited stimulation of oxygen consumption when 100 mmol/L glutamate was added to cultures respiring with glucose as the metabolic fuel (Figure 3). Since oxidative metabolism provides much more energy per glucose molecule than glycolysis, most of the required energy associated with glutamate uptake must have been obtained by oxidative metabolism (Hertz and Dienel, 2002). This conclusion is not per se in disagreement with an increase in glycolytic rate. However, six laboratories have observed either no effect or a decrease in glucose utilization, lactate formation, or glucose oxidation by exposure of cultured astrocytes to glutamate (see Table 5 in the review by Dienel and Cruz, 2004). Other relevant observations include (i) glutamate oxidation both by cultured astrocytes (e.g., Yu et al, 1982; Hertz and Hertz, 2003) and in the brain in vivo (Zielke et al, 1998); (ii) an increase in the fraction of glutamate oxidized in primary cultures of astrocytes compared with that converted to glutamine when the glutamate concentration increases (McKenna et al, 1996); (iii) stimulation of glucose phosphorylation by glutamate under anoxic, but not under normoxic conditions (Hertz et al, 1998); and (iv) stimulation of glucose phosphorylation by D-aspartate but not L-glutamate (Peng et al, 2001). As D-aspartate is accumulated by the same transporter as L-glutamate but is not metabolized, the latter finding is consistent with oxidative degradation of glutamate itself, thereby enabling glutamate uptake without an increase in glucose utilization (Peng et al, 2001). The basis for the apparently discrepant results obtained with various astrocyte preparations is not known, but different oxidative capabilities, perhaps partly dependent on glucose concentration of the culturing medium (Abe et al, 2006), could be a factor. Also, during typical cell culture assays the entire astrocyte is exposed to large amounts of bioactive compounds, and failure to specifically stimulate uptake in the peripheral astrocytic processes may limit the conclusions drawn.
To summarize, in vivo studies confirm high glucose oxidation rates in astrocytes, and a stimulation of acetate and glucose oxidation during activation. Glutamate is oxidized in the brain and in cultured astrocytes, with the fraction oxidized versus that converted to glutamine increasing at elevated glutamate concentrations. There is accordingly strong in situ evidence to support the hypothesis that glutamate stimulates astrocytic oxidative metabolism of available fuels. Glutamate does not stimulate glycogenolysis in brain slices (Magistretti et al, 1981), and it increases glycogen formation in cultured astrocytes (Swanson et al, 1990). It is not known if in vivo rates of glutamate oxidation change during activation or when activation is stopped or if glycolytic responses to glutamate occur in peripheral astrocytic processes. These are important issues for future work.
Fuel for Regulation of Extracellular K+ Concentration by Astrocytes
Differential stimulation of glucose phosphorylation by K+ in astrocytes and neurons: Clearance of excess extracellular K+ in intact brain (Figure 4A) is associated with two distinct phases of metabolic activation, first an increase in glycolysis, then a rise in oxidative metabolism (Figure 4B). Relationships between astrocytic and neuronal energy metabolism and elevated extracellular K+ concentrations ([K +]e) cannot be easily distinguished in in vivo studies, but results of in vitro experiments are useful to help to dissect out the concentration-dependent effects of [K+]e on Na+, K+ -ATPase activity, which is of considerable importance in astrocytes (Peng et al, 1998).
It has repeatedly been shown that an elevated [K+]e stimulates Na+, K+ -ATPase activity at its extracellular K+-sensitive site in cultured astrocytes and in astrocytes isolated from mature brain by gradient centrifugation, but not in corresponding neuronal preparations (Henn et al, 1972; Grisar et al, 1979; Mercado and Hernandez, 1992; Hajek et al, 1996), and elevated [K+]e reduces the ATP content in cultured astrocytes but not in neurons (Schousboe et al, 1970). In cultured astrocytes, maximal Na+, K+ -ATPase activity (Vmax) is attained at a [K+]e of ~ 12 mmol/L and it is three-fold higher than in cultured neurons; the astrocytic isoform has a lower affinity for [K+]e compared with that in neurons (Km's = 1.9 and 0.43 mmol/L, respectively) (Hajek et al, 1996). Thus, neuronal Na+, K+ -ATPase activity is saturated at normal [K+]e (~ 3 mmol/L), and ATPase activation by 5 to 12 mmol/L K+ would be astrocytic.
An elevation of [K+]e from 5 mmol/L (the normal level in culture media) to 10 to 12 mmol/L (within the range during physiological and some forms of pathological brain activation in vivo) increases deoxyglucose phosphorylation in primary cultures of mouse astrocytes but has no such effect on neuronal cultures (Figure 11A, stippled lines). A further increase in [K+]e has no additional effect on astrocytic metabolism (Peng et al, 1994), whereas a higher, strongly depolarizing concentration increases neuronal glycolysis substantially (Honegger and Pardo, 1999). Elevated [K+]e also increases lactate formation and release in cultured astrocytes (Walz and Mukerji, 1988). Sokoloff et al (1996) were unable to confirm metabolic stimulation by K+ in cultured rat astrocytes but this was subsequently verified by Abe et al (2006). Also, a similar or even larger stimulatory effect of low [K+]e on glucose utilization occurs in rat neuron–astrocyte co-cultures, whereas it was absent in purely neuronal cultures (Honegger and Pardo, 1999). The K +-dependent stimulation of glucose phosphorylation in astrocytes is completely inhibited by ouabain, a finding consistent with its arising from K+ -mediated stimulation of Na+, K+ -ATPase at the [K+]e-sensitive site (Peng et al, 1996), perhaps combined with inhibition of Na+ -mediated stimulation at its intracellular site after cotransporter activation (Figure 3).
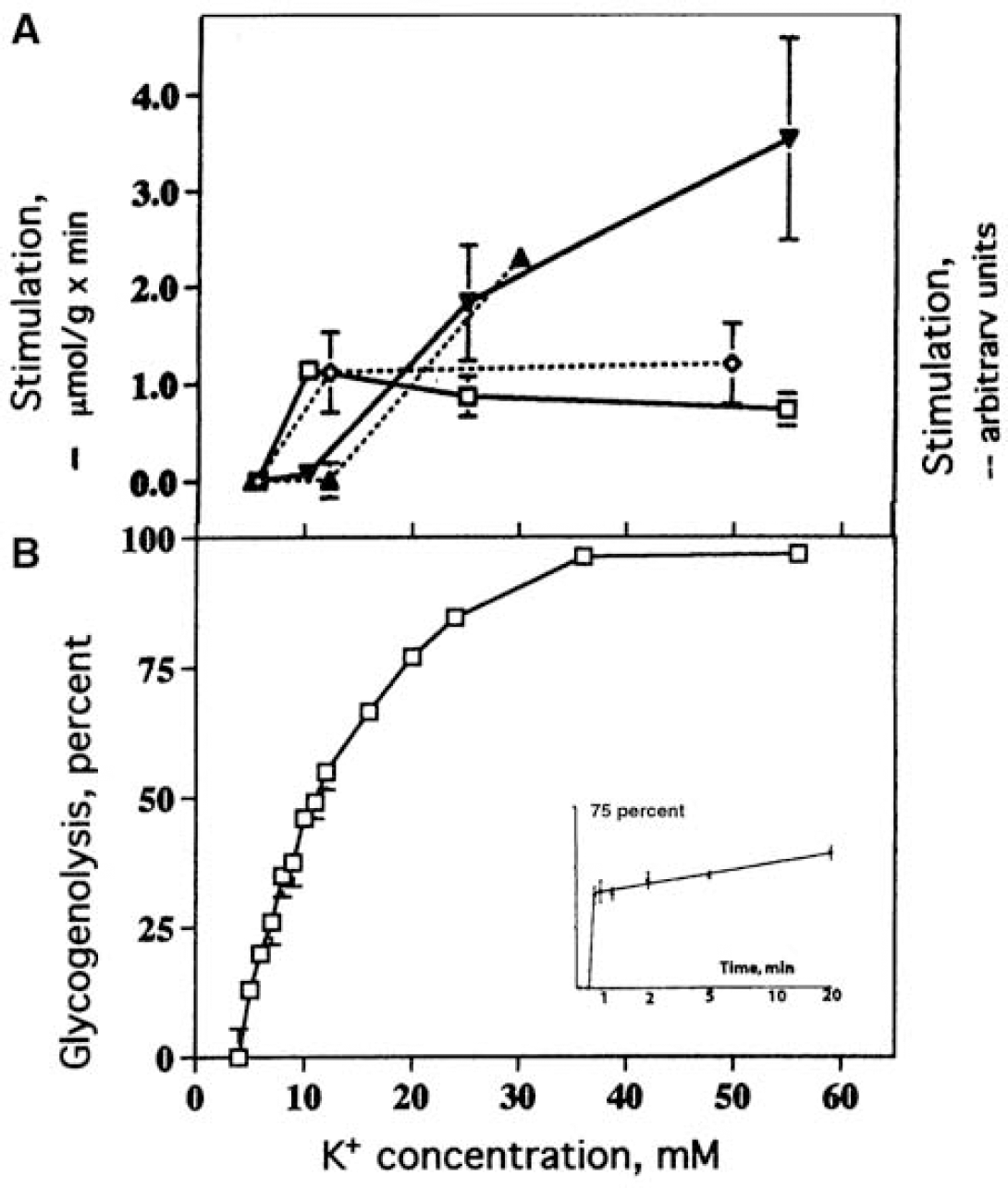
K+ effects on glucose phosphorylation and oxidative metabolism in cultured astrocytes and neurons, and on glycogenolysis in brain slices. (
Differential stimulation of oxidative metabolism by K+ in astrocytes and neurons: Low [K+]e preferentially enhances oxidative metabolism in astrocytes compared with neurons. The rate of oxygen consumption in primary cultures of mouse astrocytes is stimulated by an increase of [K+]e from 5 to 10 mmol/L, but higher levels, if anything, reduce the response (Figure 11A, solid line). High [K+]e also increases the rate of oxygen consumption in freshly isolated astrocytes (Hertz, 1966). In contrast, oxidative decarboxylation of [U-14C]glucose by cerebellar granule neurons is unaltered by doubling [K+]e from 5 to 10 mmol/L, whereas it is greatly stimulated by further increases (Figure 11A).
Oxygen consumption by brain slices also increases with elevated [K+]e when either glucose or pyruvate is the substrate, and this effect must involve astrocytic activation, because excess K+ greatly increases oxidative decarboxylation of [14C]acetate in brain slices (Gonda and Quastel, 1966; Schweigert et al, 2004). Bull and Cummins (1973) obtained evidence that elevated [K+]e stimulates brain slice respiration by two different systems, one inhibited by omission of Ca2+, the other by ouabain. These two systems are capable of substituting for each other, since K+ -mediated stimulation of respiration was not inhibited by ouabain, and it was impaired by omission of Ca2+ only at very high [K+]e; in contrast, the combination of Ca2+ omission plus ouabain almost abolished the stimulation. This dependence on the presence of Ca2+ suggests that the effect is evoked by stimulation of the Na+, K+, 2Cl− cotransporter (Figure 3). This transporter is activated by K+-mediated increase in [Ca2 +]i after depolarization-mediated opening of L-channels, as shown by its inhibition by 0.5 μmol/L nifedipine, an inhibitor of voltage-sensitive L-channels (Su et al, 2000).
The stimulation of oxygen uptake in brain slices by elevated [K+]e is also accompanied by astrocytic cell swelling (Hertz and Kjeldsen, 1985). This finding further supports a correlation between K+ -mediated stimulation of oxygen consumption and activation of the Na+, K+, 2Cl− cotransporter with its subsequent stimulation of Na+, K+ -ATPase activity, because the cotransporter and the Na+,K+ -ATPase together mediate net uptake of 2K+ plus 2Cl− into the cells (Figure 3), and the ion uptake, for osmotic reasons, is accompanied by entry of water and cell swelling. Further evidence that the K+ -mediated Ca2+ -dependent rise in respiration reflects stimulation of the cotransporter comes from the following observations: (i) the metabolic stimulation is blocked by 0.5 mmol/L ethacrynic acid, an inhibitor of the cotransporter that has no effect on the rate of O2 consumption at normal [K+]e (Hertz et al, 2004); and (ii) tetrodotoxin, chlorpromazine, and the local anesthetics procaine and cocaine, which stabilize the neuronal membrane, have no effect (Hertz, 1977). Together, these studies link increases in [K+]e to stimulation of astrocytic respiration in brain slices via the two transporter systems (Figure 3).
Pulsatile electrical stimulation of brain slices, a procedure developed by McIlwain (e.g., McIlwain, 1953), is likely to stimulate the neuronal population by causing transient depolarization, and stimulation of O2 consumption evoked by this paradigm is inhibited by tetrodotoxin, procaine, chlorpromazine, and cocaine (reviewed by Hertz, 1977), contrasting the lack of effects of these drugs on respiratory stimulation by elevated [K+]e. Electrical stimulation reduces [14C]acetate oxidation (Lindbohm and Wallgren, 1966), suggesting an inhibitory effect on astrocytic use of exogenous fuel and supporting the concept that electrical stimulation predominantly stimulates neuronal metabolism.
A comparison between metabolic substrate specificity for K+ -induced stimulation and electrical stimulation of brain slices suggests that, at least during activation, lactate is primarily oxidized in astrocytes. If lactate were predominantly a neuronal and not an astrocytic substrate, a large increase in O2 consumption would be expected during electrical stimulation when lactate was used as the exogenous substrate, whereas an elevation of [K+]e would cause a modest response under the same conditions; in fact, the opposite was found to be the case. In a review of the literature up until 1973, Hertz compared the effects of electrical stimulation versus [K+]e on rates of O2 consumption in brain slices provided with different substrates (Hertz, 1973) and found that (i) when glucose was the substrate, either activation procedure caused a stimulation of at least 60%, and (ii) when lactate was the substrate, electrical stimulation increased respiration by only 19% (McIlwain, 1953), whereas K+ caused a rise in respiration of 45% to 67% (Ghosh and Quastel, 1954; Takagaki and Tsukada, 1957). These findings in intact brain tissue support the notion that working astrocytes oxidize lactate and other fuel when [K+]e level rises.
K+ links brain activation with astrocytic biosynthetic-oxidative pathways: [K+]e enhances pyruvate carboxylation in cultured astrocytes, and the rate of CO2 fixation increases about 1.5-fold when [K+]e rises from 2.5 to 10 mmol/L (Figure 6C), more so at 25 mmol/L (Kaufman and Driscoll, 1992). The response is not affected by ouabain and may represent a direct stimulation of PC, perhaps coordinating neuronal signaling and glutamate synthesis.
K+ rapidly releases stored glucosyl units, providing energy for its uptake: K+ -activated ATPase activity creates an immediate ATP demand during activation that can be satisfied, in part, by glycogen. Hof et al (1988) showed that Ca2+ -dependent glycogenolysis in brain slices is quickly stimulated in a concentration-dependent manner by graded increases in [K+]e from 4 to 56 mmol/L K+, which covers the range occurring in brain extracellular fluid from normal to severely pathological conditions (Figure 11B). Their time course showed that the effect of 10 mmol/L K+ is almost completed within 30 secs. Glycogenolysis in cultured astrocytes is also increased by elevated [K+]e; this stimulation is inhibited by Ca2+ depletion and nifedipine (a dihydropyridine blocker of Ca2+ L-channels), indicating that it is secondary to depolarization-mediated opening of L-channels (Subbarao et al, 1996). Pentreath and Kai-Kai (1982) also found that exposure of leech and snail ganglia to slightly elevated K+ levels increases glycogenolysis and at the same time glycogen synthesis was also enhanced, consistent with the operation of a ‘glucose–glycogen shunt’.
Summary. Active K+ uptake into astrocytes occurs by two energy-requiring systems, transport of K+ via stimulation of the Na+,K+ -ATPase at its [K+]e -sensitive site and cotransport of Na+, K+, and 2Cl− into the cell. The cotransporter is activated by K+ -mediated, voltage-gated Ca2+ entry, is energized by Na+ influx along its electrochemical gradient, and is linked to Na+, K+ -ATPase activity to extrude accumulated Na+ (Figure 3). Owing to activation of these systems, [K+]e in the range occurring during neuronal activity (up to 12 mmol/L) stimulates both glycolysis and oxidative metabolism in cultured astrocytes, whereas only much higher [K+]e stimulates neuronal energy metabolism after depolarization (i.e., mimicking neuronal excitation). Both elevated [K+]e and electrical stimulation increase oxidative metabolism in brain slices but the cellular target seems to differ: excess K+ mainly or exclusively stimulates astrocytic metabolism, whereas electrical stimulation greatly increases neuronal metabolism. The K+ -induced stimulation of brain slices is a metabolic manifestation of both Na+,K+-ATPase stimulation and activation of the cotransporter and subsequent extrusion of Na+ by active transport. In brain slice experiments, lactate appears to be a better substrate to sustain the increased metabolic activity that is induced by K+ exposure and ascribed to astrocytes than the metabolic response to electrical stimuli attributed to neurons. [K+]e also appears to help provide for its own energy demands by quickly stimulating glycogenolysis via opening of voltage-dependent Ca2+ channels, and [K+]e may help integrate activities in other pathways (e.g., glutamate turnover) by stimulating PC and, at the same time, increasing energy production via oxidative pathways. Both glycolysis and glycogenolysis in astrocytes are increased by elevated [K+]e, suggesting a causal correlation between active transport of K+ and increase in ‘aerobic glycolysis'.
‘Coupling’ of Astrocytic Metabolism with Neuronal Activity by Neurotransmitters
Astrocytes ‘listen to neurons talk’: It is well established that astrocytes have many neurotransmitter receptors linked to various intracellular signaling systems that can regulate fluxes through metabolic pathways, ensuring that astrocytic and neuronal activities are integrated during activation. In contrast to glucose-derived neurotransmitters (glutamate, aspartate, GABA, glycine, acetylcholine, and D-serine), biogenic amines are degraded by oxidation and methylation. These processes do not generate ATP and are not directly involved with the major energy-yielding pathways, although the methyl groups are ultimately derived from glucose and NADPH is derived form the pentose shunt pathway (Figure 5A). Like K+, the biogenic amines stimulate energy metabolism, in part, via changes in intracellular calcium level, as illustrated below in a few examples.
Biogenic amines activate astrocytic metabolism: Some in vivo studies have clearly shown that neurotransmitters modulate local energy metabolism in brain. Thus, intracranially administered norepinephrine increases oxidative metabolic activity (MacKenzie et al, 1976), noradrenergic antagonists reduce glucose utilization in many structures (Savaki et al, 1982), and, as discussed above, the β-adrenergic antagonist propranolol abolishes the reduction of the CMRo2/CMRglc ratio (Schmalbruch et al, 2002). The cellular basis of these types of changes is difficult to establish in vivo, but in vitro experiments illustrate the fact that astrocytes constitute an important target, not only for noradrenaline, but also for many other transmitters.
Noradrenaline has strong metabolic effects that are mediated by different receptor subtypes. For example, in cultured mouse astrocytes glycolytic activation is an α1-adrenergic effect (Subbarao and Hertz, 1991). In contrast, oxidative activation is an α2-adrenergic response (Subbarao and Hertz, 1991; Chen and Hertz, 1999) secondary to increases in [Ca2+]i and intramitochondrial Ca2+ that stimulate pyruvate- and α-ketoglutarate dehydrogenases (reviewed by Hertz and Dienel, 2002), thereby accelerating both entry of pyruvate into the TCA cycle and cycle turnover (Figure 2A).
Glycogenolysis in cultured astrocytes is stimulated by many neurotransmitters and modulators, including noradrenaline, serotonin, vasoactive intestinal peptide (VIP), histamine, adenosine, and ATP (Magistretti et al, 1983; Magistretti, 1988; Subbarao and Hertz, 1990; Sorg et al, 1995; Chen et al, 1995). This mode of activation is less rapid than that evoked by K+ (Subbarao and Hertz, 1990) and is executed via specific subclasses of receptors and second messenger systems. For example, noradrenaline increases glycogen phosphorylase activity via protein kinase A activation in response to α-adrenergic receptor stimulation and by an increase in [Ca2+]i in α2-adrenergic receptor stimulation. Serotonin stimulates glycogenolysis by acting on 5-HT2A and 5-HT2B receptors coupled to intracellular Ca2+ release and an increase in [Ca2+]i; the effect on 5-HT2B receptors is extremely potent, with a half-maximum effect at 10−11 to 10−10 mol/L (Hertz et al, 2004).
Glycogen synthesis can be stimulated by a slow, protein synthesis-requiring process initiated by exposure to VIP, noradrenaline (acting at β-adrenergic receptors), or adenosine (acting at A2B receptors) (Allaman et al, 2003). However, modulation of enzyme levels after several hours does not explain the very rapid (secs to mins) changes of glycogenolysis and glycogen synthesis involved in the normal, dynamic aspects of its turnover during brain activation. Moreover, treatment of astrocyte cultures with transmitters that activate protein kinase A have nonspecific, differentiating effects on the cells, mimicking that of chronic treatment with dibutyryl cyclic AMP (Magistretti et al, 1983).
Such a nonspecific effect may not be relevant to regulation of glycogen turnover in differentiated astrocytes in the brain in situ. In contrast to the above delayed stimulation of glycogen synthesis, α2-adrenergic stimulation tends to exert an immediate stimulation of glucose incorporation into glycogen, and α2-adrenergic inhibition reduces the incorporation by 50% (Figure 12B). This effect is not shared by the β-adrenergic agonist isoproterenol, which causes a decreased incorporation of glucose into glycogen, consistent with an inhibitory effect of protein kinase A-mediated phosphorylation on glycogen synthase (Exton, 1987).
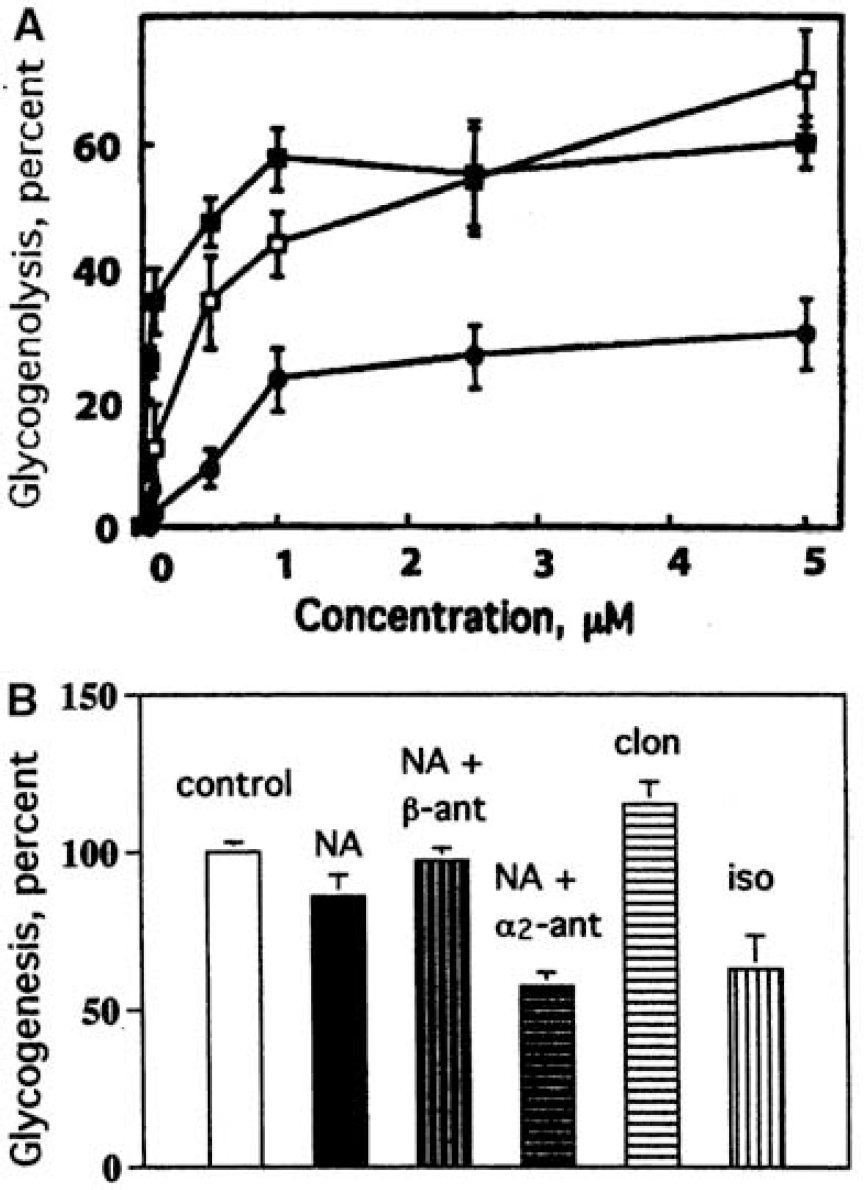
Effect of noradrenergic agonists and antagonists on glycogenolysis and glycogen synthesis from glucose (glycogenesis) in cultured astrocytes. (
Summary: Glycolysis, glycogenolysis, and oxidative metabolism in astrocytes are all stimulated by noradrenaline, but different receptor subtypes are involved. Like K+, specific neurotransmitters, with the exception of glutamate, govern glycogenolysis, strengthening the concept that glycogen turnover in astrocytes is closely linked to neuronal activity in diverse neural astrocytic preparations. α2-Adrenergic stimulation stimulates both glycogenolysis and glycogen synthesis, suggesting that it is capable of enhancing the operation of a ‘glucose–glycogen shunt’.
Concluding Remarks
Astrocytes express and use their capacity for oxidative metabolism, which per volume unit is similar to that of neurons in brain in vivo. Their biosynthetic and oxidative capabilities are essential for glutamate and GABA synthesis and glutamate-glutamine cycling, and disruption of these processes quickly alters neuronal function. The respiratory capabilities of astrocytes can be upregulated by signaling mechanisms and metabolic controls. Astrocytic respiratory fuels include glucose, glutamate, lactate, and acetate; the astrocyte-specific oxidation of acetate provides a means to assay oxidative metabolism in astrocytes and glutamate-glutamine cycling in the brain in situ.
There is consensus that many types of brain activation increase formation of pyruvate/lactate in astrocytes from glucose and glycogen, but an estimate of the magnitude in vivo is difficult and data are sparse. In these situations, the increase in astrocytic glycolysis probably exceeds that in oxidative metabolism, contributing to, or perhaps even causing, the decrease in the CMRo2/CMRglc ratio observed in intact brain. The reason for a selective increase in glycolytic and glycogenolytic activity is the ultrastructure of the astrocytes. Many energyrequiring processes occur in peripheral astrocyte processes, which are devoid of mitochondria but contain glycogen and are therefore dependent on glycolysis and glycogenolysis to meet acute energy demands. These processes seem, however, to be accessible to phosphocreatine and ATP that diffuse more slowly from the central parts of the cell and eventually fuel energy-requiring processes; diffusion of neurotransmitter glutamate from processes to mitochondria would provide oxidative fuel for working astrocytes. This concept is supported by the high rate of oxidative metabolism in astrocytes and the inhibition by hypoxia of the second phase of extracellular K+ clearance after neuronal stimulation.
Glycogenolysis enables rapid metabolism of glucosyl units without an ATP requirement for glucose phosphorylation from preformed glycogen. However, the energy recovered from glycolysis via rapid turnover of glycogen is half that from direct glycolytic metabolism, and ‘glycogen shunting’ may accelerate utilization of blood borne glucose to make up for the difference in ATP yield. The temporal relationships between glycogen synthesis and degradation during activation are difficult to measure, but prevention of the decrease in CMRo2/CMRglc ratio during activation by propranolol, a glycogenolysis inhibitor, supports a role for glycogen turnover during brain activation. However, even without a glucose–glycogen shunt, compartmentation of glycolytic/glycogenolytic metabolism in the peripheral astrocyte processes from oxidative metabolism in the central part of the cells would promote selective upregulation of astrocytic glycolysis during activation. Thus, glycolytic fluxes are likely to be high, but they cannot be inferred only from lactate or NADH levels that reflect net differences in rates of formation and disappearance.
The location where the oxidation of lactate generated from glucose and/or glycogen in excess of oxygen during brain activation occurs is a crucial, controversial, and unresolved question. Is it indeed oxidized in the brain, or released, perhaps by other routes than into circulating blood? The decrease in CMRo2/CMRglc ratio during activation is augmented further when lactate or glycogen is included in the total carbohydrate metabolized, indicating that the excess lactate is not oxidized in the activated region. However, the cellular build-up of the ‘excess’ lactate must be restrained to maintain glycolysis under aerobic conditions, consistent with the finding in many laboratories that the net accumulation of lactate in activated brain is very small, rising from 1 to ~ 2 mmol/L and corresponding to only a few percent of the total amount of glucose and glycogen consumed during the same interval. Although it has been postulated that little or no oxidation of exogenous lactate occurs in astrocytes in vivo, the evidence is far from conclusive, and brain slice experiments suggest that, at least under stimulated conditions, lactate is oxidized mainly by astrocytes. Also, in cell culture experiments, lactate is oxidized by both astrocytes and neurons. Lactate fluxes and metabolism are extremely difficult to assay in vivo, and little definitive information is available regarding its fate.
Oxidative metabolism, glycolysis and, in some cases, glycogenolysis in astrocytes increase when their work load rises. The energy requirement for glutamate uptake and metabolism to glutamine may be the most generally recognized energy-requiring process in astrocytes, but other signaling processes in astrocytes, filopodial motility, Ca2+ homeostasis, propagation of Ca2+ waves, responses to a wide variety of neurotransmitters, and signals from other astrocytes, and use of ATP as a transmitter also increase their energy demand. During brain stimulation, active K+ uptake may pose the largest demand on ATP, and especially on glycolytically generated ATP. Energy balance sheets and models of astrocyte-neuron interactions that do not include these functions greatly underestimate astrocytic energetics and portray a very limited context within which to understand astrocytic energetics.