
Abstract
Recent studies have revealed that the phosphatidylinositol 3-kinase (PI3-K) pathway is involved in apoptotic cell death after experimental cerebral ischemia. The serine—threonine kinase, Akt, functions in the PI3-K pathway and prevents apoptosis by phosphorylation at Ser473 after a variety of cell death stimuli. After phosphorylation, activated Akt inactivates other apoptogenic factors, including glycogen synthase kinase-3β (GSK3β), thereby inhibiting cell death. However, the role of Akt/GSK3β signaling in the delayed death of hippocampal neurons in the CA1 subregion after transient global cerebral ischemia (tGCI) has not been clarified. Transient global cerebral ischemia for 5 mins was induced by bilateral common carotid artery occlusion combined with hypotension. Western blot analysis showed a significant increase in phospho-Akt (Ser473) and phospho-GSK3β (Ser9) in the hippocampal CA1 subregion after tGCI. Immunohistochemistry showed that expression of phospho-Akt (Ser473) and phospho-GSK3β (Ser9) was markedly increased in the vulnerable CA1 subregion, but not in the ischemic-tolerant CA3 subregion. Double staining with phospho-GSK3β (Ser9) and terminal deoxynucleotidyl transferase-mediated uridine 5ʼ-triphosphate-biotin nick end labeling showed different cellular distributions in the CA1 subregion 3 days after tGCI. Phosphorylation of Akt and GSK3β was prevented by LY294002, a PI3-K inhibitor, which facilitated subsequent DNA fragmentation 3 days after tGCI. Moreover, transgenic rats that overexpress copper/zinc-superoxide dismutase, which is known to be neuroprotective against delayed hippocampal CA1 injury after tGCI, had enhanced and persistent phosphorylation of both Akt and GSK3β after tGCI. These findings suggest that activation of the Akt/GSK3β signaling pathway may mediate survival of vulnerable hippocampal CA1 neurons after tGCI.
Introduction
Transient global cerebral ischemia (tGCI) induces selective and delayed neuronal death (Kirino, 1982; Pulsinelli et al, 1982). Although the molecular mechanisms underlying the pathogenesis of delayed neuronal death remain unclear, recent studies have shown that this neuronal death is caused in part by apoptosis (MacManus et al, 1993; Nitatori et al, 1995). We have reported that cell survival pathways, including Akt signaling, are the focus for clarifying the apoptotic neuronal cell death machinery after cerebral ischemia (Noshita et al, 2001).
The serine—threonine kinase, Akt, which is also known as protein kinase B, plays an important role in the cell death/survival pathway (Franke et al, 1997a, Franke et al, 1997b). Growth factors such as insulin-like growth factor-1 activate Akt and promote cell survival through the phosphatidylinositol 3-kinase (PI3-K) pathway, which can be inhibited by wortmannin or LY294002 (Alessi et al, 1996; Datta et al, 1997). Phosphorylation of Ser473 residues is required for Akt activity (Alessi et al, 1996). After phosphorylation, Akt functions through phosphorylation and inhibition of Bad (Datta et al, 1997), glycogen synthase kinase-3β (GSK3β) (Cross et al, 1995), forkhead transcription factors (Brunet et al, 2001), or caspase-9 (Cardone et al, 1998). Glycogen synthase kinase-3 is involved in fundamental cellular processes that can determine developmental cell fate, tumorigenesis, and cell death (Frame and Cohen, 2001). There are two highly homologous isoforms of GSK3, GSK3α and GSK3β (Woodgett, 1990). Glycogen synthase kinase-3β is particularly abundant in the central nervous system and is neuron-specific (Leroy and Brion, 1999). Akt phosphorylates GSK3β on Ser9 to render it inactive, a proposed mechanism by which neurons become resistant to apoptotic stimuli (Pap and Cooper, 1998; Bijur et al, 2000; Hetman et al, 2000). Changes in phospho-Akt levels were reported after tGCI (Ouyang et al, 1999; Namura et al, 2000; Yano et al, 2001) and transient focal cerebral ischemia (tFCI) (Janelidze et al, 2001; Noshita et al, 2001). These reports showed a temporal increase in phospho-Akt at Ser473 and suggest that Akt phosphorylation is neuroprotective against ischemic injury. Glycogen synthase kinase-3β has also been reported to be involved in ischemic brain injury (Sasaki et al, 2001; Kelly et al, 2004). Glycogen synthase kinase-3β levels were increased in neurons after tFCI (Sasaki et al, 2001). A GSK3β inhibitor reduced infarct volume after tFCI (Kelly et al, 2004), suggesting that inhibition of GSK3β is neuroprotective against ischemic injury. However, the exact mechanism of cell survival mediated by the Akt/GSK3β signaling pathway after tGCI remains unclear.
Reactive oxygen species play important roles in the pathogenesis of central nervous system injury. We have reported that copper/zinc-superoxide dismutase (SOD1) is a crucial endogenous enzyme responsible for eliminating superoxide and that overexpression of SOD1 in transgenic (Tg) rats or mice reduces superoxide production and protects neurons from death after focal cerebral ischemia (Kinouchi et al, 1991) and global cerebral ischemia (Chan et al, 1998). The neuroprotective role of SOD1 against ischemia is partly mediated by Akt activation (Noshita et al, 2003). However, whether SOD1 affects the Akt/GSK3β signaling pathway after tGCI remains unclear. In the present study, we investigated the role of the Akt/GSK3β signaling pathway downstream of the PI3-K pathway after tGCI. We also examined the relationship between the Akt/GSK3β signaling pathway and SOD1 after tGCI.
Materials and methods
Superoxide Dismutase Copper/Zinc-Transgenic Rats
All animals were treated in accordance with Stanford University guidelines and the animal protocols approved by Stanford University's Administrative Panel on Laboratory Animal Care. Heterozygous SOD1 Tg rats with a Sprague—Dawley background, carrying human SOD1 genes, were derived from the founder stock and further bred with wild-type (Wt) Sprague—Dawley rats to generate heterozygous rats as previously described by our group (Chan et al, 1998). The phenotype of the SOD1 Tg rats was identified by isoelectric focusing gel electrophoresis as described (Chan et al, 1998). There were no observable phenotypic differences in brain vasculature between the Tg rats and Wt littermates (Chan et al, 1998).
Global Cerebral Ischemia
Transient global cerebral ischemia for 5 mins was induced by bilateral common carotid artery occlusion combined with hypotension, using the method described originally by Smith et al (1984), with some modifications (Sugawara et al, 1999). Male Sprague—Dawley rats (300 to 350 g) were anesthetized with 2.0% isoflurane in 70% nitrous oxide and 30% oxygen using a face mask. The rectal temperature was controlled at 37°C during surgery with a homeothermic blanket. The femoral artery was exposed and catheterized with a PE-50 catheter to allow continuous recording of arterial blood pressure. A midline neck skin incision was made and the right jugular vein and both common carotid arteries were exposed. After intravenous injection of 150IU/kg heparin, blood was quickly withdrawn via the jugular vein. When the mean arterial blood pressure became 30 mm Hg, both common carotid arteries were clamped with surgical clips. Blood pressure was maintained at 30 mm Hg by withdrawing or infusing blood through the jugular vein during the ischemic period. After 5 mins of ischemia, the clips were removed and the blood was reinfused. Sham-operated animals underwent exposure of vessels without blood withdrawal or clamping of carotid arteries. The animals were maintained in an air-conditioned room at 20°C with ad libitum access to food and water before and after surgery.
Drug Injection
To investigate the role of the PI3-K pathway after tGCI, we administered a PI3-K inhibitor, LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one) (Cell Signaling Technology, Beverly, MA, USA), as described previously (Noshita et al, 2001). LY294002 was dissolved in dimethyl sulfoxide and phosphate-buffered saline (PBS, pH 7.4) just before use. The scalp was incised on the midline and the skull was exposed. LY294002 (50 mmol/L in 25% dimethyl sulfoxide in PBS) and the vehicle (25% dimethyl sulfoxide in PBS) were injected intracerebroventricularly (10 μL, bregma; 1.4 mm lateral, 0.8 mm posterior, 3.6 mm deep) 1 h before tGCI.
Western Blot Analysis
Protein extraction of the cytosolic fraction was performed using a multiple centrifugation method as described previously, with some modification (Fujimura et al, 1999). Fresh brain tissue was removed after 1, 4, and 8 h and 1 and 3 days (n = 4 each) of reperfusion. The brain tissue was cut into 1-mm coronal slices using a brain matrix (Zivic Laboratories Inc., Pittsburgh, PA, USA) and the bilateral hippocampus was removed. The hippocampal CA1 subregion was quickly dissected under a microscope and used as a sample. For protein extraction, the tissue was homogenized by gently douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ, USA) in 7 volumes of cold suspension buffer (20 mmol/L Hepes-KOH, pH 7.5, 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L ethylenediamine tetraacetate (EDTA), and 1 mmol/L ethyleneglycol tetraacetate plus 0.7% protease and phosphatase inhibitor cocktails (Sigma, St Louis, MO, USA)). The homogenate was centrifuged at 750g for 10 mins at 4°C and then at 10,000g for 20 mins at 4°C. The 10,000g pellets were used to obtain the mitochondrial fraction. The supernatant was further centrifuged at 100,000g for 60 mins at 4°C and was then used for the cytosolic analysis. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, USA) to the supernatant, we loaded equal amounts of the samples per lane. The primary antibodies were 1:1000 dilution of rabbit polyclonal antibody against GSK3β and phospho-GSK3β (Ser9) (Cell Signaling Technology), 1:1000 dilution of mouse monoclonal antibody against phospho-GSK3β (Tyr216) (Upstate Cell Signaling Solutions, Lake Placid, NY, USA), 1:2000 dilution of rabbit polyclonal antibody against Akt and phospho-Akt (Ser473) (Cell Signaling Technology), and 1:10,000 dilution of mouse monoclonal antibody against β-actin (Sigma). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG (Cell Signaling Technology) using enhanced chemiluminescence Western blotting detection reagents (Amersham International, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA, USA) and the results were quantified using Multi-Analyst software (Bio-Rad).
Immunohistochemistry
Anesthetized animals were perfused with 10 U/mL heparin saline and subsequently with 4% formaldehyde in PBS 8 h and 1 and 3 days after tGCI. The brains were removed, postfixed for 24 h, sectioned at 50 μm with a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with a rabbit polyclonal antibody against phospho-GSK3β (Ser9) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and a mouse monoclonal antibody against phospho-Akt (Ser473) (Cell Signaling Technology) at dilutions of 1:50. Immunohistochemistry was performed via the avidin—biotin technique, and then the nuclei were counterstained with methyl green solution for 10 min.
Immunofluorescent Double-Labeling Staining
To evaluate colocalization of phospho-GSK3β (Ser9) and neuron-specific nuclear protein (NeuN) or phospho-Akt (Ser473), we performed double immunofluorescent staining. The sections fixed by 4% paraformaldehyde were incubated with blocking solution and reacted with the rabbit polyclonal antibody against phospho-GSK3β (Ser9) (Santa Cruz Biotechnology) as described above, followed by a Texas Red-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch, West Grove, PA, USA) at the dilution of 1:200. The sections were then incubated with a mouse anti-NeuN Alexa Fluor-conjugated monoclonal antibody (Chemicon International, Temecula, CA, USA) at a dilution of 1:200 for double staining of phospho-GSK3β (Ser9) and NeuN. The sections were incubated with a mouse monoclonal antibody against phospho-Akt (Ser473) (Cell Signaling Technology) at the dilution of 1:50, followed by a fluorescein isothiocyanate-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories) at a dilution of 1:200 for double staining of phospho-GSK3β (Ser9) and phospho-Akt (Ser473). The sections were placed on slides, which were then covered with VECTASHIELD mounting medium with 4ʼ,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). Fluorescence of fluorescein and Alexa Fluor were observed at an excitation of 495 nm and an emission of > 515nm, and fluorescence of Texas Red was observed at an excitation of 510 nm and an emission of > 580 nm. Fluorescence of DAPI was observed at an excitation of 360 nm and an emission of > 460nm.
Immunofluorescent Double Labeling with Phospho-Glycogen Synthase Kinase-3β (Ser9) Immunohistochemistry and an In Situ Staining Technique
To clarify the spatial relationship between phospho-GSK3β (Ser9) expression and DNA fragmentation, we performed double staining for phospho-GSK3β (Ser9) and terminal deoxynucleotidyl transferase-mediated uridine 5ʼ-triphosphate-biotin nick end labeling (TUNEL), using a fluorescent method. The experimental animals were killed 3 days after tGCI. The sections fixed by 4% paraformaldehyde were incubated with a blocking solution and reacted with the anti-phospho-GSK3β (Ser9) antibody as described above, followed by the Texas Red-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch). Then the sections were incubated with NeuroPore (Trevigen, Gaithersburg, MD, USA) for 30 mins. They were placed in 1 × terminal deoxynucleotidyl transferase buffer (Invitrogen) for 30 mins, followed by reaction with terminal deoxynucleotidyl transferase enzyme (Invitrogen) and biotinylated 16-deoxyuridine triphosphate (Roche Diagnostics, Indianapolis, IN, USA) at 37°C for 90 mins. The sections were washed in 2 × saline-sodium citrate (150 mol/L sodium chloride, 15 mol/L sodium citrate, pH 7.4) for 15 mins, followed by washing in PBS two times for 15 mins each. Fluorescein avidin DCS (Vector Laboratories) was applied to the sections for 30 mins. Subsequently, the slides were covered with VECTASHIELD mounting medium with DAPI. Fluorescence was assessed as described above.
Cell Death Assay
For quantification of apoptotis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals, Mannheim, Germany), which detect apoptotic but not necrotic cell death (Leist et al, 1998). Samples were obtained from the hippocampal CA1 subregion as described above in the Western blot analysis section (n = 4 each). For protein extraction, the samples were homogenized with a Teflon homogenizer in 7 volumes of ice-cold buffer (50 mmol/L KH2PO4, 0.1 mmol/L EDTA, pH 7.8). The homogenate was centrifuged at 750g for 10 mins at 4°C and then at 10,000g for 20 mins at 4°C. The supernatant was further centrifuged at 100,000g for 60 mins at 4°C. The resulting supernatant was collected and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, following the manufacturer's protocol.
Quantification and Statistical Analysis
The data obtained from each time point were compared with those from the sham-operated group using one-way analysis of variance, followed by a Scheffé post-hoc analysis (StatView; SAS Institute Inc., Cary, NC, USA). Comparisons between the two groups were achieved with Student's unpaired t-test. The data are expressed as mean ± s.d. and significance was accepted with P < 0.05.
Results
Delayed Hippocampal CA1 Neuronal Death Occurred 72 h After Transient Global Cerebral Ischemia
At 1, 8, and 24 h after 5 mins of tGCI, there was no neuronal degeneration in the hippocampal CA1 subregion in the Wt rats, confirmed by cresyl violet staining (data not shown). Three days after tGCI, however, more than 80% of the CA1 neurons were degenerated in the Wt rats, as described previously (Chan et al, 1998; Sugawara et al, 1999).
Phosphorylation of Akt (Ser473) After Transient Global Cerebral Ischemia
To investigate the phosphorylation of Akt after tGCI in the Wt rats, we performed Western blot analysis and immunohistochemistry using a specific antibody against phospho-Akt (Ser473), and Akt. Bands of phospho-Akt (Ser473) and Akt were observed at 60 kDa in the samples from the hippocampal CA1 subregion in the Western blot analysis (Figure 1A). Immunoreactivity of phospho-Akt was significantly increased 8 h after reperfusion (Figure 1B, P < 0.05), whereas it was decreased by day 3 after reperfusion. Immunoreactivity of Akt showed no significant change after reperfusion. An immunohistochemistry study showed slight immunoreactivity of phospho-Akt in both the hippocampal CA1 subregion and the CA3 subregion of the nonischemic brains (Figure 1C). Immunostaining of phospho-Akt was increased 8 h after tGCI in the hippocampal CA1 subregion and then returned to basal expression 1 day after tGCI. Three days after tGCI, the CA1 neurons degenerated and immunoreactivity could not be evaluated. In contrast, immunoreactivity of phospho-Akt in the hippocampal CA3 subregion showed no difference among all time points after reperfusion. These results suggest that phosphorylation of Akt was accelerated in the vulnerable hippocampal CA1 subregion after tGCI in the Wt rats.
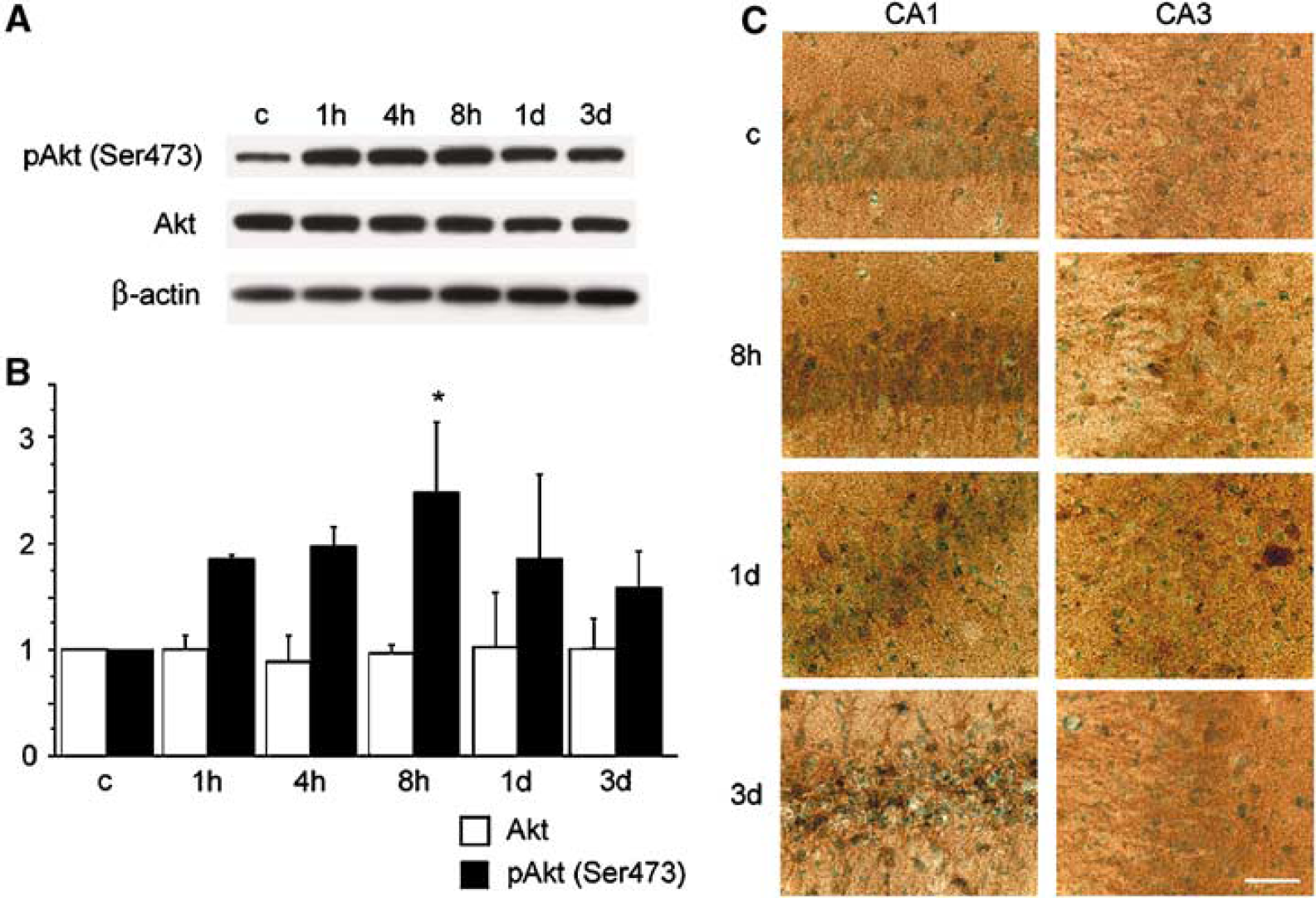
Changes in phosphorylation of Akt (pAkt) in the hippocampal CA1 subregion after tGCI in Wt rats. (
Phosphorylation of Glycogen Synthase Kinase-3β (Ser9) After Transient Global Cerebral Ischemia
Since phospho-Akt (Ser473) functions through phosphorylation and inhibition of GSK3β (Cross et al, 1995), we further examined the phosphorylation of GSK3β after tGCI in the Wt rats by Western blot analysis and immunohistochemistry. Bands of phospho-GSK3β (Ser9 and Tyr216) and GSK3β were observed at 46 kDa in the samples from the hippocampal CA1 subregion in the Western blot analysis (Figure 2A). Immunoreactivity of phospho-GSK3β (Ser9) was significantly increased 8 h and 1 day after tGCI (Figure 2B, P 0.05), whereas immunoreactivity of GSK3β and phospho-GSK3β (Tyr216) showed no significant change after tGCI. The immunohistochemistry study showed slight immunoreactivity of phospho-GSK3β (Ser9) in both the hippocampal CA1 subregion and the CA3 subregion of the nonischemic brains (Figure 2C). Immunostaining of phospho-GSK3β (Ser9) was increased 8 h after tGCI in the hippocampal CA1 subregion. Immunoreactivity of phospho-GSK3β (Ser9) 1 day after tGCI still remained stronger than the basal expression. Three days after tGCI, the CA1 neurons degenerated and immunoreactivity could not be evaluated. In contrast, immunoreactivity of phospho-GSK3β (Ser9) in the hippocampal CA3 subregion showed no difference among all time points after tGCI. These results suggest that phosphorylation of GSK3β was accelerated in the vulnerable hippocampal CA1 subregion after tGCI in the Wt rats.
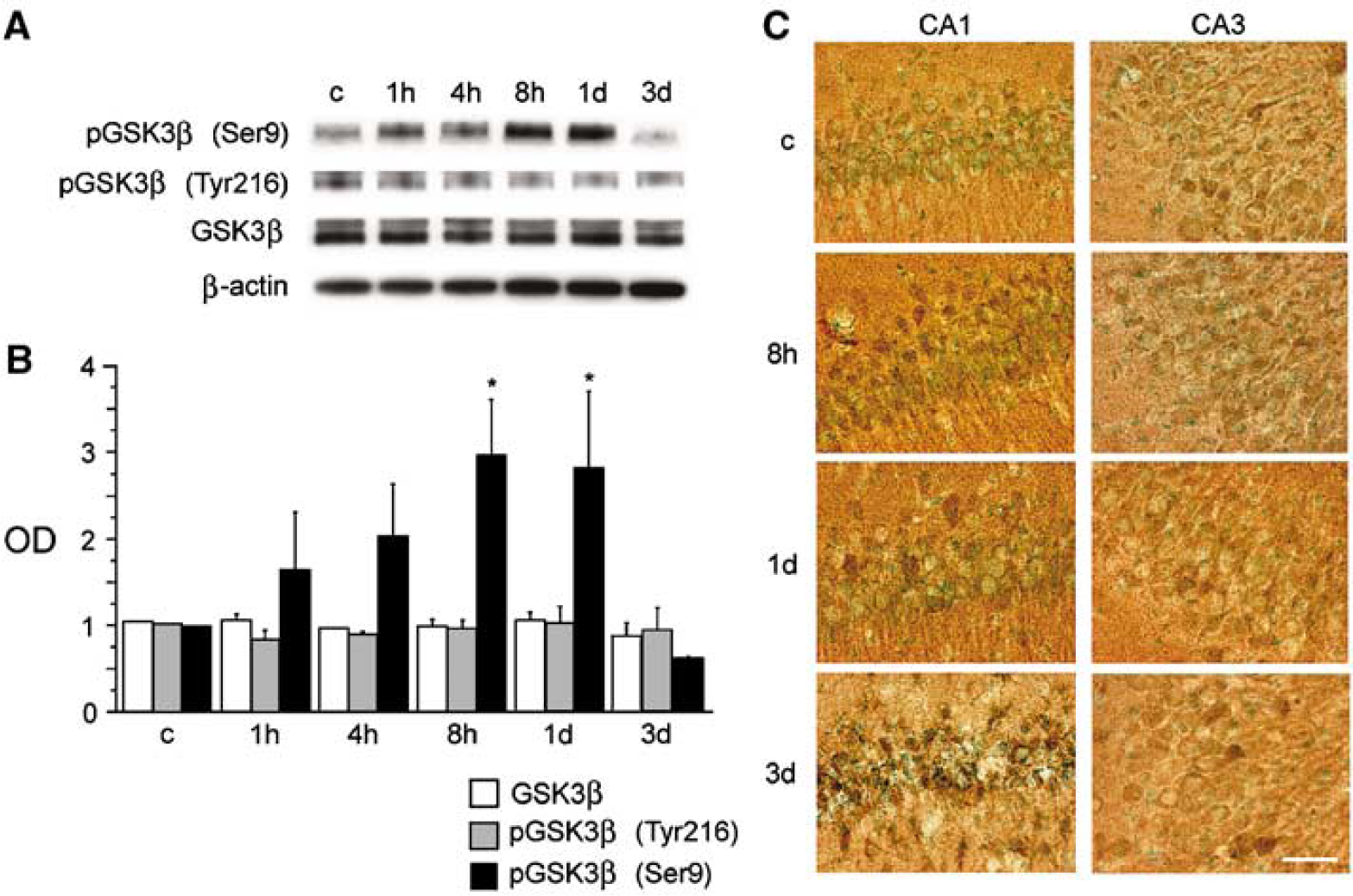
Changes in phosphorylation of GSK3β (pGSK3β) in the hippocampal CA1 subregion after tGCI in Wt rats. (
Neuronal Expression of Phospho-Akt (Ser473) and Phospho-Glycogen Synthase Kinase-3β (Ser9) After Transient Global Cerebral Ischemia
To investigate whether phosphorylation of Akt (Ser473) and GSK3β (Ser9) occurs in hippocampal CA1 neurons after tGCI in the Wt rats, we performed a double immunofluorescence study. Double immunofluorescence for phospho-GSK3β (Ser9) (Figure 3A) and NeuN (Figure 3B) showed that phospho-GSK3β-positive cells in the hippocampal CA1 subregion colocalized mainly with neurons 8 h after tGCI (Figure 3C). Double immunofluorescence for phospho-GSK3β (Ser9) (Figure 3D) and phospho-Akt (Ser473) (Figure 3E) showed that phospho-GSK3β-positive cells in the hippocampal CA1 subregion colocalized with phospho-Akt-positive cells 8 h after tGCI. These results suggest that phosphorylation of both Akt and GSK3b occurred mainly in the hippocampal CA1 neurons after tGCI in the Wt rats.
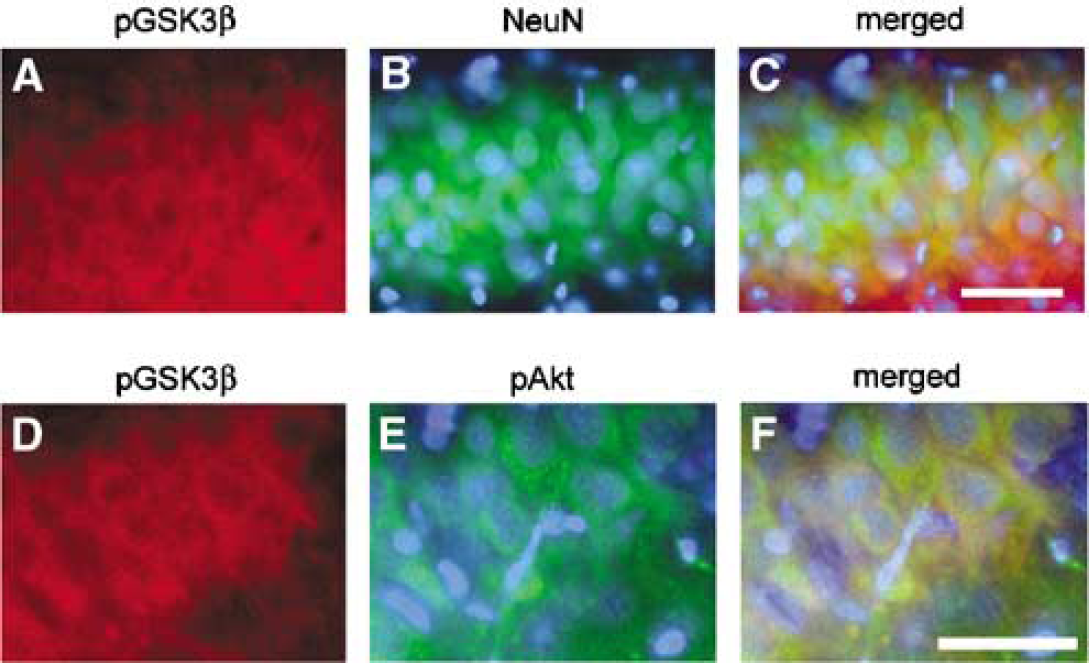
Representative photomicrographs show fluorescent double staining of phospho-GSK3β (pGSK3β) (Ser9) (red) and NeuN (green), and phospho-GSK3β (Ser9) (red) and phospho-Akt (pAkt) (Ser473) (green) in the hippocampal CA1 subregion 8 h after tGCI in the Wt rats. Nuclei were counterstained with DAPI (blue). Phospho-GSK3β-positive cells were observed in the hippocampal CA1 subregion (
Double Immunofluorescence for Phospho-Glycogen Synthase Kinase-β (Ser9) and Terminal Deoxynucleotidyl Transferase-Mediated Uridine 5ʼ-Triphosphate-Biotin Nick End Labeling After Transient Global Cerebral Ischemia
To investigate the cellular population of phospho-GSK3b (Ser9) and DNA fragmentation in the hippocampus of the Wt rats after tGCI, we performed double immunofluorescence for phospho-GSK3b (Ser9) and TUNEL. Three days after tGCI, TUNEL-positive cells were seen in the hippocampal CA1 subregion (Figure 4A), though no TUNEL-positive cells were observed in the CA3 subregion at the same time point (Figure 4D). Some phospho-GSK3β (Ser9)-positive cells seen in the CA1 subregion (Figure 4B) did not colocalize with TUNEL-positive cells (Figure 4C). In contrast, a significant number of phospho-GSK3b (Ser9)-positive cells were seen in the CA3 subregion (Figure 4E), but were not positive for TUNEL (Figure 4F). These results suggest that the cellular population of phospho-GSK3β (Ser9) is different from that of DNA fragmentation after tGCI in the Wt rats.
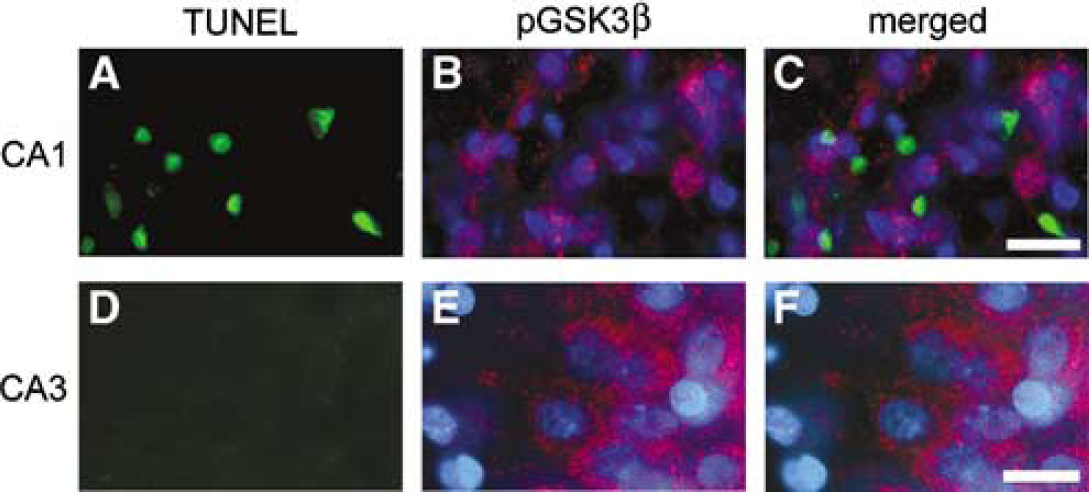
Representative photomicrographs show immunofluorescent staining for phospho-GSK3ʲ (pGSK3ʲ) (Ser9) and TUNEL after tGCI in the Wt rats. Three days after tGCI, a significant number of TUNEL-positive cells was observed in the hippocampal CA1 subregion (
Phosphatidylinositol 3-Kinase Inhibitor Blocked Phosphorylation of Akt and Glycogen Synthase Kinase-β and Accelerated DNA Fragmentation After Transient Global Cerebral Ischemia
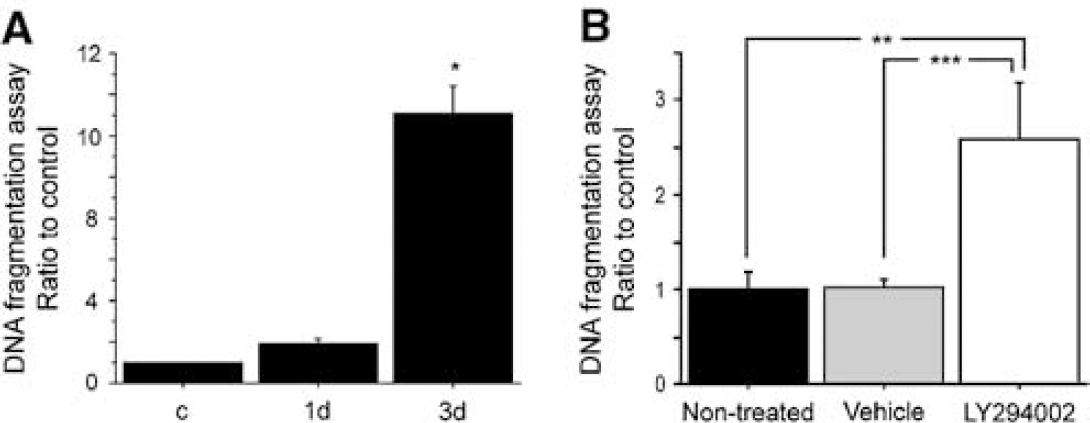
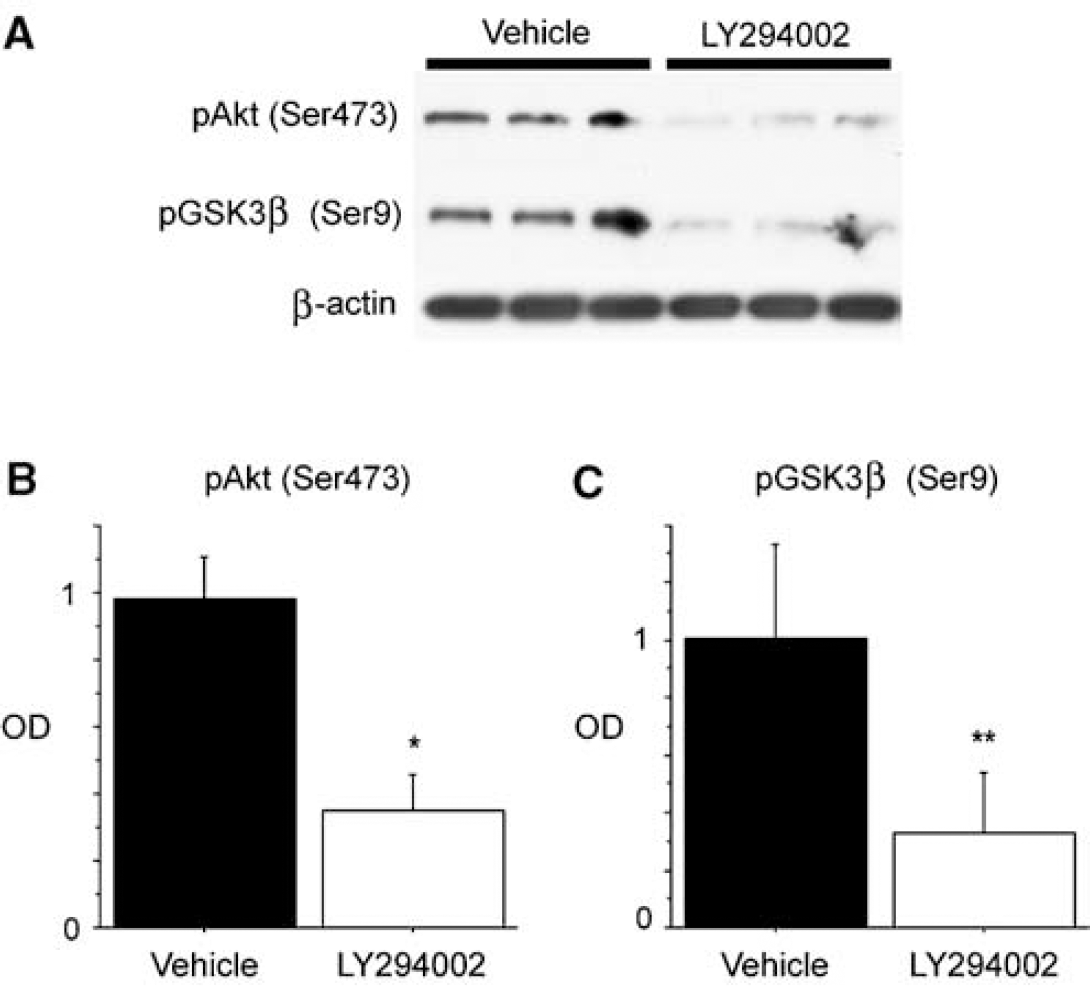
Phosphorylation of Akt (Ser473) is downstream of the PI3-K pathway (Alessi et al, 1996) and functions through phosphorylation of GSK3β (Ser9) (Cross et al, 1995). To investigate the involvement of the Akt/GSK3b signaling pathway as a downstream effector of the PI3-K pathway in the Wt rats after tGCI, we performed a study with LY294002, a specific inhibitor of PI3-K. First, we investigated the effect of LY294002 on apoptotic neuronal death after tGCI in the Wt rats. Apoptotic-related DNA fragmentation after ischemia was analyzed with a commercial cell death detection kit. In the nontreated Wt animals, DNA fragmentation was significantly increased in the hippocampal CA1 subregion 3 days after tGCI compared with the nonischemic brains (Figure 5A) (n = 4, * P < 0.001). This result was consistent with our TUNEL-positive cell-counting study (Sugawara et al, 1999). Then, we examined DNA fragmentation of the non-treated animals, vehicle-treated animals and LY294002-treated animals 3 days after tGCI to investigate the role of the PI3-K pathway in DNA damage after tGCI. DNA fragmentation 3 days after tGCI was significantly increased in the hippocampal CA1 subregion of the LY294002-treated animals compared with both the nontreated and vehicle-treated animals at the same time point (Figure 5B) (n = 4, ** P = 0.0018, *** P = 0.0020). We then examined expression of phospho-Akt and phospho-GSK3/ after administration of LY294002. Eight hours after tGCI, both phospho-Akt and phospho-GSK3/ were decreased in the LY294002-treated animals compared with the vehicle-treated animals (Figure 6A). Statistical analysis showed a significant difference between the two groups in the hippocampal CA1 subregion (Figures 6B and 6C) (n = 4, * P = 0.0025, **P = 0.0392). These results suggest that the PI3-K signaling pathway is involved in delayed CA1 neuronal death after tGCI and that Akt/GSKβ signaling functions downstream of the PI3-K signaling pathway.
(
Phospho-Akt (Ser473) and Phospho-Glycogen Synthase Kinase-3β (Ser9) Expression Persistently Increased in the Copper/Zinc-Superoxide Dismutase Transgenic Rats After Transient Global Cerebral Ischemia
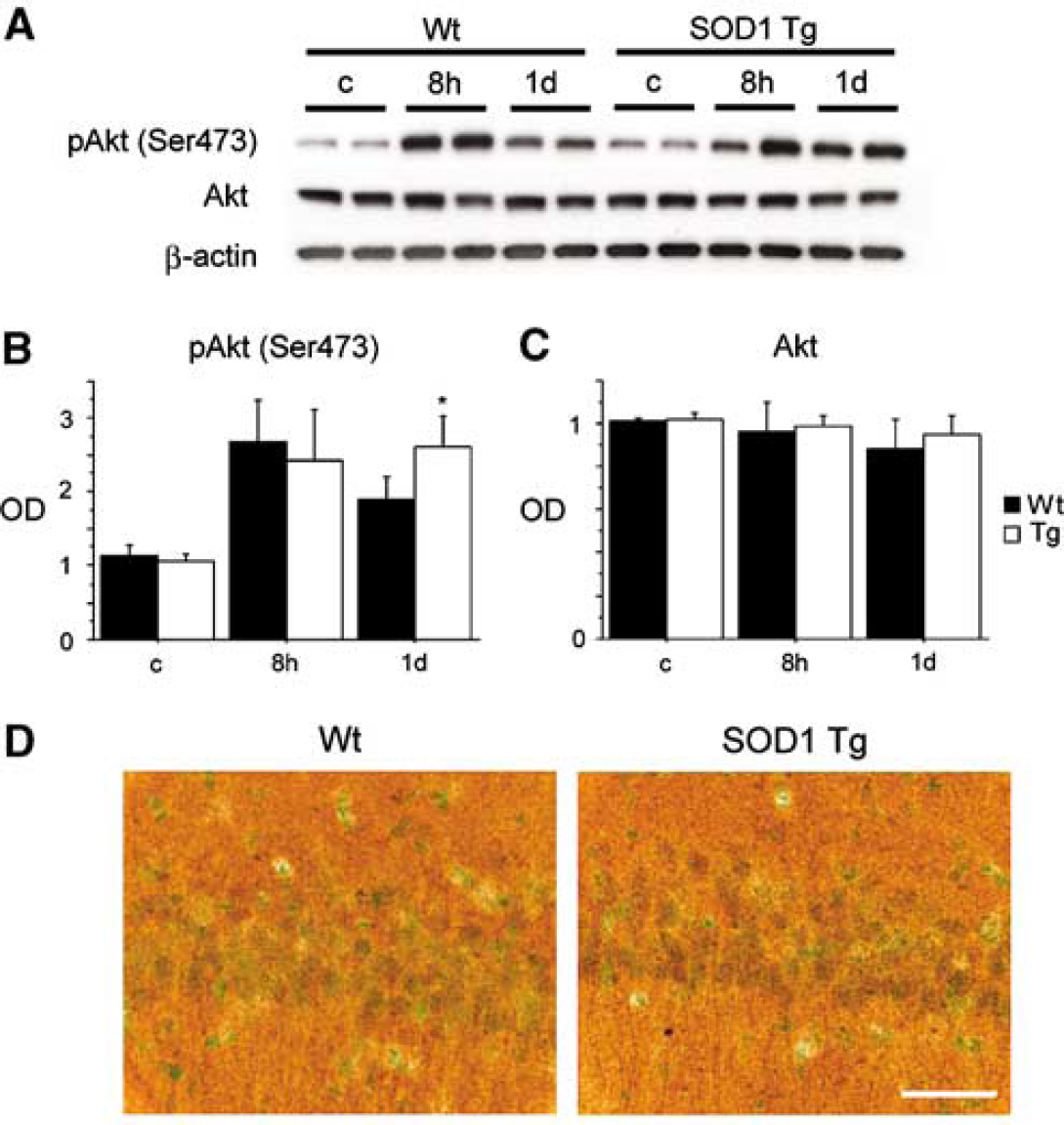
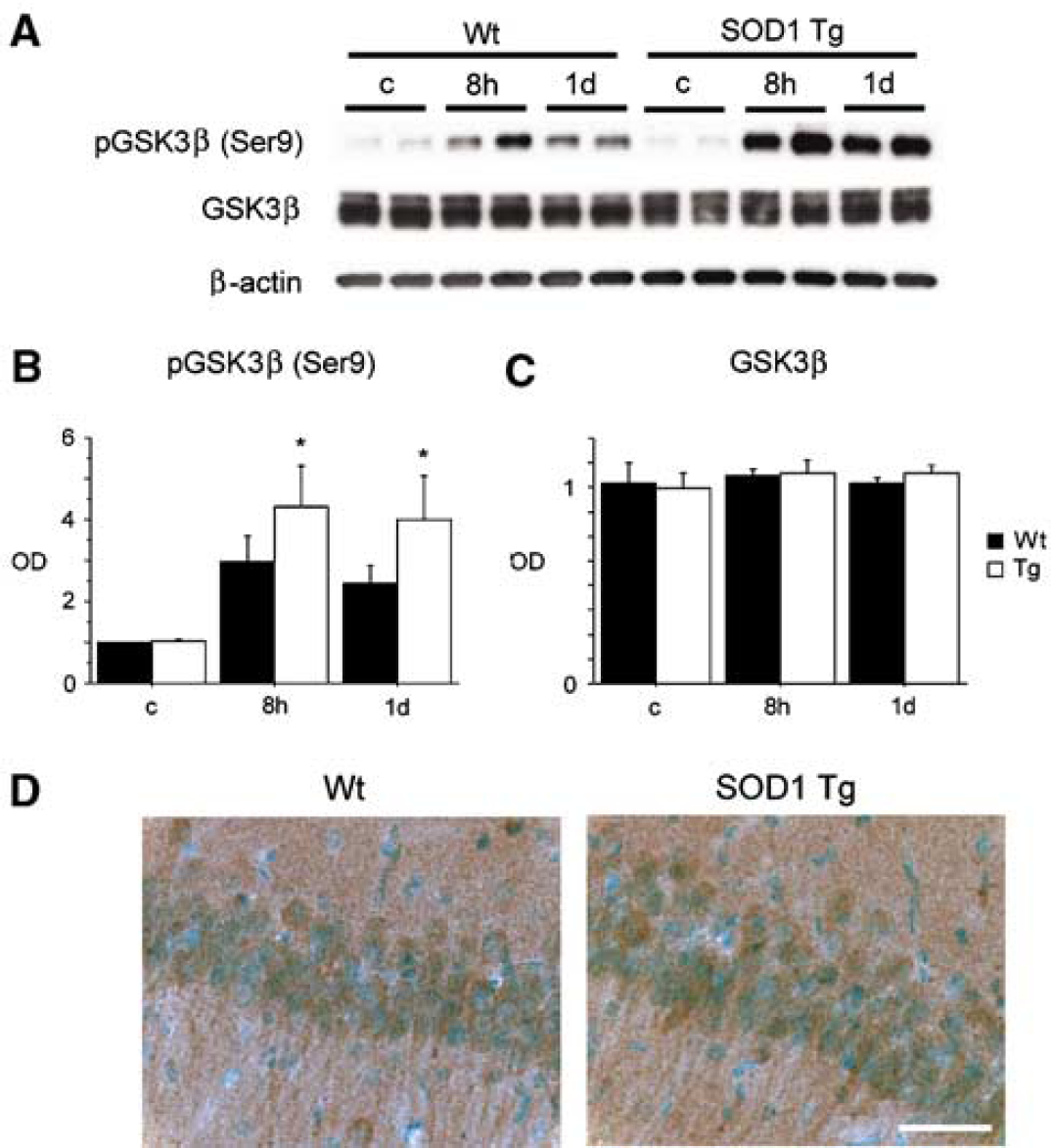
We have reported on the neuroprotection of SOD1 overexpression against delayed hippocampal CA1 neuronal death after tGCI in rats (Chan et al, 1998). In the present study, we used SOD1 Tg rats to investigate whether this neuroprotection is mediated by Akt/GSK3β signaling. Western blot analysis showed that phospho-Akt (Ser473) was significantly increased 1 day after tGCI in the SOD1 Tg rats compared with the Wt rats (Figures 7A and 7B) (P < 0.05). There was no prominent modification of Akt after tGCI (Figures 7A and 7C). Furthermore, the immunostaining study showed that phospho-Akt (Ser473) expression was enhanced in the SOD1 Tg rats 1 day after tGCI in the hippocampal CA1 subregion (Figure 7D). Next, we examined the expression of phospho-GSK3β (Ser9) after tGCI. Western blot analysis showed that phospho-GSK3β (Ser9) was significantly increased 8 h and 1 day after tGCI in the SOD1 Tg rats compared with the Wt rats (Figures 8A and 8B) (P< 0.05). There was no prominent modification of GSK3β after tGCI (Figures 8A and 8C). Moreover, the immunostaining study showed that phospho-GSK3β (Ser9) expression was enhanced in the SOD1 Tg rats 1 day after tGCI in the hippocampal CA1 subregion (Figure 8D). These results suggest that the neuroprotection of SOD1 against tGCI is partly mediated by enhanced and persistent activation of the Akt/GSK3β signaling pathway.
(
Discussion
Accumulating evidence suggests that the PI3-K/Akt signaling pathway plays a crucial role in neuronal survival after cerebral ischemia. We have reported that activation of Akt signaling has neuroprotective effects against ischemic neuronal injury (Noshita et al, 2001, 2003). Several potential substrates for Akt that are related to cell survival after cerebral ischemia include Bad (Saito et al, 2003), prolinerich Akt substrate (Saito et al, 2004), Forkhead transcription factors (Kawano et al, 2002), and endothelial nitric oxide synthase (Hashiguchi et al, 2004). The current study shows that GSK3β is also one of the targets of Akt in the PI3-K signaling pathway after cerebral ischemia. Phosphorylation of Akt (Ser473) and GSK3β (Ser9) was temporally increased in the vulnerable hippocampal CA1 subregion, but not in the CA3 subregion. Administration of the PI3-K inhibitor decreased phosphorylation of these proteins and facilitated delayed death of the hippocampal CA1 neurons after tGCI. Some phospho-GSK3β (Ser9)-positive neurons survived in the vulnerable CA1 subregion as well as in the CA3 subregion 3 days after tGCI. Moreover, SOD1 Tg rats, which are known to have neuroprotection against vulnerable CA1 damage after tGCI (Chan et al, 1998), had enhanced and persistent phosphorylation of both Akt and GSK3β after tGCI. These results suggest that activation of Akt/GSK3β signaling, which means phosphorylation at Ser473β activation of Akt and phosphorylation at Ser9/ inactivation of GSK3β, may mediate survival of vulnerable hippocampal CA1 neurons after tGCI.
neuroprotective (
(
Changes in the phospho-Akt levels were reported after tGCI (Ouyang et al, 1999; Namura et al, 2000; Yano et al, 2001) and tFCI (Janelidze et al, 2001; Noshita et al, 2001; Zhao et al, 2005). These reports showed a temporal increase in phospho-Akt at Ser473. Phosphorylation of Akt at Ser473 was increased in the hippocampal CA1 subregion after tGCI (Namura et al, 2000; Yano et al, 2001), which was consistent with our study. Sublethal global ischemia induced persistent Akt phosphorylation in the hippocampal CA1 subregion, and Yano et al (2001) concluded that this persistent Akt activation resulted in neuronal survival against lethal global ischemia. In the tFCI model, the Akt phosphorylation observed in the ischemic penumbra after ischemia was prevented by LY294002, a PI3-K inhibitor, which facilitated ischemic injury (Noshita et al, 2001). This Akt phosphorylation was enhanced and persistent after tFCI in SOD1 Tg mice, which are known to have neuroprotection against ischemia (Noshita et al, 2003). Moreover, hypothermic neuroprotection against ischemia was in part mediated by Akt activation (Zhao et al, 2005). These reports strongly support our results showing that enhanced and persistent Akt activation induces neuroprotection against ischemic neuronal injury.
Glycogen synthase kinase-3β is also involved in the mechanisms of ischemic neuronal injury. Changes in GSK3β expression were reported after cerebral ischemia. Strong immunoreactivity for GSK3β was observed in the cerebral cortex and dorsal caudate after tFCI, though the authors of these reports did not mention the phosphorylation of GSK3β (Wang et al, 2000; Sasaki et al, 2001). Phosphorylation regulates GSK3b activity in apoptotic cell death. Phosphorylation of GSK3β at Tyr216 is necessary for its activation, whereas phosphorylation at Ser9 induces its inactivation in the apoptotic neuronal death machinery (Bhat et al, 2000). These authors also reported that phosphorylation of GSK3β at Tyr216 was increased in cortical neurons after tFCI, though phosphorylation of GSK3β at Ser9 was not altered. In our study, Western blot analysis showed that phosphorylation of GSK3β at Ser9 was increased in the vulnerable hippocampal CA1 subregion after tGCI, though there was no change in expression in either GSK3β or phospho-GSK3β (Tyr216). A possible explanation for this discrepancy is the difference in ischemic severity and ischemic models. Under severe ischemic conditions, such as in models of 90 mins of tFCI, activation of GSK3β which facilitates apoptotic cell death, might be emphasized. In contrast, inactivation of GSK3β, which enhances cell survival, might be emphasized in mild ischemia, such as in our model of 5 mins of tGCI. From the point of view that activation of GSK3β has a detrimental effect on ischemic neuronal injury, a GSK3β inhibitor may ameliorate neuronal death after ischemia. In fact, several GSK3β inhibitors have been reported to be effective in the treatment of cerebral ischemia. Selective, but not specific, GSK3β inhibition by lithium has been shown to decrease infarct volume after tFCI (Ren et al, 2003). Insulin-like growth factor-1, also a nonspecific inhibitor of GSK3β, significantly reduced infarct volume after tFCI and reduced the number of GSK3β-immunoreactive neurons (Wang et al, 2000). Moreover, Chir025, a selective and specific inhibitor of GSK3β, reduced infarct size by nearly 20% after tFCI (Kelly et al, 2004). Two selective GSK3β inhibitors, SB216763 and SB415286, are adenosine triphosphate-dependent and have selectivity and preference for GSK3β inhibition without inhibition of Akt (Coghlan et al, 2000). These inhibitors have been shown to be cardioprotective after cardiac ischemia (Gross et al, 2004), though there is no report regarding cerebral ischemia. The neuroprotective effect of these drugs against cerebral ischemia needs to be investigated in a future study.
In the molecular pathway of ischemic neuronal death, mechanisms downstream of the Akt/GSK3β signaling pathway are largely unknown, but we may speculate on a few possibilities. Glycogen synthase kinase-3β-mediated phosphorylation of β-catenin has been reported to enhance cell death in hippocampal neurons (Zhang et al, 1998). However, it has also been reported that phosphorylation of GSK3β (Ser9) is not critical for the regulation of β-catenin phosphorylation after in vivo cerebral ischemia (Cappuccio et al, 2005; Zhao et al, 2005). Another possibility is the effect of GSK3β on tau phosphorylation. GSK3β phosphorylates the tau protein and makes it insoluble, resulting in neuronal cell death (Takashima et al, 1998). The tau protein has been shown to be involved in ischemic neuronal injury in humans (Uchihara et al, 1995). However, spatial distribution of GSK3β and tau expression were different after tFCI (Sasaki et al, 2001). Further investigation of effectors downstream of GSK3β could help to clarify the mechanisms of ischemic neuronal injury.
Our previous studies showed that overexpression of intrinsic SOD1 has protective effects against ischemic damage (Kinouchi et al, 1991; Chan et al, 1998). In this study, we focused on the role of the Akt/GSK3β signaling pathway in the neuroprotective effect of SOD1. Our results show that overexpression of SOD1 activated Akt/GSK3β signaling after tGCI and it shifted the balance between survival and death to cell survival. Overexpression of SOD1 resulted in increased phosphorylation of Akt at Ser473 after tFCI (Noshita et al, 2003). Thus, in this study, SOD1 overexpression might have affected phosphorylation of GSK3β via phosphorylation of Akt. Another possibility is that SOD1 overexpression directly affects phosphorylation of GSK3β. Though the exact mechanisms of how oxidative stress is involved with Akt/GSK3β signaling after tGCI remain unclear, it is obvious that neuroprotection against tGCI by SOD1 overexpression is partly mediated by enhanced and persistent activation of Akt/GSK3β signaling. Further study is required to clarify the link between Akt/GSK3β signaling and oxidative stress after tGCI.
Conclusion
Our results show that phosphorylation of Akt (Ser473) and GSK3β (Ser9) was accelerated in the vulnerable hippocampal CA1 subregion, but not in the resistant CA3 subregion, and that these proteins functioned downstream of the PI3-K signaling pathway after tGCI. Moreover, neuroprotection of SOD1 against ischemia was partly mediated by enhanced and persistent activation of the Akt/GSK3β signaling pathway. These findings suggest that activation of the Akt/GSK3β signaling pathway may mediate survival of vulnerable hippocampal CA1 neurons after tGCI.
Footnotes
Acknowledgements
We thank Liza Reola, Bernard Calagui, and Trisha Crandall for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.