
Abstract
Activation of the complement cascade contributes to brain injury after intracerebral hemorrhage (ICH). However, a recent study found that complement C5 deficient mice had enhanced ICH-induced brain injury. The present study, therefore, investigated the role of complement C3 (which is upstream from C5) in ICH. Male complement C3 deficient and sufficient mice had an intracerebral infusion of 30-μL autologous whole blood. The mice were killed and the brains were sampled for edema, Western blotting, immunohistochemistry and histologic analysis. Behavioral tests including forelimb use asymmetry test and corner turn were also performed before and after ICH. Compared to complement C3 sufficient mice, C3 deficient mice had less brain edema, lower hemeoxygenase-1 levels, less microglia activation and neutrophil infiltration around the clot after ICH. In addition, the C3-deficient mice had less ICH-induced forelimb use asymmetry deficits compared with C3-sufficient mice. These results suggest complement activation may affect heme metabolism and the inflammatory response after ICH suggesting that complement C3 is an important factor causing ICH-induced brain injury.
Introduction
Complement-mediated brain injury has been found in many central nervous system diseases, including intracerebral hemorrhage (ICH) and cerebral ischemia (Hua et al, 2000; Huang et al, 1999; Morgan et al, 1997; Xi et al, 2001). Our previous studies have demonstrated that complement depletion or complement inhibition reduces perihematomal brain edema in a rat model of ICH (Hua et al, 2000; Xi et al, 2001), but the mechanisms underlying complement-mediated brain injury after ICH are not well understood. Recently, we found more (rather than less) ICH-induced brain injury in complement C5 deficient mice (Nakamura et al, 2004b). This suggests that factors upstream from C5, such as C3a and C3b derived from C3, have a detrimental effect in ICH. Complement C3a is involved in regulating inflammation, while C3b is an opsonization agent (i.e. it is involved in phagocytosis) (Morgan et al, 1997).
While inflammation and phagocytosis are involved in hematoma resolution, they may also be involved in brain injury. There is evidence that microglia activation and neutrophil infiltration contribute to brain injury after ICH (Wang et al, 2003; Wang and Tsirka 2005; Xi et al, 2006). In rat ICH models, neutrophil infiltration develops within 2 days and activated microglial cells persist for at least a month (Gong et al, 2004; Jenkins et al, 1989).
There is also evidence that constituents formed during hematoma breakdown are involved in brain injury (Huang et al, 2002; Xi et al, 2006). Heme oxygenases are key enzymes in heme degradation. Brain heme oxygenase-1 (HO-1) can be induced by various stimuli such as heme and hemoglobin (Maines 1988; Wagner et al, 2003). Brain HO-1 levels are increased after intracerebal hemorrhage (Huang et al, 2002; Matz et al, 1997; Wu et al, 2003). Although the role of HO-1 upregulation in brain injury after ICH is still unclear, heme oxygenase inhibitors reduce ICH-induced brain damage (Huang et al, 2002; Koeppen et al, 2004; Wagner et al, 2000).
The aim of the present study was to examine ICH-induced brain damage in complement C3 deficient and sufficient mice. Brain edema and neurological deficits were examined. Brain HO-1 levels in the brain, microglia activation and neutrophil infiltration were also analyzed.
Materials and methods
Animals Preparation and Intracerebral Hemorrhage Model
The University of Michigan Committee on the Use and Care of Animals approved the animal protocols. A total of 30 male C3 deficient mice (originally obtained from Jackson Laboratory, Bar Harbor, ME, USA, and bred at the University of Michigan by Dr John Younger; Ben-David et al, 2005) and 30 male C57BL/6J C3 sufficient mice (from Jackson Laboratory, Bar Harbor, ME, USA), all approximately 2 to 3 months of age, were used in this study. Pairs of homozygote C3 deficient mice (C57BL/ 6J-C3tm1Crr, also called B6.129S4-C3tm1Crr/J) were obtained from Jackson Laboratories, and these were used to establish the breeding colony for these experiments. The homozygote C3 deficient mice had been backcrossed onto C57BL/6J mice for six generations by Jackson Laboratories and mice of that strain were used as controls (C3 sufficient mice) as recommended by Jackson Laboratories. All mice were allowed free access to food and water. Mice were anesthetized with ketamine (90 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.). The right femoral artery was catheterized to monitor arterial blood pressure and blood glucose levels, and to sample blood for intracerebral autologous blood infusion. Rectal temperature was maintained at 37.5°C using a feedback-controlled heating pad. The mice were then positioned in a stereotaxic frame (Model 500, Kopf Instruments, Tujunga, CA, USA) and a cranial burr hole (1 mm) was drilled near the right coronal suture 2.5 mm lateral to midline. A 26-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates: 0.2 mm anterior, 3.5 mm ventral and 2.5 mm lateral to the bregma). A total of 30-μL autologous whole blood was infused at a rate of 2 μL/min. The needle was removed, the burr hole was filled with bone wax, and the skin incision was closed with suture.
Experimental Groups
There were two parts in this study. In the first part, the mice were killed 3 days after infusion for brain water and ion content measurement (n = 6). Behavioral tests including forelimb use asymmetry test and corner turn were also performed before ICH and 1 day, 3 day after ICH. In the second part, the mice were killed 1 day and 3 days after ICH. The brains were sampled for Western blotting (n = 4, per time point), immunohistochemistry (n = 4, per time point) and histology (n = 4, per time point).
Brain Water and Ion Content
The mice were anesthetized (ketamine 120 mg/kg and xylazine 5 mg/kg i.p.) and decapitated 3 days after ICH to determine brain water and ion contents. The brains were removed and a coronal brain slice (2 mm thick) was cut 2 mm posterior to the frontal pole. The brain slice was divided into two hemispheres along the midline. Each hemisphere was then divided into the cortex and the basal ganglia. The cerebellum served as a control. Five samples from each brain were obtained: the ipsilateral and contralateral cortex, the ipsilateral and contralateral basal ganglia, and the cerebellum. Brain samples were weighed immediately on an electronic analytical balance (Model AE 100, Mettler Instrument Co., Highstown, NJ, USA) to obtain the wet weight. The brain samples were then dried at 100°C for 24 h to obtain the dry weight. The formula for edema calculation was the following: (wet weight—dry weight)/wet weight × 100. The dehydrated samples were digested in 1 ml of 1 mol/L nitric acid for 1 week. The Na+ and K + contents of this solution were measured using a flame photometer (Model IL 943, Instrumentation Laboratory, Inc., Lexington, MA, USA). The ion content was expressed in milli-equivalents per kilogram of dehydrated brain tissue (mEq/kg dry wt).
Behavioral Tests
Behavioral deficits were assessed using a forelimb use asymmetry test and a corner turn test (Hua et al, 2002a; Nakamura et al, 2004b). Forelimb use during exploratory activity was analyzed in a standing transparent cylinder. Behavior was quantified by determining the number of times the normal ipsilateral (I) forelimb, the impaired contralateral (C) forelimb, and both (B) forelimbs were used as a percentage of total number of limb usage. A single, overall limb-use asymmetry score was calculated as follows: forelimb-use asymmetry score = {I/(I + C + B)} –{C/(I + C + B)}.
During the corner turn test the mouse was allowed to proceed into a corner, the angle of which was 30°. To exit the corner, the animal could turn either left or right. When the mouse turned, its choice of direction was recorded. This was repeated 10 to 15 times, and the percentage of right turns was calculated.
Western Blot Analysis
Animals were anesthetized before undergoing intracardiac perfusion with 0.1 mol/L phosphate-buffered saline. The brains were then removed and a 2-mm thick coronal brain slice was cut approximately 2 mm from the frontal pole. The slice was separated into the ipsilateral and contralateral basal ganglia. Western blot analysis was performed as previously described (Xi et al, 1999). Briefly, 50 μg proteins for each were separated by sodium dodecyl sulfate polyacrymide gel electrophoresis and transferred onto a hybond-C pure nitrocellulose membrane (Amersham). The membranes were blocked in Carnation nonfat milk. Membranes were probed with the primary and secondary antibodies. The primary antibodies were rabbit anti-HO-1 polyclonal antibody (1:2000 dilution, Stressgen, Victoria, Canada) and rat anti mouse F4/80 (1:1000 dilution, Serotec Ltd, Oxford, UK). An antibody to b-actin (monoclonal anti-b-actin, Sigma, St Louis, MO, USA) was used to examine equal loading of gels. The mouse F4/80 is a 160 kDa glycoprotein expressed by most murine macrophages and microglia. The antigen—antibody complexes were visualized with a chemiluminescence system and exposed to Kodak X-OMAT film (Rochester, NY, USA). The relative densities of bands were analyzed with the NIH Image (Version 1.62, Bethesda, MD, USA).
Immunohistochemical Staining and Histologic Examination
The immunohistochemistry method has been described previously (Hua et al, 2002b). Briefly, the mice were reanesthetized and perfused with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). The brains were removed and kept in 4% paraformaldehyde for 6 h, then immersed in 25% sucrose for 3 to 4 days at 4°C. After embedding in the mixture of 25% sucrose and OCT (SAKURA, Finetek, USA), 18-μm sections were taken on a cryostat. The avidin—biotin complex technique was used in the staining and hematoxylin was used for counter staining. The primary antibodies were rabbit anti-rat HO-1 (1:200 dilution, StressGen, Victoria, Canada) and mouse anti-rat CD11b (MRC OX42, 1:200 dilution, Serotec Ltd, Oxford, UK), Normal rabbit or mouse serum and the absence of primary antibody were used as negative controls. Hematoxylin and eosin staining were used for histologic examination. To assess the effects of complement C3 on neutrophil infiltration, we used 18-μm thick coronal sections from 1 mm anterior to the blood injection site. Neutrophils in the ipsilateral basal ganglia were counted by a blinded observer.
Statistical Analysis
All data in this study are presented as mean ± s.d. Data were analyzed with Student's t-test and analysis of variance (ANOVA), followed by Scheffe's post hoc test. Significance levels were measured at P < 0.05.
Results
Perihematomal brain edema was less in complement C3 deficient mice compared to complement C3 sufficient mice (the ipsilateral basal ganglia: 79.5% 70.4% versus 80.6% 71.0%, P < 0.05; the ipsilateral cortex: 78.6% ± 0.6% versus 79.6% ± 0.6%, P < 0.05; Figure 1) 3 days after ICH. The reduced perihematomal edema in complement C3 deficient mice was associated with less accumulation of sodium ion (Figure 2A) and less loss of potassium ion (Figure 2B). Water and ion contents in the contralateral basal ganglia, the contralateral cortex and the cerebellum were the same in complement C3 deficient and complement C3 sufficient mice (Figures 1 and 2).
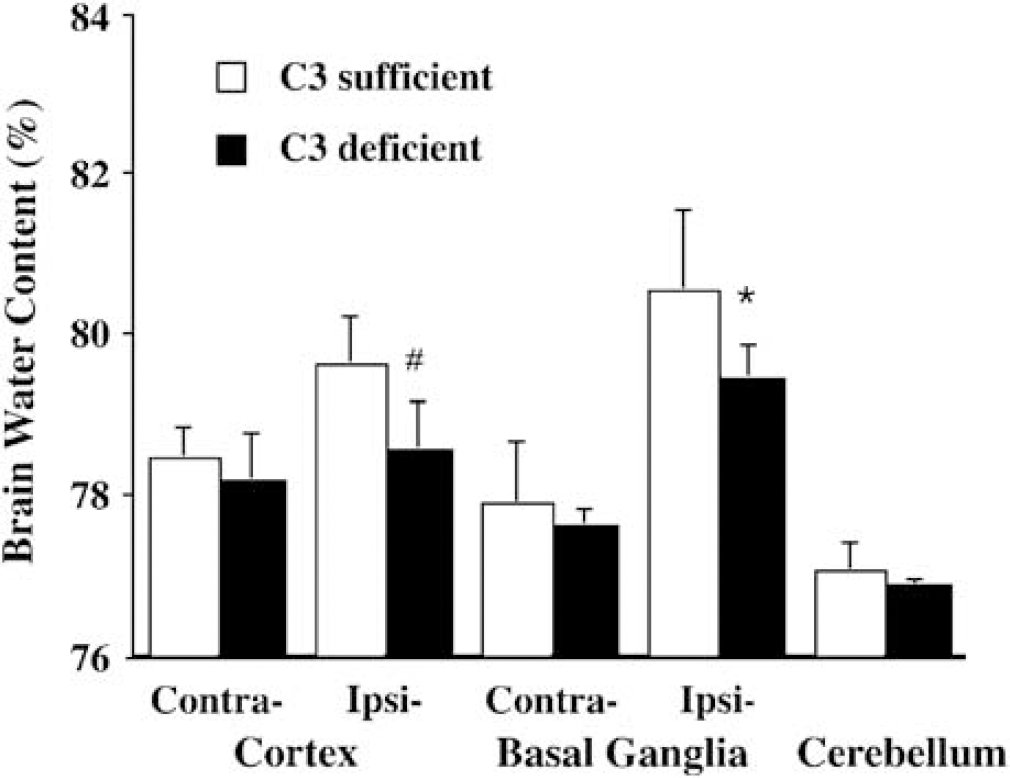
Brain water content 3 days after intracerebral infusion of 30-μL autologous whole blood in complement C3 sufficient and deficient mice. Values are expressed as mean ± s.d., n = 6. *P < 0.05, #P < 0.01 versus C3 sufficient mice.
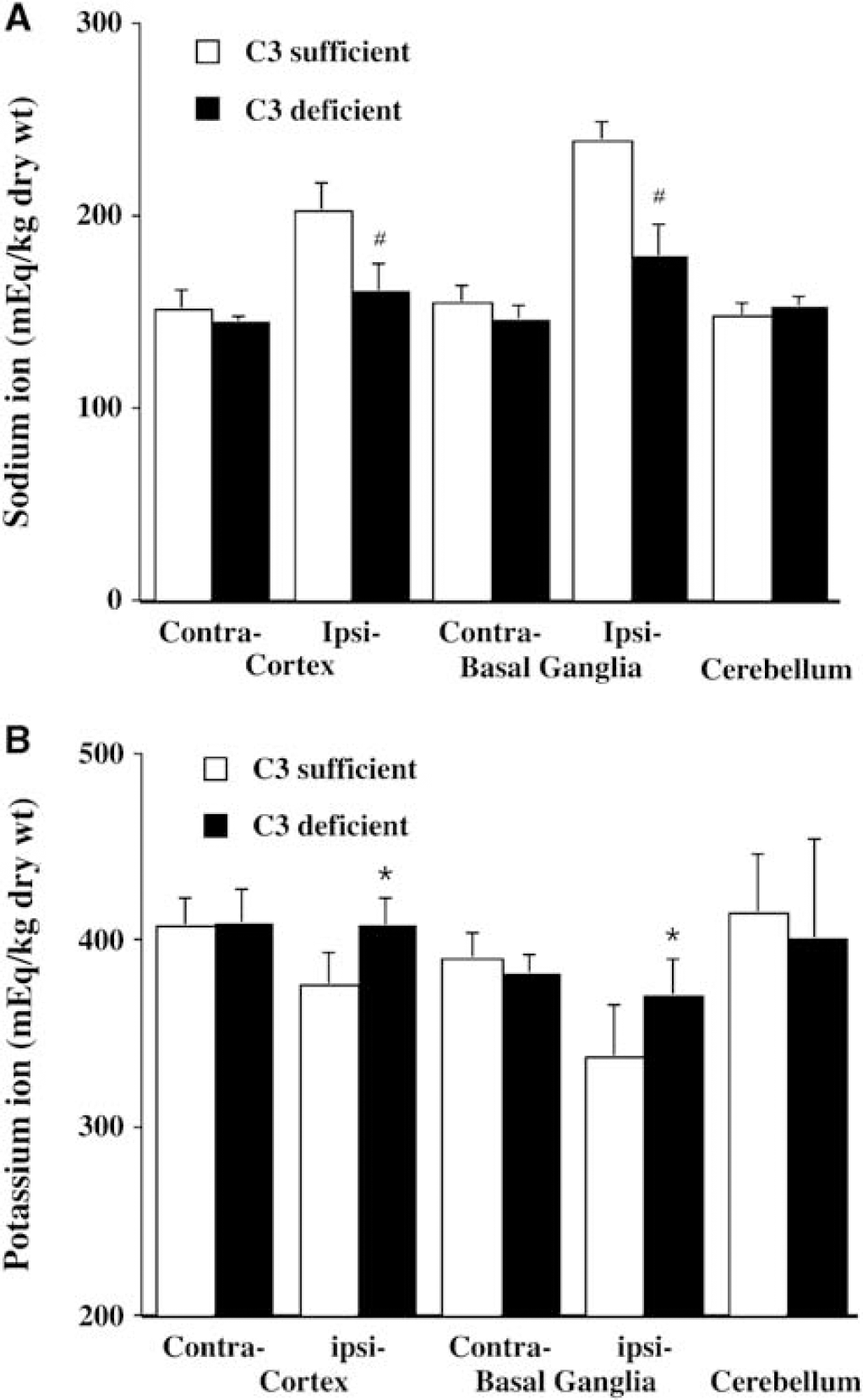
Brain sodium (
Forelimb use asymmetry test and corner turn test were performed before, and 1 and 3 days after ICH. Forelimb use asymmetry deficits in complement C3-deficient mice were less than those in complement C3-sufficient mice (P < 0.05; Figure 3). However, there was no difference in the corner turn score at day 1 (90.0% ± 10.0% versus 97.5% ± 14.0% in complement C3 deficient and sufficient mice, respectively; P > 0.05) and day 3 (82.0% ± 11.0% versus 80.0% ± 10.7% in deficient and sufficient mice, respectively; P> 0.05).
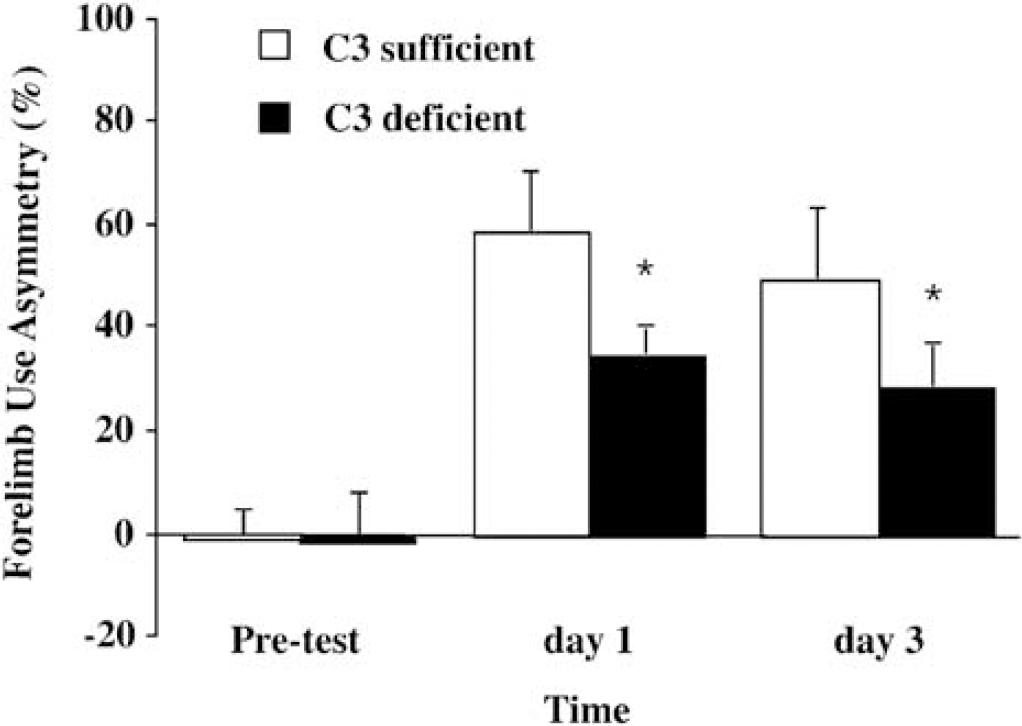
Forelimb use asymmetry scores before and after intracerebral hemorrhage in C3 sufficient and deficient mice. Values are expressed as mean ± s.d., n = 6. * P < 0.05 versus C3 sufficient mice.
Microglial activation contributes to ICH-induced brain damage. Immunoreactivity of perihematomal microglia (OX-42) was less in complement C3 deficient compared to complement C3 sufficient mice (Figures 4A and 4B). Western blot analysis also showed that the levels of F4/80 protein, another marker of microglia, were less in complement C3 deficient mice in the ipsilateral basal ganglia after ICH (479 ± 270 versus 1468 ± 104 pixels in complement C3 sufficient mice, n = 3 each group, P < 0.05).
Neutrophil infiltration may exacerbate brain injury after ICH. At 3 days after ICH, perihematomal neutrophils were less in complement C3 deficient mice compared with complement C3 sufficient mice (46 ± 40 versus 261 ± 77 cells/mm2, P < 0.01; Figure 4C and 4D).
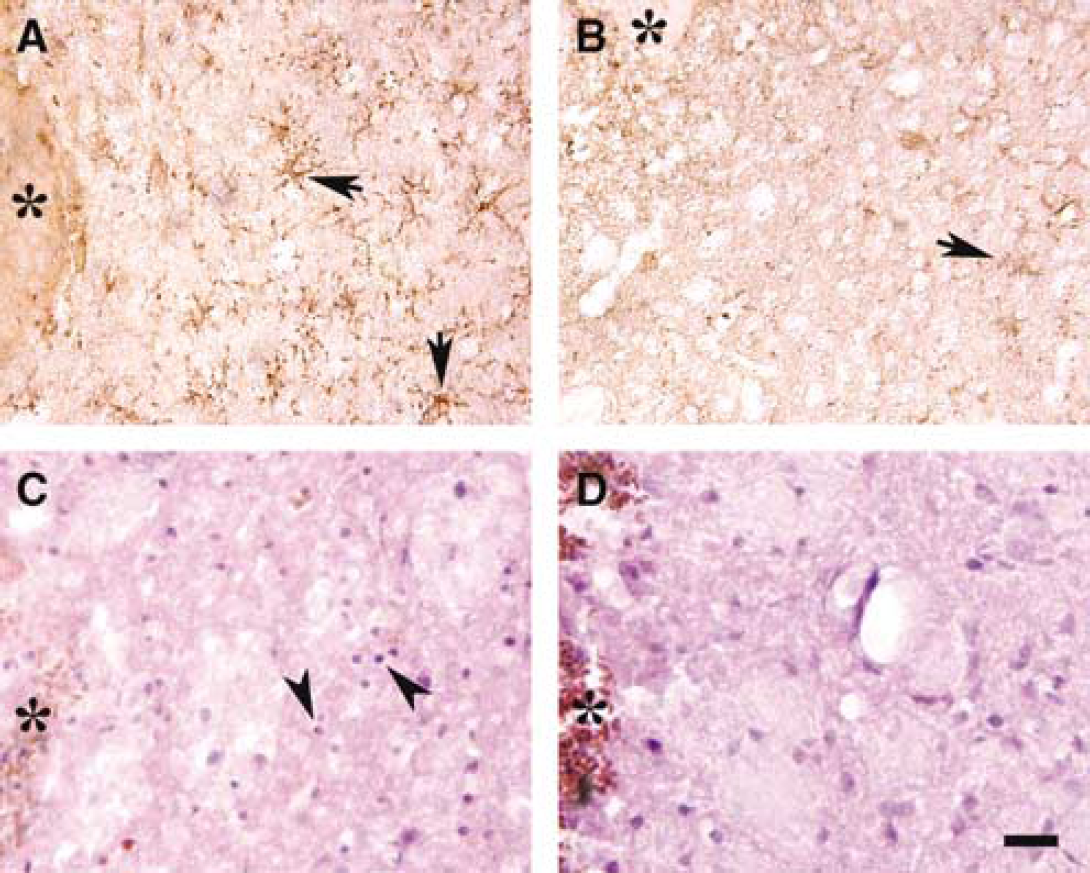
OX-42 immunoreactivity around the hematoma in complement C3 sufficient (
Brain HO-1 levels were measured by Western blot analysis and the cellular location of HO-1 was examined by immunohistochemistry. HO-1 levels in the ipsilateral basal ganglia were lower in complement C3 deficient mice (2024 ± 560 versus 5140 ± 1151 pixels in complement C3 sufficient mice, P < 0.05; Figure 5) 3 days after ICH. Most HO-1 positive cells in the perihematomal zone were glialike cells. The imunoreactivity of HO-1 around the hematoma was also less in the complement C3 deficient mice than that in the complement sufficient mice (Figure 6).
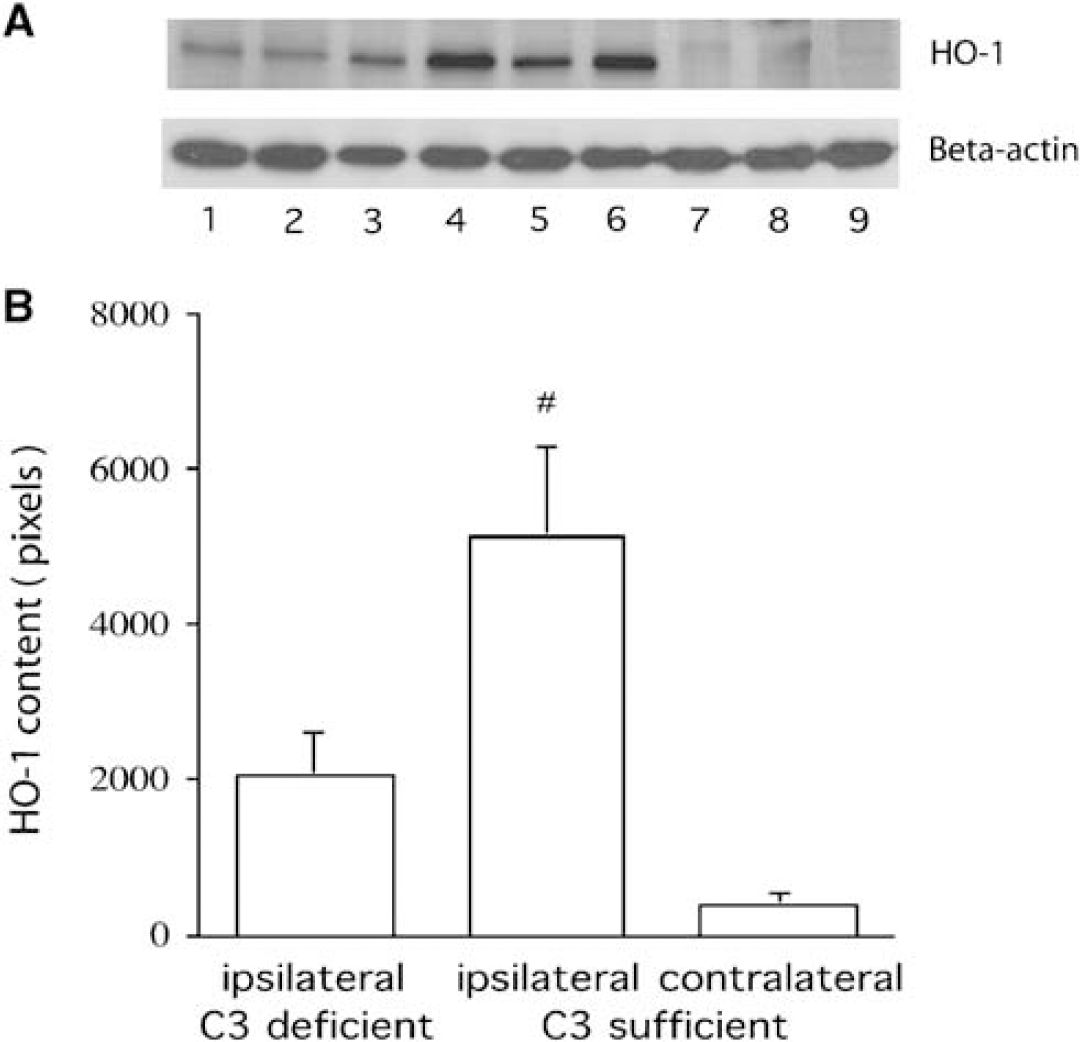
Western blot analysis of HO-1 protein levels in the basal ganglia 3 days after intracerebral infusion of autologous blood. (
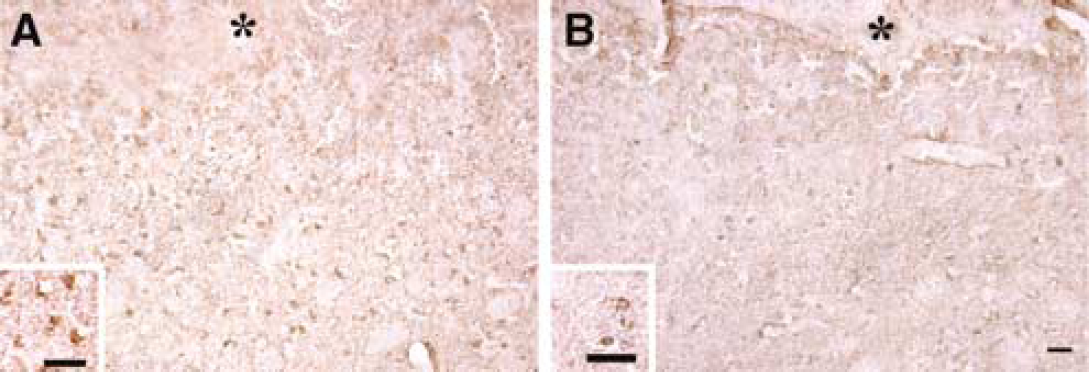
HO-1 immunoreactivities around the hematoma in complement C3 sufficient (
Discussion
The main findings of this study are: (1) ICH-induced brain edema and forelimb use asymmetry deficits are less in the complement C3 deficient mice compared to complement C3 sufficient mice. (2) Microglial activation and neutrophil infiltration around the hematoma are less in complement C3 deficient mice. (3) Intracerebral hemorrhage-induced increases in brain HO-1 in the ipsilateral basal ganglia are less in complement C3 deficient mice. These results indicate that complement C3 is an important factor causing brain damage following ICH and suggest that inflammation and enhanced heme metabolism contribute to complement-mediated perihematomal brain injury.
The development of mice deficient in elements of the complement system are stimulating investigators to study the role of different complement components in systemic injury (Holers 2000). Such experiments suggest that not all components of the complement system may enhance injury but some may, in fact, be beneficial (Mukherjee and Pasinetti, 2000). We found that ICH-induced forelimb use asymmetry deficit and brain edema are less in complement C3 mice, but our earlier study showed that neurological deficits and brain edema were worse in complement C5 deficient mice (Nakamura et al, 2004b). These controversial results indicate different complement factors have different effects on brain injury after ICH, that is, either beneficial or harmful. One study has shown that, for example, kainic acid-induced hippocampal damage increases in C5 deficient mice (Pasinetti et al, 1996). C5 deficient mice fail to form C5a fragments and member attack complex (MAC), but complement C3 deficient mice fail to form of C3a, C3b, C5a and MAC. Thus, our present results suggest that C3a and C3b may be the main factors causing brain damage after ICH.
Complement C3a production may enhance the inflammatory response after ICH. Inflammation in the perihematomal zone has been found after experimental ICH and is probably complement-mediated (Gong et al, 2000; Xi et al, 2001). C3a can attract leukocytes and cause blood-brain barrier disruption. We have demonstrated that systemic complement depletion by cobra venom factor reduces tumor necrosis factor-alpha (TNF-α) levels, myeloperoxidase positive cells and brain edema (Xi et al, 2001). In the present study, we have shown reduced leukocyte infiltration around the clot in complement C3 deficient mice.
Intracerebral hemorrhage-induced microglial activation was less in our complement C3 deficient mice. Microglia within the brain are activated in response to injury. Although microglial activation is a brain injury marker for many CNS diseases and is associated with neuronal death in the ischemic penumbra (Dirnagl et al, 1999; Schwartz 2003), depending on specific conditions they have neurotrophic actions (Streit et al, 1999). Activation of microglia around the hematoma occurs after ICH (Gong et al, 2000, 2004). We have found that microglial activation enhances brain injury in aged rats after ICH(Gong et al, 2004). Microglia secrete many toxic materials such as free radicals (Klegeris and McGeer 2000). In addition, tuftsin fragment 1 to 3, an inhibitor of macrophage/microglia activation, reduces brain injury in a mouse model of ICH (Wang et al, 2003).
This study demonstrates that less perihematomal edema is associated with lower brain HO-1 levels after ICH in C3 deficient mice. HO-1 can degrade heme into iron, carbon monoxide and biliverdin (Kutty and Maines 1981). Studies have shown that heme oxygenases are key enzymes in ICH-induced brain injury since injury is reduced by heme oxygenase inhibition (Huang et al, 2002; Koeppen et al, 2004; Wagner et al, 2000). A smaller increase in HO-1 may result in less accumulation of iron in the brain. Brain iron overload has been found after ICH and contributes to ICH-induced brain injury (Nakamura et al, 2004a; Wu et al, 2003). Furthermore, the levels of HO-1 are critical for ICH-induced brain injury. A limited increase in HO-1 protected against cell damage in vitro, but a greater increase caused significant oxidative cytotoxicity (Suttner and Dennery 1999). Whether a moderate increase of HO-1 is neuroprotective in ICH should be studied further.
We found that forelimb use asymmetry deficits but not corner turn scores were less in C3-deficient mice. The corner test may be more sensitive to injury because it reflects multiple asymmetries, including postural, vibrissae sensory, forelimb and hindlimb use asymmetries, which all combine at nearly the same time to bias turning. In a rat model of ICH, we have reported that hirudin treatment can improve forelimb use asymmetry score after ICH but not corner turn score (Hua et al, 2002a).
The effects of an element of the complement system may depend on the absolute level of activity. Partial inhibition may have different effects from total deficiency. The same can be said about the role of heme oxygenase 1 in brain injury, which may be protective or detrimental depending on its concentration (Suttner and Dennery 1999).
In summary, ICH results in less brain injury in complement C3 deficient mice. This is associated with reduced HO-1 upregulation, less microglial activation and diminished neutrophil infiltration. These results indicate that C3 is a key detrimental factor in complement-mediated brain injury following ICH.