
Abstract
Cholinesterase inhibitors (ChEIs) have been used to treat Alzheimer's disease (AD). The efficacy of these drugs, however, is less than satisfactory. The possibility that ChEIs may have effects unrelated to ChE activity, such as negatively modulate neuronal nicotinic acetylcholine receptors (nAChRs) was evaluated. Since α7-nAChRs on cerebral perivascular sympathetic neurons mediate cerebral parasympathetic-nitrergic vasodilation, effects of physostigmine, neostigmine, and galantamine on α7-nAChR-mediated dilation in isolated porcine basilar arterial rings denuded of endothelium was examined using in vitro tissue bath technique. The results indicated that these ChEIs blocked vasodilation induced by choline (0.3 mmol/L), nicotine (0.1 mmol/L), and transmural nerve stimulation (TNS). The ChEl inhibition of dilation induced by TNS but not by choline or nicotine was prevented by atropine (0.1 μmol/L) pretreatment. Furthermore, using confocal microscopy, significant calcium influx induced by choline and nicotine in cultured porcine superior cervical ganglion (SCG) cells was attenuated by ChEIs. In α7-nAChR-expressed Xenopus oocytes, nicotine-induced inward currents were attenuated by α-bungarotoxin and ChEIs. Moreover, ChEI inhibition of nicotine- and choline-induced dilation was prevented by pretreatment with mevastatin and lovastatin (10 μmol/L), which did not affect ChEI inhibition of TNS-induced relaxation. These findings suggest that ChEIs inhibit the α7-nAChRs located on postganglionic sympathetic nerve terminals of SCG origin, causing a decreased release of nitric oxide in the neighboring nitrergic nerves and cerebral vasodilation. Inhibition of α7-nAChRs leading to a potential cerebral hypoperfusion may contribute to the limitation of ChEIs and question the validity of using a ChEI alone in treating AD. The efficacy of ChEIs may be improved by concurrent use of statins.
Introduction
The cholinergic hypothesis of Alzheimer's disease (AD) has advocated a decrease in cholinergic markers such as choline acetyltransterase and acetylcholinesterase in the brains of patients with AD (Bartus et al, 1982). This hypothesis is further supported by reduced choline uptake (Rylett et al, 1983) and acetylcholine (ACh) release (Nilsson et al, 1986). Also, decreased levels of ACh are present in the cerebrospinal fluid of AD and vascular dementia patients, and this decrease positively correlates to the degree of severity of dementia (Tohgi et al, 1996).
The development of cholinesterase inhibitors (ChEIs) for the treatment of AD was based on the cholinergic hypothesis of AD that the ChEIs would decrease breakdown of the synaptic ACh. Only less than halt of the patients receiving ChEIs, however, achieve a clinically significant response (Ibach and Haen, 2004; Grady, 2004; Courtney et al, 2004). Furthermore, these inhibitors have been shown to be effective in the early stages of AD, although there have been reports that cholinergic deticits may not be present in the early stages of AD (Davis et al, 1999). These findings suggest that besides increasing available synaptic ACh, ChEIs may have other actions unrelated to inhibition of ChE enzyme.
We have shown recently that nicotine-induced nitric oxide (NO)-mediated neurogenic vasodilation in porcine basilar arteries and feline middle cerebral arteries depends on intact perivascular postganglionic sympathetic, adrenergic innervation originating in the superior cervical ganglion (SCG) (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2002). We have further demonstrated in porcine basilar arteries that nicotine and choline act on α7-nicotinic acetylcholine receptors (nAChRs) located on perivascular postganglionic sympathetic nerve terminals to release norepinephrine (NE), which then acts on presynaptic β2-adrenoceptors located on the neighboring nitrergic nerve terminals, resulting in release of NO and vasodilation (Lee et al, 2000; Si and Lee, 2001, 2002).
The α-bungarotoxin-sensitive α7-nAChR is a predominant nAChR subtype in the brain and is a prototype of the ligand gated ion channels. The activity of the α7-nAChR can be allosterically potentiated by ChEIs (Pereira et al, 1993; Dajas-Bailador et al, 2003). The ChEIs have also been shown to block the nAChRs in the striatum (Clarke et al, 1994). Effect of ChEIs on the role of α7-nAChR in mediating cerebral nitrergic vasodilation, however, has not been reported. This is particularly important as the cerebral perivascular sympathetic α7-nAChR can be activated by choline, a preferential α7-nAChR agonist and a major endogenous metabolite of the natural transmitter ACh cleaved by ChE enzymes. Cholinesterase inhibitorss by inhibiting metabolism of ACh are expected to decrease the concentration of synaptic choline, possibly resulting in decreased α7-nAChR-mediated vasodilation in the cerebral circulation.
Statins are another group of drugs that have been approved for the treatment of AD. Various studies have shown that these 3-hydroxy-3-methylglutarylcoenzyme A reductase inhibitors reduce the incidence and progression of AD by 40% to 70% (Jick et al, 2000; Rockwood et al, 2002). Besides reducing serum cholesterol and low-density lipoproteins, statins possess cholesterol-independent actions. They have been shown to upregulate endothelial nitric oxide synthase production (Endres et al, 1998), inhibit inducible nitric oxide synthase (Pahan et al, 1997), and possess antioxidant properties (Aviram et al, 1998). These pleiotropic effects of statins may be beneficial in managing AD, and it is intriguing to determine if statins alone modulate the α7-nAChR or its modulation by ChEIs.
Hence, effects of ChEIs and statins on perivascular α7-nAChR-mediated neurogenic vasodilation in porcine basilar arteries were examined in the present study using in vitro tissue bath, double electrode voltage clamp, and calcium fluorescence confocal microscopic techniques. Our results indicated that ChEIs attenuated α7-nAChR-mediated nitrergic vasodilation by directly blocking these receptors. This ChEI blockade was prevented by pretreatment with statins.
Materials and methods
General Procedure
Fresh heads of adult pigs (60 to 100 kg) of either sex were collected at local packing companies (Excel, Beardstown, IL, USA; Y-T, Springfield, IL, USA). The entire brain, with dura matter attached, was removed and placed in Krebs' bicarbonate solution equilibrated with 95% O2 and 5% CO2 at room temperature. The composition of the Krebs' solution was as follows: 122.0 mmol/L NaCl, 5.16 mmol/L KCl, 1.31 mmol/L CaCl2, 1.22 mmol/L MgSO4, 25.6 mmol/L NaHCO3, 0.03 mmol/L ethylenediamine-tetraacetic acid, 0.1 mmol/L L-ascorbic acid, and 11.0 mmol/L glucose (pH 7.4). The basilar artery was dissected and cleaned of surrounding tissue under a dissecting microscope.
In Vitro Tissue Bath Studies
The ring segment (4 mm long) was cannulated with a stainless-steel rod (30-gauge hemispherical section) and a short piece of platinum wire and mounted horizontally in a plastic tissue bath containing 6 ml of Krebs' bicarbonate solution. The platinum wire was bent into a U shape and anchored to a gate. The stainless-steel rod was connected to a strain gauge (UC2; Gould Instrument Systems Inc., Cleveland, OH, USA) for isometric recording of changes in force, as described in our previous report (Lee et al, 1976). The temperature of the Krebs' solution equilibrated with 95% O2 and 5% CO2 was maintained at 37°C. Tissues were equilibrated in the Krebs' solution for an initial 30 mins and then mechanically stretched to a resting tension of 750 mg (Zhang et al, 1998). The basilar arterial ring segments were then precontracted with 9,11-dideoxy-9,11-methanoepoxy prostaglandin F2 (U-46619) (0.3 to 3 μmol/L) to induce an active muscle tone of 0.5 to 0.75 g. Transmural nerve stimulation (TNS) at 2, 4, and 8 Hz, nicotine (0.1 mmol/L), and choline (0.3 mmol/L) were applied to induce a relaxation. After relaxation induced by TNS, nicotine, or choline, the arteries were washed with prewarmed Krebs' solution. A similar magnitude of active muscle tone was induced with U-46619 again, and TNS was repeated (to serve as a control compared with the relaxation elicited by TNS before the wash). Effects of different concentrations of physostigmine, neostigmine (3 to 30 μmol/L), and galantamine (10 to 100 μmol/L) were then administered, and TNS and nicotine/choline at the same concentration before the wash were repeated. To avoid possible development of tachyphylaxis on repeated applications of nicotinic agonists, at least 90 mins with six washes was allowed before the next application of nicotinic agonists (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2002, 2003). Experimental drugs were added at least 15 mins before TNS and application of nicotinic agonists. After this, the arteries were washed with prewarmed Krebs' solution again. A similar magnitude of active muscle tone was induced with U-46619 again, and TNS and nicotine/choline were repeated (to serve as a second control compared with the relaxation elicited by TNS and nicotine before the drug application).
For TNS, tissues were electrically, transmurally stimulated with a pair of platinum electrodes through which 100 biphasic square-wave pulses of 0.6 ms in duration and 200 mA in intensity were applied at various frequencies (Zhang et al, 1998). Stimulation parameters were continuously monitored on a Tektronix oscilloscope. The neurogenic origin of this TNS-induced response was verified by its complete blockade by tetrodotoxin (0.3 μmol/L). At the end of each experiment, papaverine (300 μmol/L) was added to induce a maximum relaxation. The magnitude of a vasodilator response was expressed as a percentage of the maximum response induced by papaverine (Zhang et al, 1998).
For examining effects of experimental drugs on relaxation induced by isoproterenol or sodium nitroprusside, concentration—response relationships for these two vasodilators were obtained by a cumulative technique in arteries without endothelial cells in the presence of active muscle tone induced by U-46619. After the arterial rings were washed with prewarmed Krebs' solution, a similar magnitude of active muscle tone was again induced by U-46619. The experimental drugs were then added, and 15 mins later, concentration—response relations for isoproterenol or sodium nitroprusside were repeated. EC50 values (the concentration that produces 50% of the maximum relaxation) were determined for each arterial ring. From these values, the geometric means EC50 values with 95% contidence intervals (Fleming et al, 1972) were calculated.
The endothelial cells of all arterial ring segments were mechanically removed by a standard briet gentle rubbing of the intimal surtace with a stainless-steel rod having a diameter (25 to 30 gauge) equivalent to the lumen of the arteries (Zhang et al, 1998, Lee et al, 2000). A complete removal of endothelial cells was verified by lack of effect of nitro-L-arginine in increasing basal tone (Zhang et al, 1998, Lee et al, 2000).
Superior Cervical Ganglion Cell Culture
Freshly dissected SCGs from animals were placed in cold Hibernate A (Invitrogen, Carlsbad, CA, USA) solution (Liu et al, 2000). After being cut into smaller pieces, the ganglia were transterred to Mg2+/Ca2+ -tree Hanks‘-balanced salt solution containing papain (2 U/ml; Sigma-Aldrich, St Louis, MO, USA), collagenase D (1.2 mg/ml; Roche Diagnostics, Indianapolis, IN, USA), and dispase (4.8 mg/ml; Invitrogen), and were incubated for 50 mins at 37°C. Cells were released by gentle trituration at the end of the incubation. The cell suspension was centrituged at 300g for 5 mins. The pellet was gently resuspended in Neurobasal culture medium (Invitrogen) containing B27 (1:50 dilution; Invitrogen), 0.5 mmol/L L-glutamine, 25 μmol/L L-glutamate, and nerve growth factor (50 ng/ml; Alomone Labs, Jerusalem, Israel). All media and Hanks‘-balanced salt solution contained 100 U/ml penicillin and 100 U/ml streptomycin. The cell suspension was plated into a fourwell culture plate with a poly (D-lysine)-coated (50 μg/ml; Sigma-Aldrich) glass coverslip (12 mm diameter; Fisher Scientitic Co., Fair Lawn, NJ, USA) in each well and incubated with air containing 5% CO2 at 37°C. The growth medium was changed on days 2 and 6. The SCG cells were stained with anti-rabbit neurotilament 200 (Sigma-Aldrich) as a marker of neuronal cells (Liu et al, 2000).
Intracellular Calcium Imaging
Between 3 and 7 days in culture, the SCG cells were used to examine effects of nicotine and choline on calcium influx in these cells by contocal microscopy. The cells were washed with physiologic buffer (130 mmol/L NaCl, 5 mmol/L KCl, 10 mmol/L HEPES, 5 mmol/L glucose, 2 mmol/L CaCl2, and 2 mmol/L MgCl2, pH 7.3) and were loaded with 3 μmol/L fluo-4 acetoxymethyl ester (AM) in physiologic buffer and incubated at room temperature for 30 min. The cells were washed with calcium indicator-free buffer to remove any dye that is nonspecitically associated with the cell surtace, and then incubated for a further 30 mins to allow complete de-esteritication of intracellular AM esters. Nicotine (0.1 mmol/L) or choline (0.3 mmol/L) was then applied, and the calcium influx was measured. Physostigmine (10 and 30 μmol/L), neostigmine (10 and 30 μmol/L) and galantamine (30 and 100 μmol/L) were added 15 mins before application of nicotine, choline, or KCl (40 mmol/L). At the end of each experiment, KCl was added and responses to nicotine and choline were normalized to KCl-induced calcium intlux. Calcium fluorescence images were examined with a Fluoview contocal microscope (Olympus, Melville, NY, USA). Fluo-4 was excited at 488 nm, and emitted fluorescence was filtered with a 535 ± 25-nm bandpass filter and read into a computer running Fluoview software and quantitied using this software (Si and Lee, 2002).
Double-Electrode Voltage Clamp
Stage V and VI oocytes from Xenopus leavis were harvested and injected with the α7-nAChR RNA using a nanoinjector (Drummond, Broomall, PA, USA). The oocytes were maintained at 18°C. Membrane currents were recorded 2 days After the injection. During the recording, oocytes were continuously pertused with the ND96 bath solution containing (in mmol/L): NaCl 96, KCl2, MgCl2 1.0, CaCl2 1.8, and HEPES 5.0, pH 7.5, at a rate of 10 ml/min. All drugs were diluted in the ND96 bath solution. Nicotine (0.1 mmol/L) was applied directly on the oocytes and nicotine-induced current were measured using an OC-725C amplifier (Warner, Hamdem, CT, USA; Chen and Chen, 2001, 2003). The oocytes were continuously clamped at −60 mV and the maximum inward current was determined as the current amplitude. Electrodes were prepared from borosilicate glass capillaries (1.5 mm) (World Precision Instruments, Saratota, FL, USA) using a P-97 microelectrode puller (Sutter, Novato, CA, USA) and had 0.1 to 1 MΩ resistance when filled with 3 M KCl. Data acquisition and analysis were performed with pClamp 9.0 and Digidata 1322A (Axon Instruments, Union City, CA, USA). The traces were filtered at 1 kHz and sampled at 2 kHz. To compensate for the variable expression of the α7-nAChR, the data were normalized as a percentage of nicotine-induced response.
Drugs and Statistical Analysis
The following drugs were used: (−)-nicotine, choline chloride, N-nitro-L-arginine, tetrodotoxin, papaverine, isoproterenol, sodium nitroprusside, atropine, physostigmine, neostigmine (all from Sigma-Aldrich), U-46619 (Upjohn, Kalamazoo, MI, USA), galantamine (Tocris, Ellisville, MO, USA), mevastatin sodium, lovastatin sodium (Calbiochem, La Jolla, CA, USA) and Fluo-4 AM (Molecular Probes, Eugene, OR, USA). All drugs, unless otherwise stated, were dissolved in deionized water and added directly to the tissue baths. The drug concentrations reported were the final concentration in the bath.
Results were expressed as means ± s.e.m. Statistical analysis was evaluated by analysis of variance, and Student's t-test for paired or unpaired samples as appropriate. The P < 0.05 level of probability was accepted as significant.
Results
Choline-, Nicotine-, and Transmural Nerve Stimulation-Induced Neurogenic Vasodilation in Porcine Basilar Arteries
Consistent with our previous reports (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2002, 2003), the porcine basilar arteries without endothelial cells, which in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), was relaxed exclusively on TNS (2, 4, and 8 Hz), and applications of nicotine (0.1 mmol/L) or choline (0.3 mmol/L) (Figures 1A and 1B). The relaxation induced by TNS, nicotine, and choline was significantly blocked by tetrodotoxin (0.3 μmol/L; n =7), and was abolished by N-nitro-L-arginine (30 μmol/L; n = 6) and cold-storage denervation (n = 4; data not shown). These results suggest that the relaxation induced by TNS and nicotinic agonists was because of the release of neurogenic NO.
Cholinesterase Inhibitors-Inhibited Nicotine- and Choline-Induced Neurogenic Vasodilation
Since nicotine at 0.1 mmol/L, and choline at 0.3 mmol/L induced maximum relaxation, these parameters, which have previously been used by us and many others (Toda and Okamura, 1998; Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2002), were used in the subsequent studies. As reported previously by many investigators, neurogenic vasodilation induced by nicotinic agonists diminished on repeated applications of this agonist within short time intervals (Zhang et al, 1998; Si and Lee, 2002). Accordingly, in the present study, a 90-mins interval was allowed before repeating each application of nicotine and choline. Three consecutive, reproducible relaxations induced by nicotine (0.1 mmol/L) or choline (0.3 mmol/L) were obtained, which were not significantly different (Zhang et al, 1998; Si and Lee, 2002).
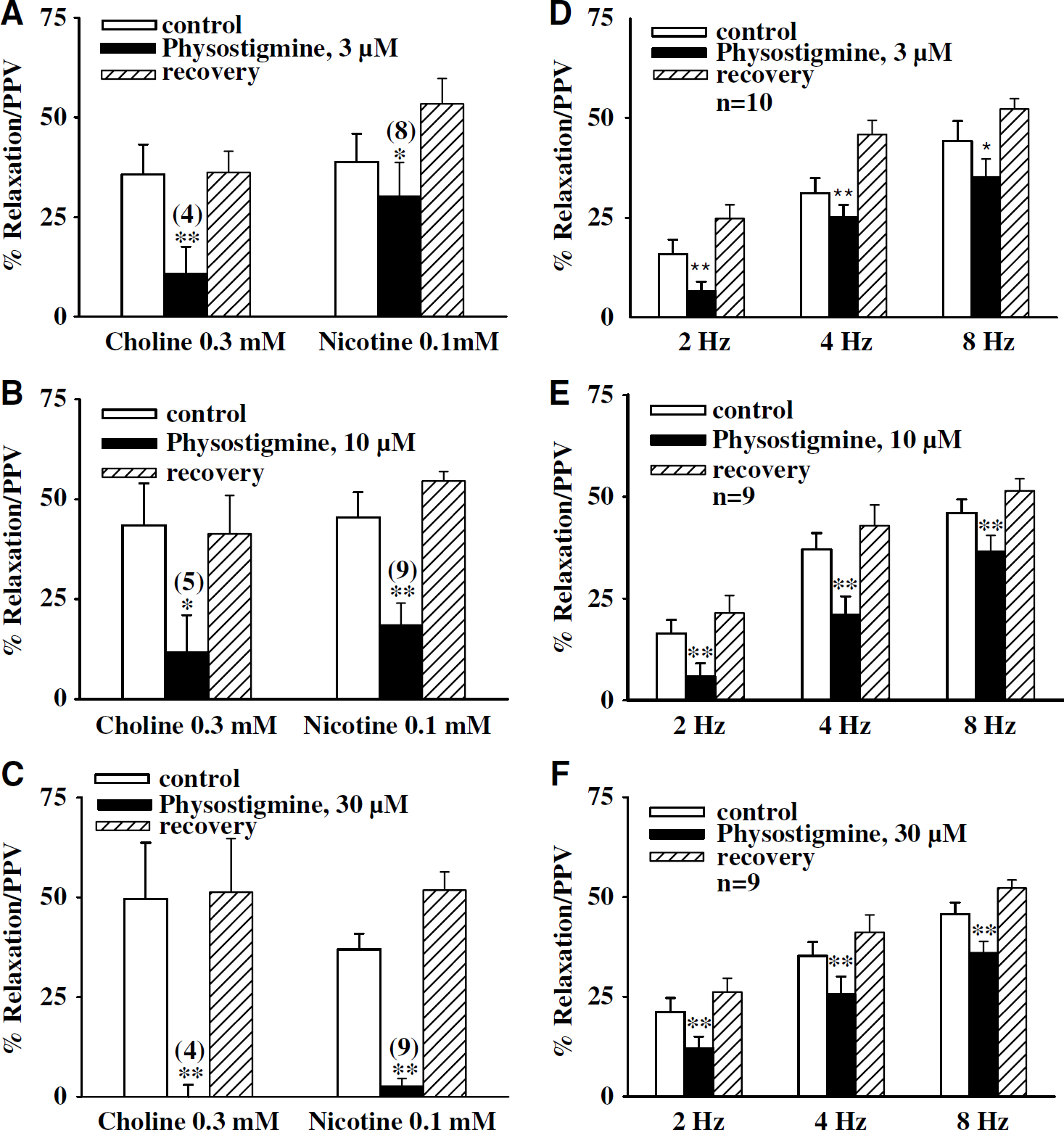
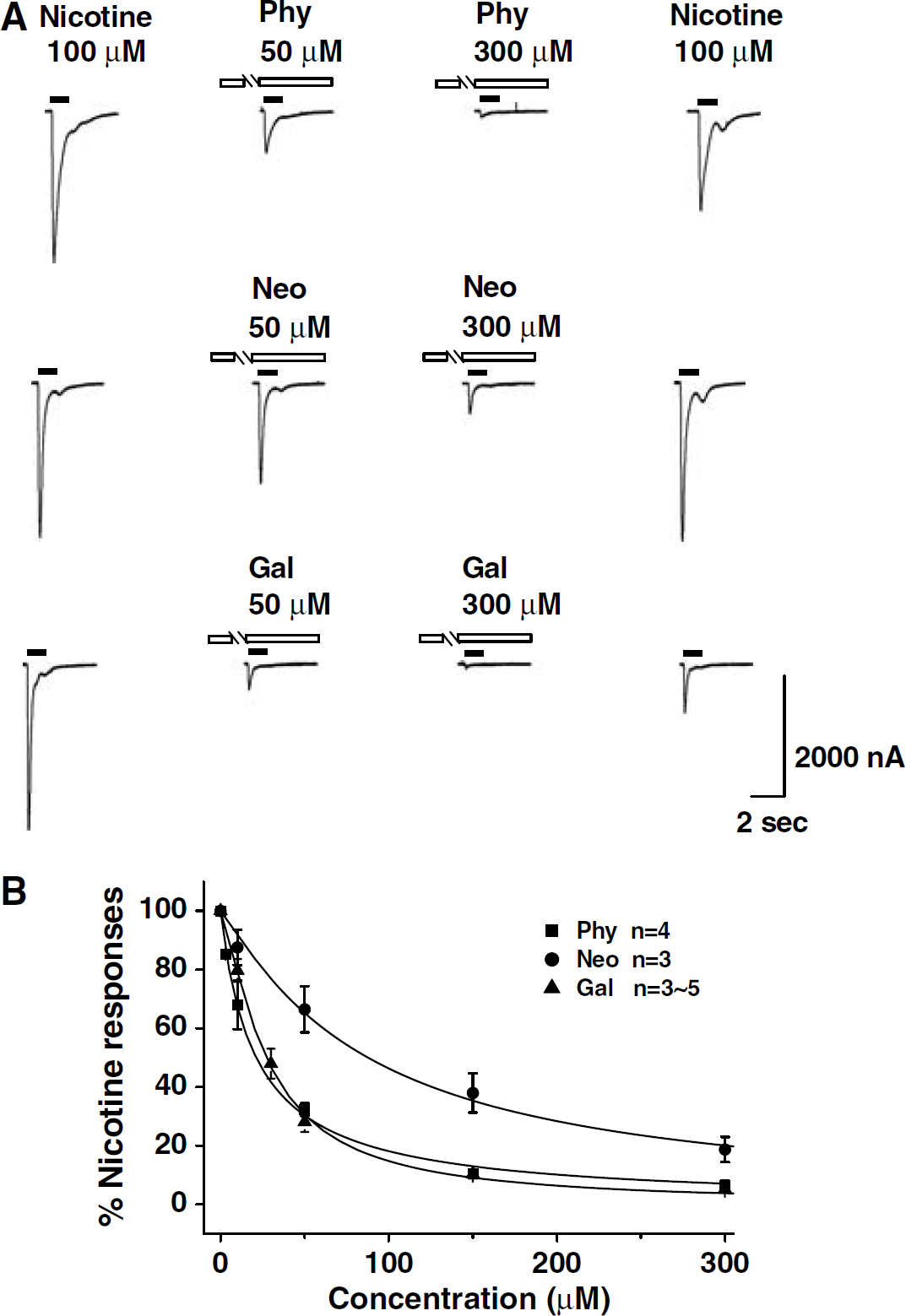
In basilar arteries (without endothelial cells) in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), relaxation induced by nicotine (0.1 mmol/L) and choline (0.3 mmol/L) was significantly inhibited by physostigmine (3 to 30 μmol/L; n > 4 for each concentration; Figures 2A–2C) and galantamine 10 to 100 μmol/L, n > 6 for each concentration; Figures 3A–3C) in a concentration-dependent manner. The IC50 values for physostigmine against nicotine- and choline-induced relaxation were 8.69 (6.52 to 11.58) × 10−6 and 6.27 (2.59 to 15.14) × 10−6 mol/L respectively, which were not significantly different (P < 0.05). The IC50 values for galantamine against nicotine- and choline-induced relaxation were 3.18 (1.4 to 7.18) × 10-5 and 4.42 (1.26 to 15.56) × 10−5 mol/L, respectively, which were not significantly different (P < 0.05). On the other hand, neostigmine at 3 to 30 μmol/L had variable effects on relaxation induced by choline and nicotine. Relaxation induced by choline was significantly inhibited by 10 and 30 μmol/L neostigmine (Figures 4A and 4B; n = 8 to 9), while nicotine induced relaxation was significantly inhibited by only 30 μmol/L neostigmine. (Figure 4C; n = 11). The IC50 values for neostigmine against nicotine- and choline-induced relaxation were 4.14 (1.48 to 11.57) × 10−5 and 4.26 (1.99 to 9.09) × 10−5 mol/L, respectively, which were not significantly different (P < 0.05). The physostigmine, neostigmine, and galantamine inhibition of relaxation induced by nicotine and choline was completely reversible after washing off physostigmine, neostigmine, and galantamine (Figures 1A and 1B, 2A–2C, 3A–3C, and 4A–4C).
Cholinesterase Inhibitors Inhibited Transmural Nerve Stimulation-Induced Neurogenic Vasodilation
The relaxation elicited by repeated TNS at 2, 4, and 8 Hz, like other reports in the porcine basilar arteries (Zhang et al, 1998; Lee et al, 2000), was reproducible on repeated application. In basilar arteries (without endothelial cells) in the presence of active muscle tone induced by U-46619 (0.3 μmol/L), relaxation induced 2, 4, and 8 Hz TNS was significantly inhibited by all three ChEIs at each concentration tested; physostigmine (3 to 30 μmol/L), neostigmine (3 to 30 μmol/L) and galantamine (10 to 100 μmol/L) (Figure 1A and 1B, 2D–2F, 3D–3F and 4D–4F).
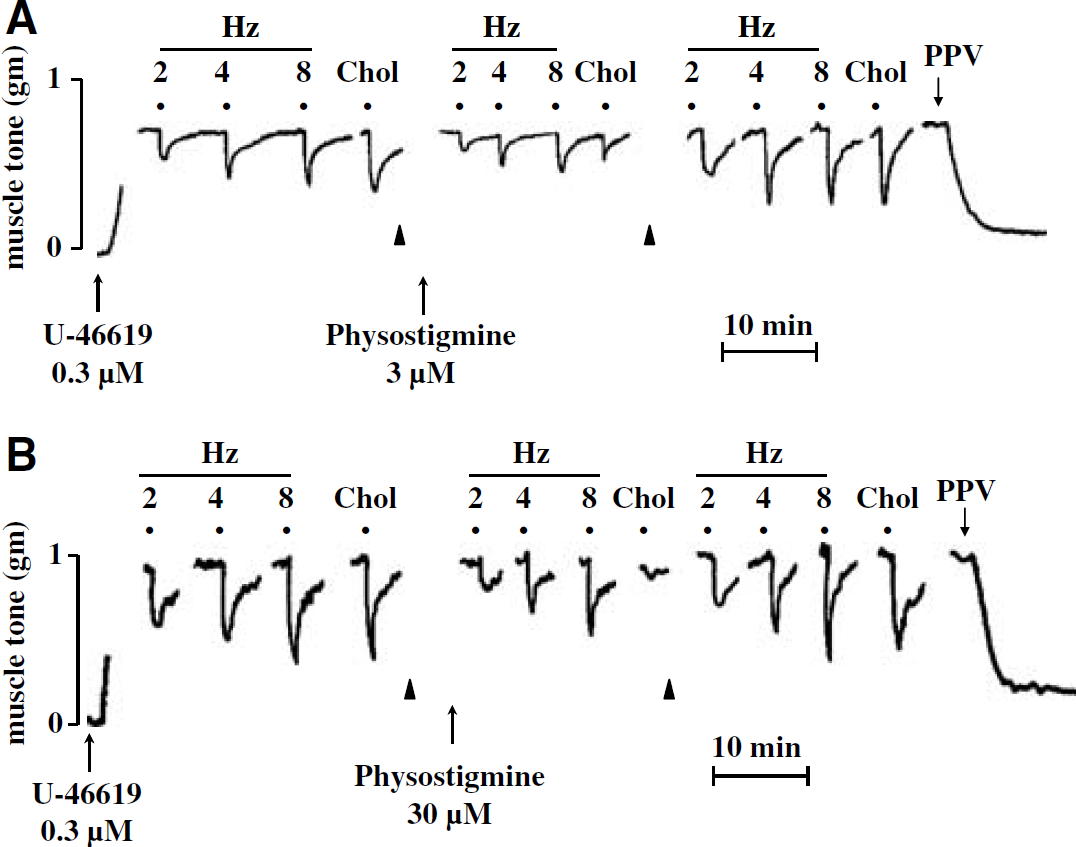
Effects of physostigmine on transmural nerve stimulation (TNS)- and choline-induced relaxation of porcine basilar arterial rings without endothelial cells. Representative tracings showing effect of physostigmine 3 μmol/L (
Effects of physostigmine on choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. The effect of physostigmine on relaxation elicited by choline (0.3 mmol/L), nicotine (0.1 mmol/L), and TNS at 2, 4, and 8 Hz was examined in endothelium-denuded basilar arterial rings in the presence of active muscle tone induced by 9,11-dideoxy-9,11-methanoepoxy prostaglandin F2 (U-46619) (0.3 μmol/L). Physostigmine in a concentration-dependent manner inhibited choline- and nicotine-induced relaxation (estimated as a percentage of papaverine/PPV-elicited maximum relaxation). The inhibition by physostigmine was fully reversible after the arteries were washed with prewarmed fresh Krebs' solution (
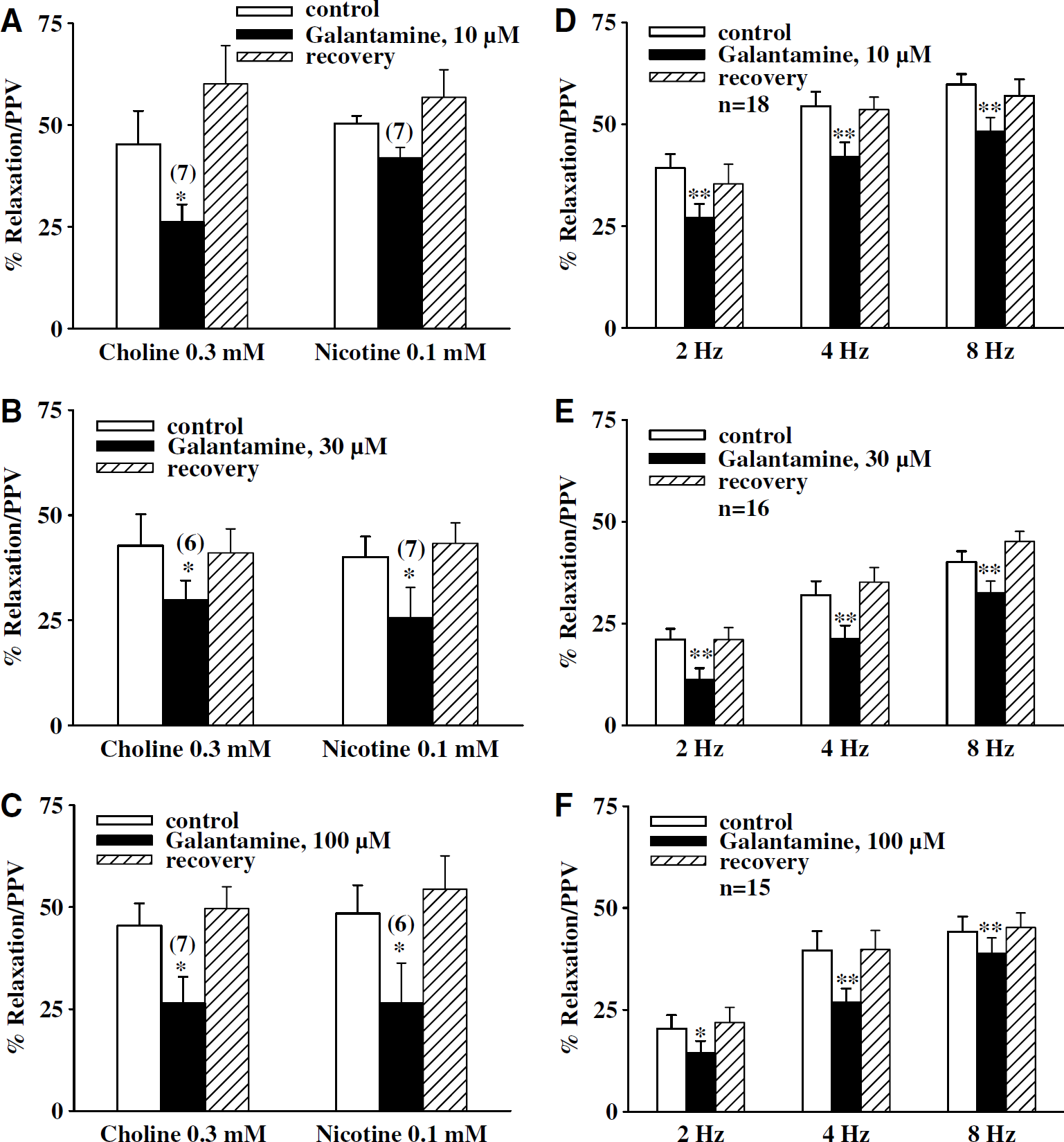
Effects of galantamine on choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. The effect of galantamine on relaxation elicited by choline (0.3 mmol/L), nicotine (0.1 mmol/L), and TNS at 2, 4, and 8 Hz was examined in endothelium-denuded basilar arterial ring in the presence of active muscle tone induced by 9,11-dideoxy-9,11-methanoepoxy prostaglandin F2 (U-46619) (0.3 μmol/L). Galantamine in a concentration-dependent manner inhibited choline- and nicotine-induced relaxation (estimated as a percentage of papaverine/PPV-elicited maximum relaxation). The inhibition by galantamine was fully reversible after the arteries were washed with prewarmed fresh Krebs' solution (
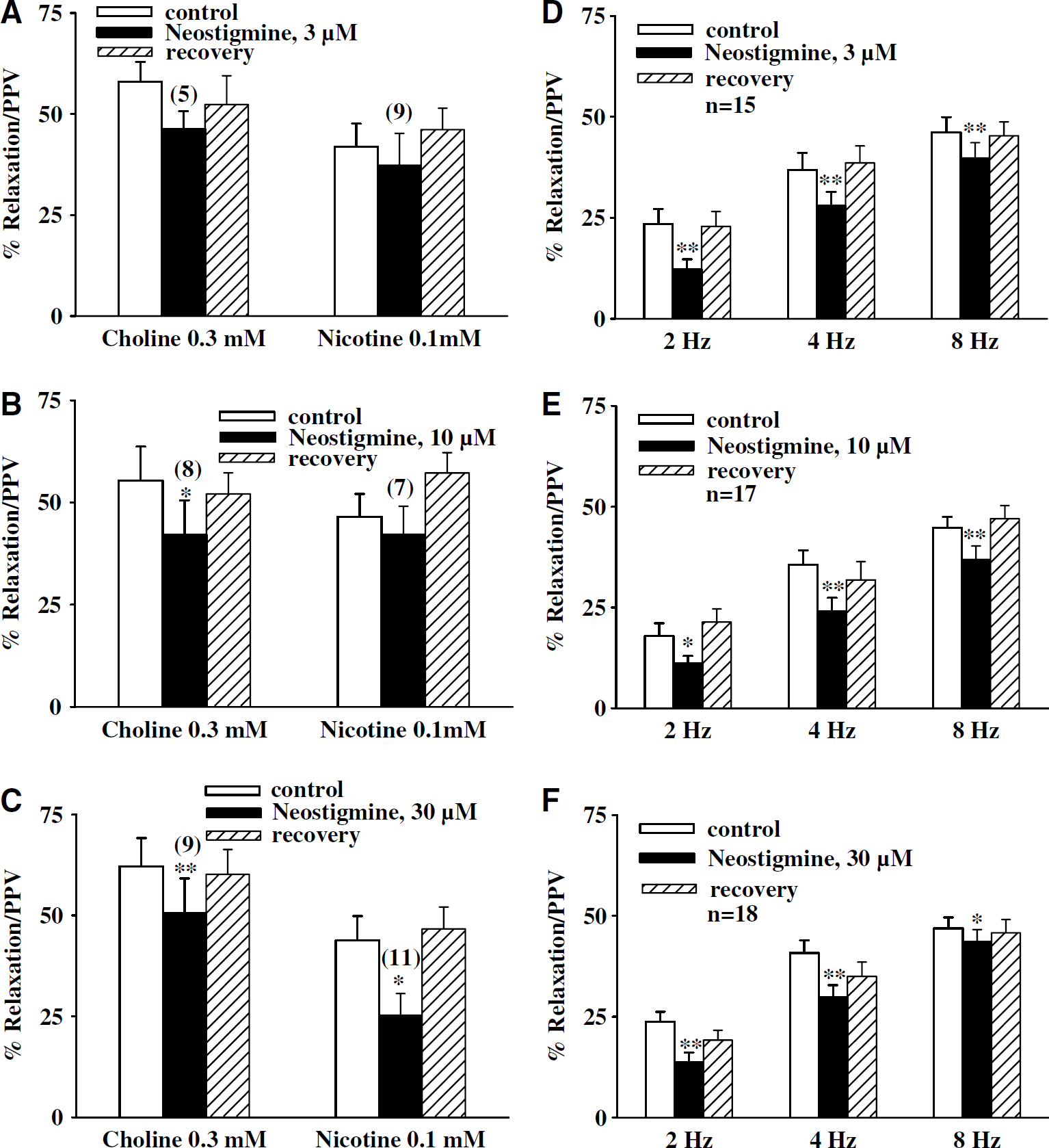
Effects of neostigmine on choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. The effect of neostigmine on relaxation elicited by choline (0.3 mmol/L), nicotine (0.1 mmol/L), and TNS at 2, 4, and 8 Hz was examined in endothelium-denuded basilar arterial ring in the presence of active muscle tone induced by 9,11-dideoxy-9,11-methanoepoxy prostaglandin F2 (U-46619) (0.3 μmol/L). Neostigmine in a concentration-dependent manner inhibited choline- and nicotine-induced relaxation (estimated as a percentage of papaverine/PPV-elicited maximum relaxation). The inhibition by neostigmine was fully reversible after the arteries were washed with prewarmed fresh Krebs' solution (
Atropine prevented ChEI inhibition of TNS-induced neurogenic vasodilation without affecting that of nicotinic agonist-induced neurogenic vasodilation. In the presence of atropine (0.1 μmol/L), relaxation induced by TNS at 2, 4, and 8 Hz was enhanced while that induced by nicotine and choline remained unchanged (n = 4; data not shown); a result similar to our previous reports (Liu and Lee, 1999; Si and Lee, 2001, 2002). Meanwhile, this atropine treatment prevented the inhibition of TNS-induced relaxation by phy-sostigmine (30 μmol/L), neostigmine (30 μmol/L), and galantamine (100 μmol/L). Atropine at the same concentration, however, did not prevent ChEI inhibition of nicotine- or choline- induced relaxation. The effect of atropine and the ChEIs on TNS- and nicotinic agonist-induced relaxation was reversible after washing off these antagonists (Figures 5A–5C).
Cholinesterase Inhibitors did not Affect Isoproterenol- or Sodium Nitroprusside-Induced Relaxation in Basilar Arteries
In the presence of active muscle tone induced by U-46619 (0.3 μmol/L), porcine basilar arteries without endothelial cells relaxed on application of isoproterenol and sodium nitroprusside in a concentration-dependent manner, a result similar to that reported previously (Lee et al, 2000). Physostigmine (3 to 30 μmol/L), neostigmine (3 to 30 μmol/L), and galantamine (10 to 100 μmol/L) did not affect relaxation induced by isoproterenol (1 nmol/L to 3 μmol/L) (Figures 6A–6C) sodium nitroprusside (10 nmol/L to 0.1 mmol/L) (Figures 6D–6F; data for other concentrations not shown) in arteries denuded of endothelial cells. The EC50 values for isoproterenol and sodium nitroprusside-induced relaxation were not significantly different between control and in the presence of drug (P < 0.05).
Cholinesterase Inhibitors Inhibited Choline- and Nicotine-Induced Calcium Influx in Cultured Superior Cervical Ganglion
Cultured SCG cells, like cerebral perivascular sympathetic neurons in whole-mount arterial preparations, have been shown to contain dense α7-nAChRs (Si and Lee, 2001), which form membrane cation channels possessing high Ca2+ permeability (Sargent, 1993). Using the intracellular calcium imaging indicator Fluo-4 AM to examine calcium influx, quantitative analysis on single cells indicated that both nicotine (0.1 mmol/L) and choline (0.3 mmol/L) significantly increased calcium influx in the SCG cells (Figure 7) as reported previously (Si and Lee, 2002). Addition of KCl (40 mmol/L) did not further increase intracellular calcium. The nicotine- and choline-induced calcium influx was attenuated in cells pretreated with physostigmine (10 and 30 μmol/L), neostigmine (10 and 30 μmol/L) and galantamine (30 and 100 μmol/L) in a concentration-dependent manner (Figures 7 and Figure 8).
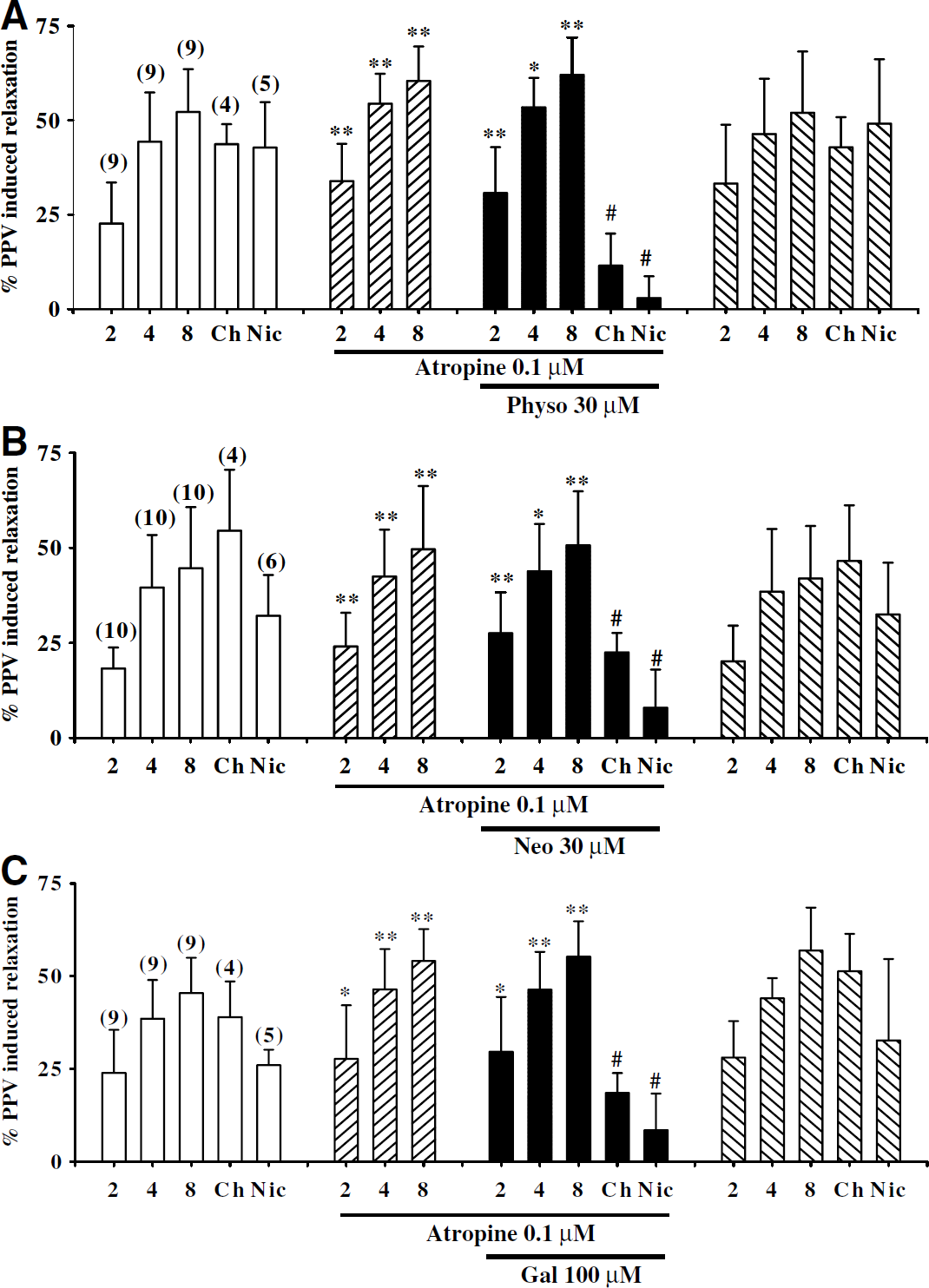
Effects of atropine on cholinesterase inhibitor (ChEI) inhibition of choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. The relaxation induced by TNS at 2, 4, and 8 Hz, was potentiated by atropine (0.1 μmol/L) (
The IC50 values for physostigmine against nicotine-and choline-induced calcium influx were 2.65 (1.83 to 3.84) × 10−5 and 2.1 (1.75 to 2.52) × 10−5 mol/L, respectively, which were not significantly different (P > 0.05). The IC50 values for neostigmine against nicotine- and choline-induced calcium influx were 7.29 (6.89 to 7.71) × 10−5 and 14.79 (5.61 to 132.04) × 10−5 mol/L, respectively, which were not significantly different (P < 0.05). The IC50 values for galantamine against nicotine- and choline-induced calcium influx were 6.88 (3.78 to 12.54) × 10−5 and 7.97 (5.51 to 11.53) × 10−5 mol/L, respectively, which were not significantly different (P < 0.05). Physostigmine, neostigmine, or galantamine alone did not affect calcium influx (n = 6, data not shown). In the presence of blockade of calcium influx by physostigmine, neostigmine, and galantamine, KCl (40 mmol/L) still induced a significant calcium influx (Figures 8A–8C), which was comparable with that seen in preparations in the absence of physostigmine, neostigmine, or galantamine. In this latter study, KCl-induced intracellular calcium increases over the basal concentration in control and after physostigmine, neostigmine (30 μmol/L) and galantamine (100 μmol/L) were 258.5% ± 13.1%; 247.6% ± 18.4%; 243.1% ± 7.7% and 287.2% ± 29.9%, respectively, and were not statistically different (P < 0.05).
Cholinesterase inhibitors inhibited choline- and nicotine-induced inward currents mediated by α7-nAChRs expressed in Xenopus oocytes. The α7-nAChRs were expressed as homopentamers in Xenopus oocytes, and the direct effect of ChEIs on currents mediated by these receptors were examined. As described by others (Couturier et al, 1990) the α7-nAChR-mediated current desensitized rapidly. Nicotine (0.1 mmol/L) was applied several times in a 10-mins interval till a stable response was achieved, and its maximal negative deflection served as control. Inward currents induced by nicotine were abolished by α-bungarotoxin (100 nmol/L, n = 3, data not shown). Physostigmine, neostigmine, or galantamine alone did not change the holding currents on oocytes. These ChEIs, however, profoundly inhibited nicotine-induced inward currents in a concentration-dependent manner (Figures 9A and Figure 9B). The IC50 values for physostigmine, neostigmine, and galantamine were 1.78 (1.25 to 2.53) × 10−5, 8.37 (3.33 to 20.91) × 10−5, and 2.70 (2.02 to 3.61) × 10−5 mol/L, respectively (Figure 9B). After washing off ChEIs, blockade of the nicotine-induced currents was partially reversed (Figure 9A).
Statins prevented ChEI inhibition of nicotine- and choline-induced neurogenic vasodilation, but did not affect that of TNS-induced neurogenic vasodilation. Mevastatin (10 μmol/L) prevented the inhibition of nicotine- and choline-induced relaxation by physostigmine (30 μmol/L), neostigmine (30 μmol/ L), and galantamine (100 μmol/L). Cholinesterase inhibitors inhibition of relaxation induced by TNS at 2, 4, and 8 Hz, however, was not prevented by mevastatin (10 μmol/L) (Figures 10A–10C). Lovastatin (10 μmol/L) had a similar effect (Figures 11A–11C). Mevastatin and lovastatin alone did not affect nicotinic agonist- (n = 4; data not shown) or TNS-elicited relaxation, nor did the solvent DMSO used to dissolve these statins have any effect on TNS- or nicotinic agonist-induced relaxation or the basal tone of the artery (n = 3; data not shown). The effect of mevastatin, lovastatin and the ChEIs on TNS- and nicotinic agonist-induced relaxation was reversible after washing off these drugs (Figures 10A–10C and 11A–11C).
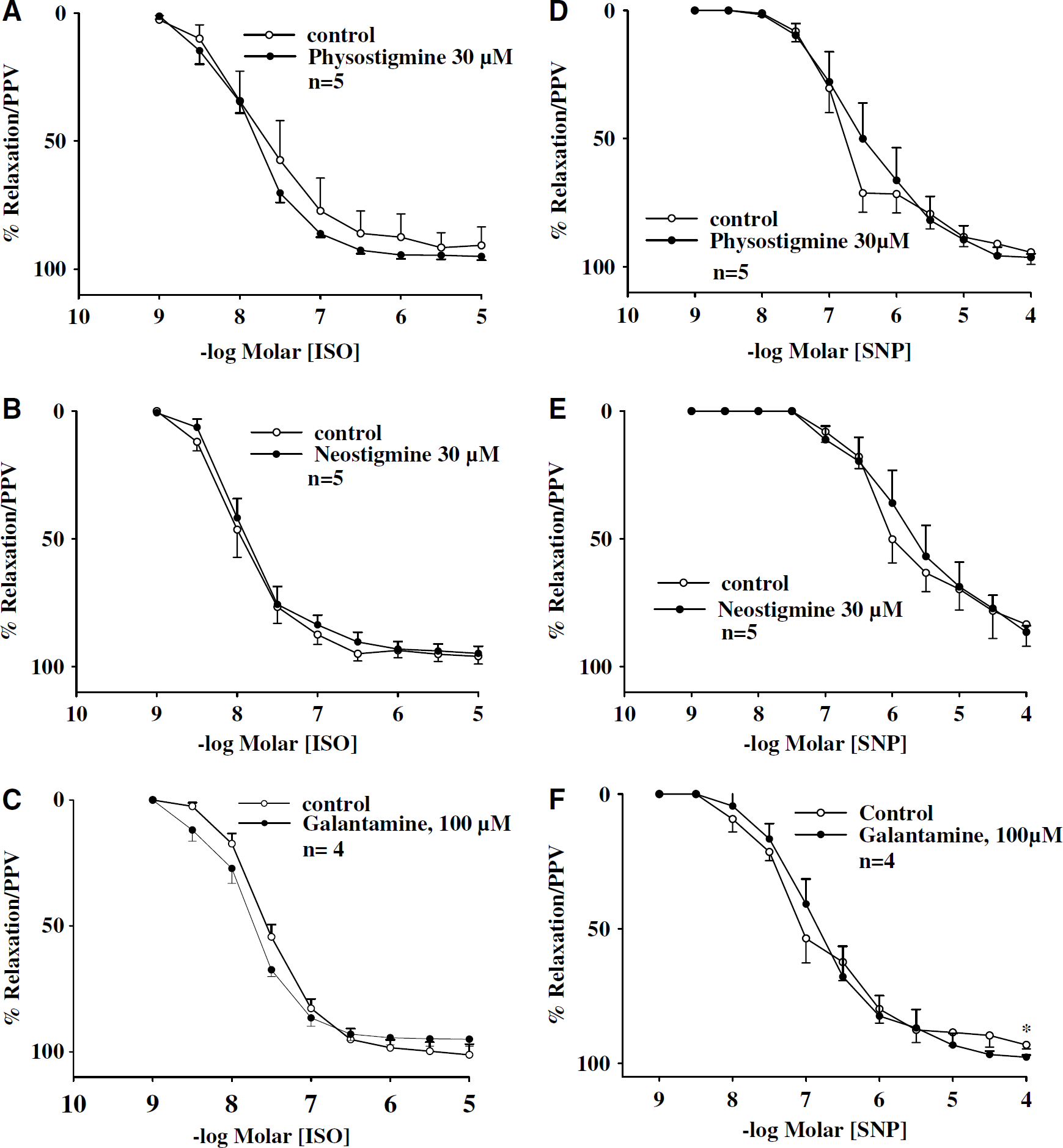
Effects of physostigmine, galantamine, and neostigmine on isoproterenol- and sodium nitroprusside relaxation in basilar arteries. In the presence of active muscle tone induced by 9,11-dideoxy-9,11-methanoepoxy prostaglandin F2 (U-46619) (0.3 μmol/L), porcine basilar arterial rings without endothelial cells relaxed on application of isoproterenol (ISO) (
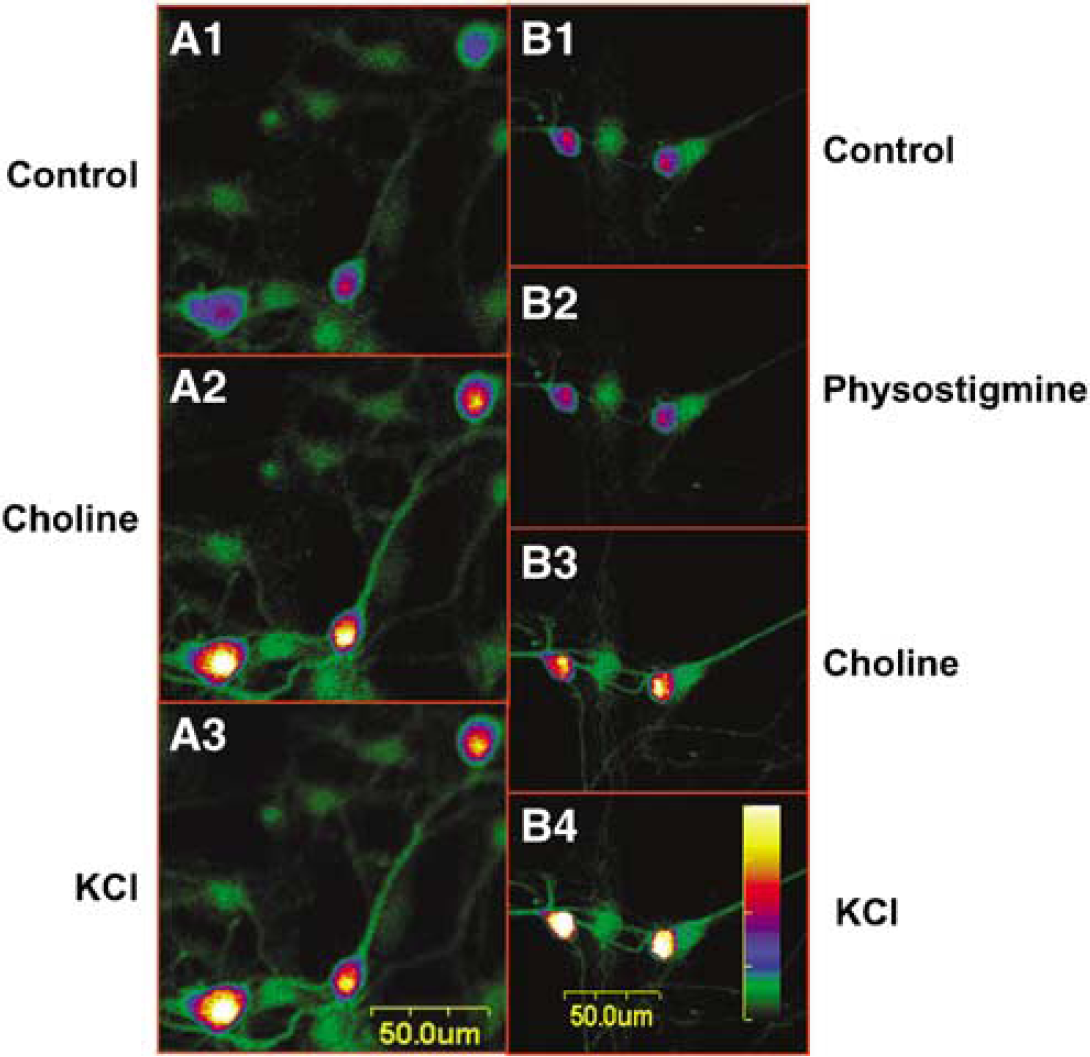
Effects of physostigmine on choline-induced calcium influx in cultured porcine superior cervical ganglion (SCG) cells. Cultured SCG cells were loaded with fluo-4 acetoxymethyl ester (AM) (3 μmol/L) in physiological buffer and incubated at room temperature for 30 mins (
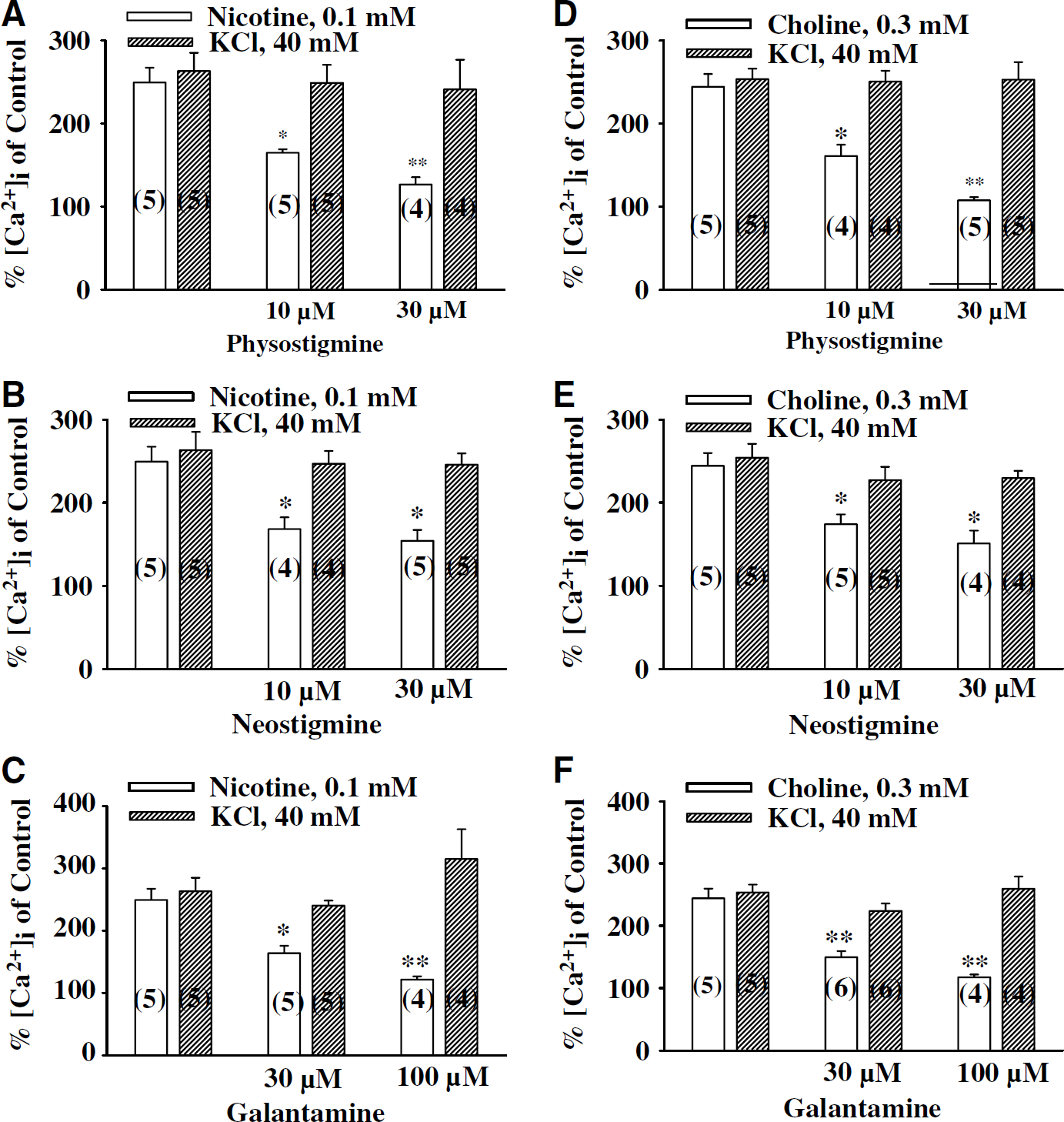
Summary of physostigmine, galantamine, and neostigmine inhibition of choline- and nicotine-induced calcium influx in the porcine superior cervical ganglions (SCGs). Changes in intracellular calcium was compared with the basal calcium concentration, which served as control (100%). Physostigmine (10 and 30 μmol/L; A and D), neostigmine (10 and 30 μmol/L; B and E), and galantamine (30 and 100 μmol/L; C and F) in a concentration-dependent manner inhibited choline (0.3 mmol/L)- and nicotine (0.1 mmol/L)-induced calcium influx. In the presence of these cholinesterase inhibitors (ChEIs), KCl (50 mmol/L) still induced calcium influx that was comparable with that in the absence of ChEIs. The values are means ± s.e.m. Number in parenthesis represents number of experiments; *P < 0.05 significant difference from respective controls.
Effect of physostigmine, galantamine, and neostigmine on inward currents mediated by α7-nAChR expressed in Xenopus oocytes. (
Discussion
Earlier evidence indicates that functional α7-nAChRs are found on the cerebral perivascular sympathetic, but are not on the nitrergic, neurons (Si and Lee, 2002). Binding of these receptors by nicotinic agonists results in release of NE, which then diffuses to act on β2-adrenoceptors located on the neighboring nitrergic nerves, causing release of NO from these nerves and, therefore, vasodilation (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2001, 2002) (Figure 12). The present study demonstrated that acute administration of physostigmine, galantamine, and neostigmine inhibited neurogenic relaxation of basilar arteries elicited by TNS at all frequencies examined; a result similar to previous findings (Liu and Lee, 1999; Liu et al, 2002). These ChEIs also inhibited nicotine- and choline-induced, α7-nAChR-mediated, calcium influx in cultured porcine SCG neurons, inward currents in α7-nAChR-expressed Xenopus oocytes, and neurogenic nitrergic dilation in isolated porcine basilar arteries. Cholinesterase inhibitors inhibition of α7-nAChR-mediated, but not TNS-elicited, neurogenic vasodilation, however, was prevented by mevastatin and lovastatin (Figure 12).
The finding that ChEIs inhibited the α7-nAChR and associated cerebral vasodilation raises concerns of the validity of applying ChEIs in the treatment of AD. Various studies have shown decreased regional cerebral blood flow in dementia patients, and this decrease was correlated with the degree of cognitive impairment (Jagust et al, 1987; O'Brien et al, 1992). Since ChEIs inhibit the α7-nAChR-mediated and TNS-induced vasodilation, these inhibitors may cause a further reduction in the cerebral blood flow. This may decrease the efficacy of ChEIs. In this regard, it has been noted that tobacco smoking which includes inhalation of nicotine, an α7-nAChR agonist, may have a protective effect in AD (van Duijn and Hofman, 1991; Hillier and Salib, 1997).
In addition, by inhibiting the breakdown of transmitter ACh, ChEIs may cause a reduction in extracellular concentration of choline, an endogenous agonist for the α7-nAChR (Si and Lee, 2002), and an increase in that of synaptic ACh. This classical transmitter, besides acting on the α7-nAChRs to elicit nitrergic vasodilation, induces vasoconstriction by acting on the Gq-coupled postsynaptic receptors present on the vascular smooth muscle, and inhibits NO release by activating the Gi-coupled presynaptic muscarinic M2 receptors (Lee et al, 1982; Miao and Lee 1990; Kimura et al, 1997; Yu et al, 1998; Liu et al, 2002). This latter finding provides an explanation for the decreased TNS-induced neurogenic nitrergic vasodilation in the presence of ChEIs while an increased nitrergic neurogenic vasodilation by blocking the muscarinic M2 receptor with atropine. Accordingly, ChEIs can decrease choline/ACh ratio and may indirectly cause a reduction in cerebral neurogenic nitrergic vasodilation.
In the presence of atropine, ChEI inhibition of TNS-induced relaxation was prevented, while its inhibition of nicotine- or choline-induced relaxation was not affected. These results further support the hypothesis of different mechanisms for ChEI inhibition of relaxations induced by TNS and nicotinic agonists. These results also indicate that the inhibition of TNS-induced relaxation by the ChEIs is not due to a possible local anesthetic effect of these enzyme inhibitors at the concentrations examined.
According to the axo-axonal or sympathetic-nitrergic interaction hypothesis (Figure 12), β2-adrenoceptors located on the nitrergic nerves play a key role in mediating NE-induced NO release from these nerves, leading to a NO—cGMP coupling and vasodilation (Zhang et al, 1998; Lee et al, 2000; Si and Lee, 2001, 2002). Any possible effect of ChEIs on β-adrenoceptors and/or NO-coupling mechanism, therefore, may alter NO-mediated relaxation. In the present study, relaxation of the basilar arteries induced by isoproterenol that acts on both presynaptic and postsynaptic β-adrenoceptors, however, was not appreciably affected by physostigmine, galantamine, or neostigmine. Therefore, ChEI inhibition of vasodilation induced by nicotine or choline in the present study was not likely due to any possible ChEI effect on presynaptic β2-adrenoceptors or postsynaptic β1-adrenoceptors (Lee et al, 1982; Lee, 2000). Moreover, relaxation induced by sodium nitroprusside, which is known to release NO on entering the smooth muscle cells (Ignarro et al, 1981), was not affected by these three ChEIs either. These results suggest that ChEI inhibition of nitrergic neurogenic vasodilation induced by nicotine and choline was not due to blockade of NO—cGMP pathway in the smooth muscle. A similar result was found in the rat aorta (Purdy et al, 1997).
Thus, by excluding their effects on the presynaptic and postsynaptic β-adrenoceptors and the NO—cGMP pathway, ChEIs most likely block the α7-nAChR located on the cerebral perivascular sympathetic neurons leading to a decreased nicotine- and choline-induced relaxation. The SCG is the origin of the cerebral perivascular sympathetic nerves in all species including the pig (Lee et al, 1982). Cultured porcine SCG neurons have been found to contain α7-nAChRs, which mediate calcium influx on activation by nicotinic agonists, like those found in cerebral perivascular sympathetic nerve terminals (Si and Lee, 2002). In the cultured SCG cells, choline- and nicotine-induced α7-nAChR-mediated calcium influx was blocked specifically by physostigmine, galantamine, and neostigmine in a concentration-dependent manner. Together with the findings from experiments in Xenopus oocytes that the inward currents mediated by the injected α7-nAChR was blocked by all ChEIs examined, these data suggested that ChEIs directly blocked the α7-nAChR.
It has been reported that L-arginine, the substrate for NO synthesis, is known to restore the responsiveness of endothelial cells of aged and damaged arterioles to vasodilators which in turn prevents memory impairment (Pautler, 1994). Furthermore, inhibitors of nitric oxide synthase prevent memory formation, which is overcome by administration of L-arginine (Schuman and Madison, 1991; Holscher and Rose, 1992). Thus, vasodilation induced by physiological concentrations of NO of different sources seems to have a beneficial effect on cognitive function. Cholinesterase inhibitors inhibition of cerebral nitrergic neurogenic vasodilation by directly inhibiting α7-nAChR and indirectly decreasing synaptic concentration of choline with increased ACh concentrations, therefore, may decrease the clinical efficacy of this class of drugs in treating dementia. This information is important, since ChEIs are still considered as the mainstream therapy for AD (Marksteiner and Schmidt, 2004).
The finding that statins prevent the ChEI-mediated inhibition of nicotine- and choline-induced relaxation but not the TNS-induced relaxation further suggests that statins preferentially affect α7-nAChR-mediated but not muscarinic receptor-mediated responses. This finding also excludes the possibility of a direct inactivation of ChEIs by statins. Others have reported that statins do not affect the ChE enzyme levels (Zdrenghea et al, 2002; Darvesh et al, 2004) or nNOS expression (Endres et al, 1998). Although there is a possibility that statins modify α7-nAChR or downstream coupling mechanism (Wei et al, 2005), these results suggest that statins act specitically by preventing ChEI inhibition on the α7-nAChR. The exact mechanisms of action of ChEI inhibition and its prevention by statins either or not at the receptor level remain to be fully examined.
Cholinesterase inhibitors inhibition of α7-nAChR-mediated nitrergic vasodilation, and its prevention by statins in porcine basilar artery as demonstrated in the present study may be a useful platform for providing information in understanding a more widespread inhibition of α7-nAChRs throughout the CNS. Nicotinic receptor agonists have been shown to improve (Wilson et al, 1995) while their receptor antagonists (Newhouse et al, 1994) impair cognitive function. This is consistent with the findings that α7-nAChRs in the hippocampus are important for cognitive function (Whiteaker et al, 1999; Fabian-Fine et al, 2001). Furthermore, nicotine, a nonspecitic α7-nAChRs agonist, has been shown to affect memory consolidation (Colrain et al, 1992), and activation of α7-nAChRs by various agonists has been shown to have beneficial effects on memory and learning (Meyer et al, 1997; Levin et al, 1999), while inhibition of these receptors by α7-nAChRs antagonist methyllycaconitine results in impaired memory (Felix and Levin, 1997). Moreover, the α7-nAChR has important neuroprotective properties (Kihara et al, 1997, Jonnala and Buccafusco, 2001). Beta-amyloid, a major constituent of senile plaques in AD causes neurodegeneration, and nicotinic agonists have been shown to be neuroprotective against beta-amyloid-induced neurotoxicity in cortical neurons through activation of the α7-nAChR (Felix and Levin, 1997). The α7-nACR has recently been shown to express on endothelial cells (Hsu et al, 2005; Lips et al, 2005) which may mediate endothelial functions. Thus, besides inhibiting neuronal α7-nACRs, ChEIs may also cause adverse effects on vascular functions by blocking endothelial α7-nACRs. It appears that the α7-nAChR plays an important role in the CNS vascular function, and that its blockade paralleling that in perivascular sympathetic neurons, therefore, may cause serious consequences on normal functioning.
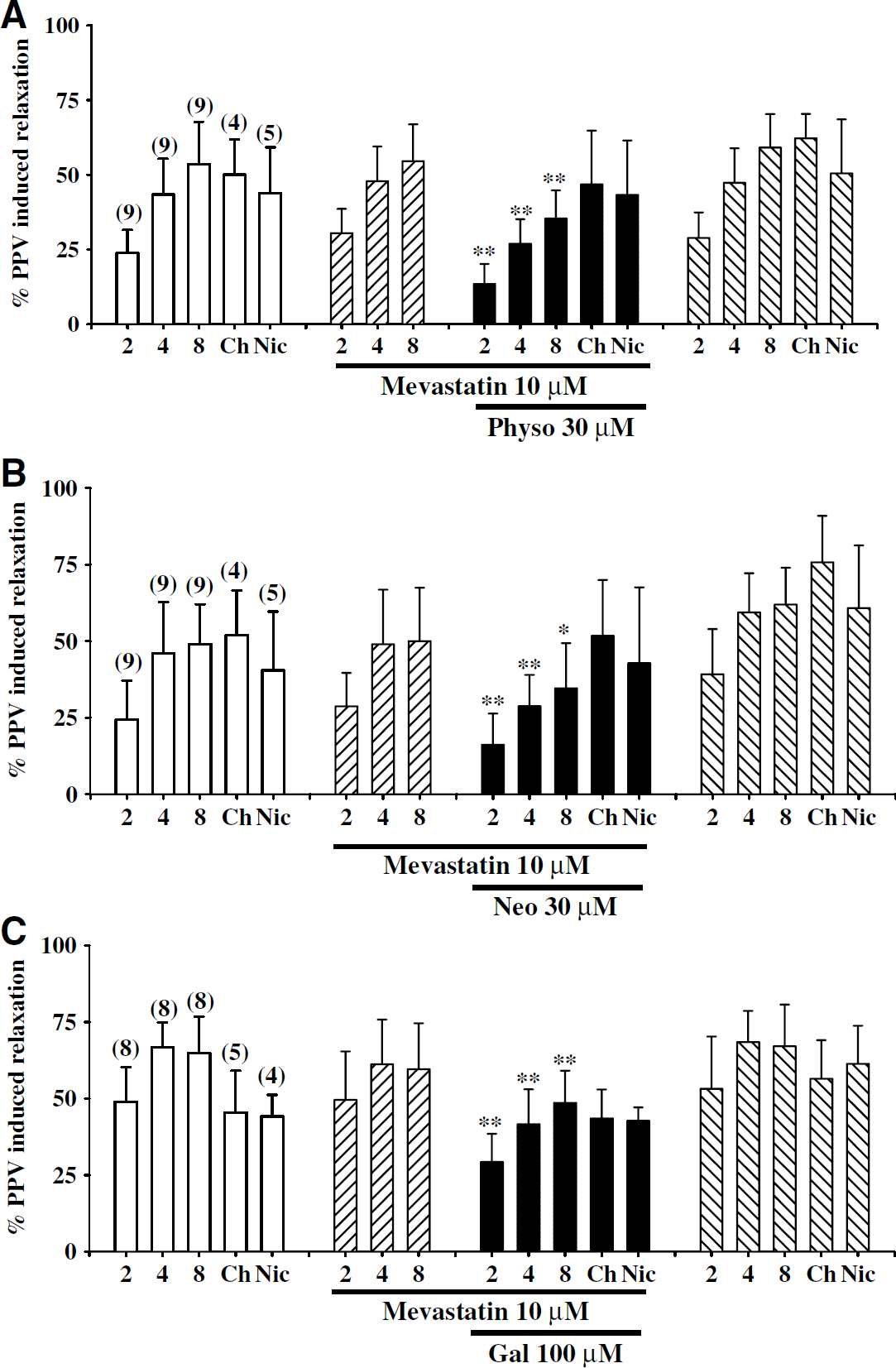
Effects of mevastatin on cholinesterase inhibitor (ChEI) inhibition of choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. In the presence of mevastatin (10 μmol/L), inhibition of nicotine- and choline-induced relaxation by physostigmine (30 μmol/L), neostigmine (30 μmol/L), and galantamine (100 μmol/L) was prevented (
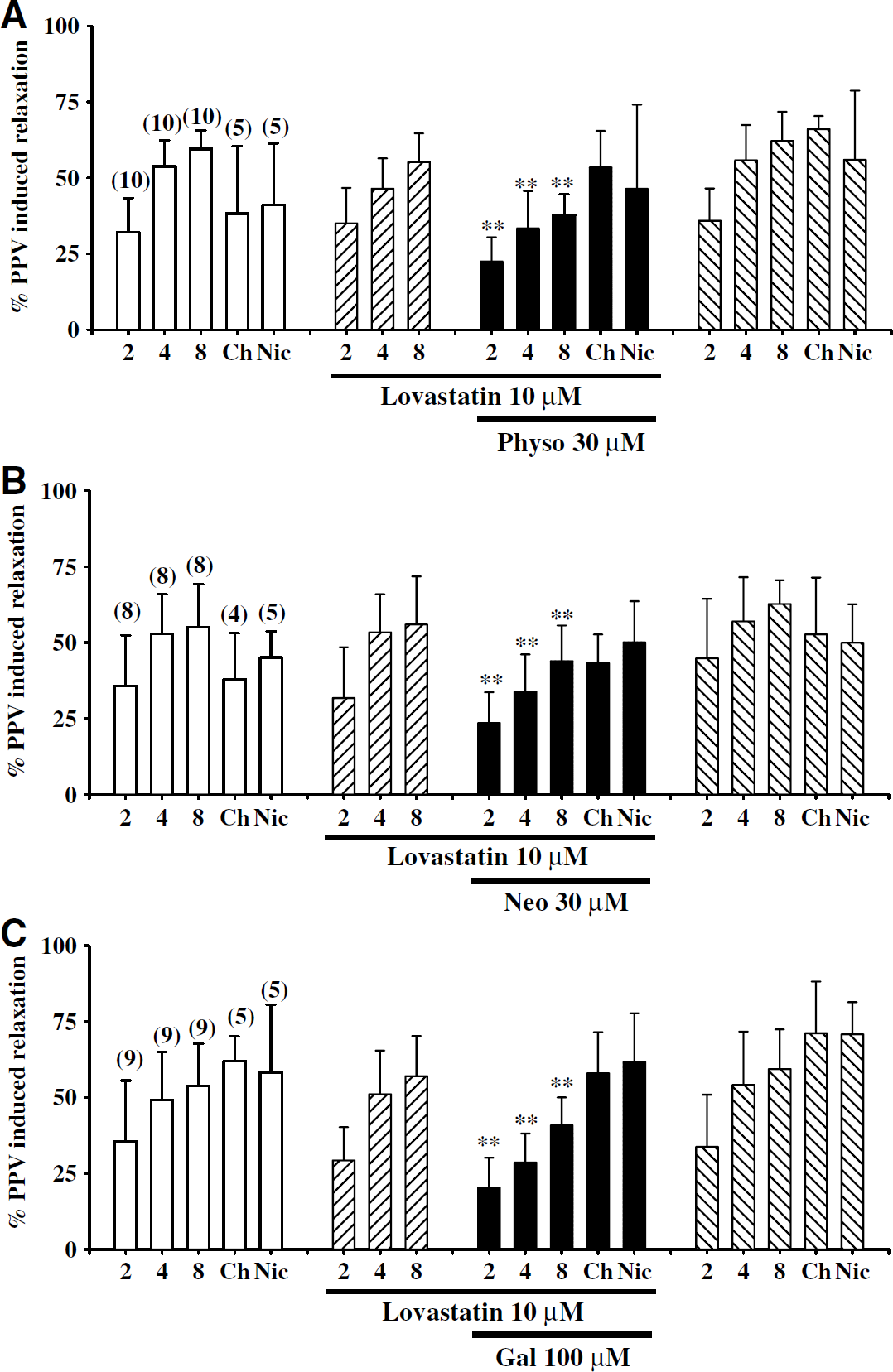
Effects of lovastatin on cholinesterase inhibitor (ChEI) inhibition of choline-, nicotine-, and transmural nerve stimulation (TNS)-induced relaxation of porcine basilar arterial rings without endothelial cells. In the presence of lovastatin (10 μmol/L), inhibition of nicotine- and choline-induced relaxation by physostigmine (30 μmol/L), neostigmine (30 μmol/L), and galantamine (100 μmol/L) was prevented (
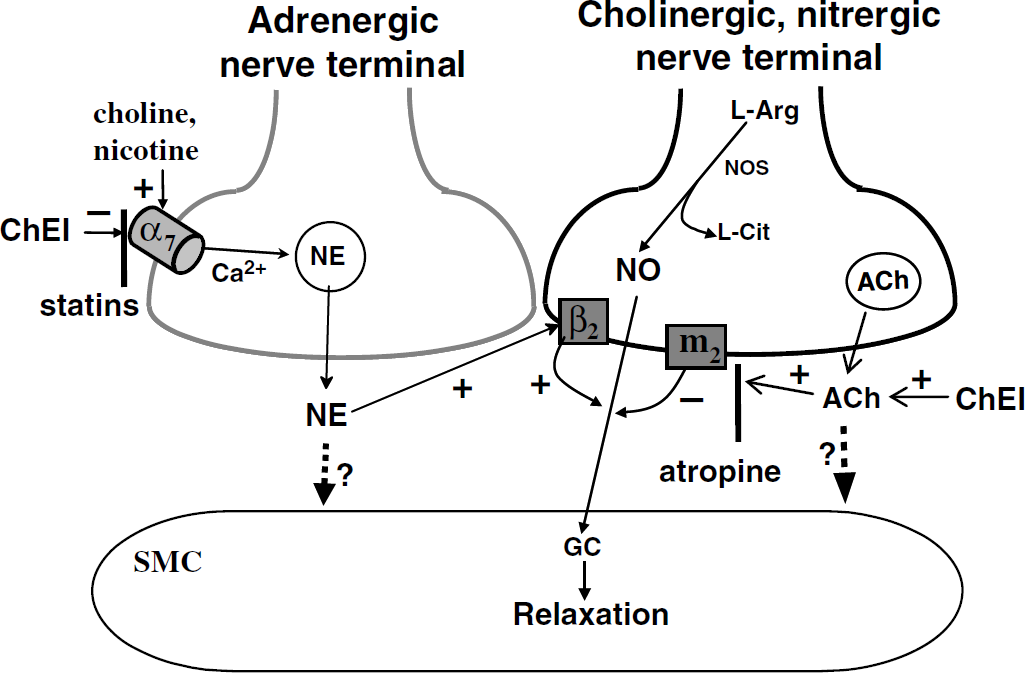
A summary diagram showing close apposition of an adrenergic and a cholinergic—nitrergic nerve terminal, and the possible mechanism of cholinesterase inhibitor (ChEI) blockade of choline- and nicotine-induced nitrergic neurogenic vasodilation in large cerebral arteries at the base of the porcine brain. The axo-axonal distance between these two different nerve terminals is closer than that between the nerves and the smooth muscle. Choline and other nicotinic agonists act on presynaptic α7-nicotinic acetylcholine receptors (α7-nAChRs) located on the adrenergic nerve terminal, causing calcium influx, and resulting in release of norepinephrine (NE) which then acts on presynaptic β2-adrenoceptors located on the adjacent cholinergic-nitrergic nerve terminal (Si and Lee, 2002). This effect of NE results in stimulating (+) synthesis and release of NO, which activates GC (Guanylate cyclase), increases cGMP synthesis, and relaxes the smooth muscle cell (SMC). NE released from the sympathetic adrenergic nerves, however, is a weak or insignificant postsynaptic transmitter (as indicated by dotted line with a question mark). NO is not stored in vesicles, and is synthesized from L-arginine (L-Arg) by nitric oxide synthase (NOS) with L-citrulline (L-Cit) as the by-product, providing evidence for the neuronal source of NO (Lee, 2000). Furthermore, NO is colocalized and coreleased with ACh which is stored in neuronal vesicles. ACh released from cholinergic-nitrergic nerves acts on presynaptic muscarinic M2 receptors (M2) resulting in G-protein-mediated negative coupling (−)to the N-type Ca2+ channels leading to suppression of Ca2+ influx through this type of Ca2+ channels (Liu et al, 2002). This will result in a decreased NOS activity accompanied by a decrease in NO formation and release, and diminished relaxation of the smooth muscle cell. ACh like NE is a very weak or negligible postsynaptic transmitter (Lee, 2000) (as indicated by dotted line with a question mark) based on animal studies. ChEIs increase synaptic concentration of ACh by attenuating its metabolism. These inhibitors also inhibit (−) α7-nAChRs, resulting in blocking calcium influx and nitrergic neurogenic vasodilation induced by choline and nicotine. Statins, which do not appear to affect ChE activity and therefore the synaptic ACh, reverse the blockade induced by the ChEI probably by preventing its binding to α7-nAChRs on the adrenergic perivascular neuron. The exact mechanism of action of statins, however, remains to be determined.
In summary, the present study demonstrated that α7-nAChRs located on the cerebral postganglionic sympathetic neurons were specifically blocked by ChEIs, resulting in inhibition of neuronal calcium influx and diminished nicotine- and choline-induced sympathetic-dependent nitrergic dilation in isolated porcine cerebral arteries and inward currents in α7-nAChR-expressed oocytes. This ChEI inhibition of the α7-nAChR was prevented by pretreatment with statins. Inhibition by ChEIs of the α7-nAChRs and the synthesis of choline (an endogenous α7-nAChR agonist), therefore, can cause diminished vasodilation (i.e., vasoconstriction), in the cerebral circulation, which may be detrimental in management of dementia especially vascular dementia. Besides their effects on cerebrovascular function, the α7-nAChRs in the CNS play intimate role in neuroprotection, memory and cognitive function. Our present findings, therefore, may provide important information in advancing our understanding of the role of α7-nAChRs in the therapeutic and/or secondary effects of AD, and raise an important concern regarding the validity of using a ChEI alone in the treatment of this disease. Our results suggest that potential adverse effects due to α7-nAChR blockade by ChEIs may be prevented by concurrent therapy of ChEIs with statins.