
Abstract
In rat brain, dopaminergic D2-like but not D1-like receptors can be coupled to phospholipase A2 (PLA2) activation, to release the second messenger, arachidonic acid (AA, 20:4n-6), from membrane phospholipids. In this study, we hypothesized that D-amphetamine, a dopamine-releasing agent, could initiate such AA signaling. The incorporation coefficient, k∗ (brain radioactivity/integrated plasma radioactivity) for AA, a marker of the signal, was determined in 62 brain regions of unanesthetized rats that were administered i.p. saline, D-amphetamine (2.5 or 0.5 mg/kg i.p.), or the D2-like receptor antagonist raclopride (6 mg/kg, i.v.) before saline or 2.5 mg/kg D-amphetamine. After injecting [1-14C]AA intravenously, k∗ was measured by quantitative autoradiography. Compared to saline-treated controls, D-amphetamine 2.5 mg/kg i.p. increased k∗ significantly in 27 brain areas rich in D2-like receptors. Significant increases were evident in neocortical, extrapyramidal, and limbic regions. Pretreatment with raclopride blocked the increments, but raclopride alone did not alter baseline values of k∗. In independent experiments, D-amphetamine 0.5 mg/kg i.p. increased k∗ significantly in only seven regions, including the nucleus accumbens and layer IV neocortical regions. These results indicate that D-amphetamine can indirectly activate brain PLA2 in the unanesthetized rat, and that activation is initiated entirely at D2-like receptors. D-Amphetamine's low-dose effects are consistent with other evidence that the nucleus accumbens, considered a reward center, is particularly sensitive to the drug.
Introduction
D-Amphetamine has been reported to ameliorate mood in patients suffering from depression (Maes et al, 1990), to enhance cognition in schizophrenics (Daniel et al, 1991), to ameliorate attention problems and impulsivity in attention deficit hyperactivity disorder (Lopez et al, 2004) and, when taken chronically, to lead to addiction (Di Chiara et al, 2004; White and Kalivas, 1998). In animals, D-amphetamine produces hyperactivity, stereotypy, and extrapyramidal motor symptoms. Such behavioral effects have been ascribed to its ability to increase synaptic dopamine (DA) and indirectly activate brain DA receptors (Butcher et al, 1988; Houston et al, 2004; Kuczenski and Segal, 1989; Laruelle et al, 1997; Sharp et al, 1987). D-Amphetamine does this by causing vesicular DA to enter the cytoplasm of presynaptic DA terminals, and by changing the direction of DA transport by the presynaptic DA reuptake transporter (DAT) (Creese and Iversen, 1975; Fuller and Hemrick-Luecke, 1980; Jones et al, 1998; Segal et al, 1980). D-Amphetamine also can activate adrenergic and serotonergic receptors by increasing extracellular synaptic concentrations of norepinephrine and serotonin, respectively (Berridge and Stalnaker, 2002; Kuczenski and Segal, 1989; Pascoli et al, 2005; Vanderschuren et al, 2003).
Dopaminergic, serotonergic and noradrenergic receptors can be coupled by G-proteins to different effector enzymes, including phospholipase A2 (PLA2). Phospholipase A2 activation releases arachidonic acid (AA, 20:4n-6) from membrane phospholipids (Axelrod, 1990; Garcia and Kim, 1997; Nilsson et al, 1998; Pavoine et al, 2003; Vial and Piomelli, 1995). Arachidonic acid and its eicosanoid products are important second messengers that can influence membrane excitability, regional cerebral blood flow (rCBF), gene transcription, mood, and sleep (Chen and Bazan, 2005; Fitzpatrick and Soberman, 2001; Niwa et al, 2000; Rapoport and Bosetti, 2002; Shimizu and Wolfe, 1990; Wang et al, 2005).
We have developed a method to image this process in vivo. A fraction of the AA released by PLA2 is lost by metabolism to eicosanoids such as prostaglandin E2 (PGE2), β-oxidation or other products, whereas the remainder is recycled into phospholipids (DeGeorge et al, 1991; Jones et al, 1996; Rapoport, 2003; Robinson et al, 1992; Sun and MacQuarrie, 1989). Arachidonic acid cannot be synthesized de novo in mammalian brain (Youdim et al, 2000), nor elongated significantly from its linoleic acid (18:2n-6) precursor (DeMar et al, 2004), but the AA lost is almost instantaneously replaced by unesterified AA from plasma (Washizaki et al, 1994). Replacement, proportional to the quantity of AA released by PLA2 activation, can be imaged following the intravenous injection of radiolabeled AA, as an increased incorporation coefficient k* for AA (regional brain radioactivity divided by integrated plasma radioactivity). Importantly, k*is independent of changes in rCBF, thus reflecting only brain metabolic events (Chang et al, 1997; DeGeorge et al, 1991; Robinson et al, 1992; Robinson and Rapoport, 1986).
Dopaminergic D2-like receptors can be coupled specifically to Ca2+-dependent AA-selective cytosolic PLA2 (cPLA2) (Clark et al, 1991; Dennis, 1994; Nilsson et al, 1998; Vial and Piomelli, 1995), which is localized on excitatory post-synaptic neuronal membranes and dendrites (Ong et al, 1999; Pardue et al, 2003). D1-like receptors have not been reported to be coupled to PLA2 in normal brain (Bhattacharjee et al, 2005b), but rather to phospholipase C or adenylate cyclase (Cooper et al, 2003). Thus, administration to unanesthetized rats of 1 mg/kg i.v. quinpirole, a D2-like receptor agonist, increased k* for AA in 37 of 61 examined brain regions, generally having high densities of D2-like receptors, whereas the D1-like receptor agonist, SKF-38393, did not affect k* for AA in any region (Bhattacharjee et al, 2005b).
We thought it of interest to use our in vivo fatty acid method to try to image D-amphetamine's effect on PLA2 signaling via AA, and to see how much of the signal was mediated via D2-like receptors. In this study, we quantified k* for AA in unanesthetized rats acutely administered saline, D-amphetamine, or the selective D2-like receptor antagonist, raclopride (Gerlach and Casey, 1984), before either saline or D-amphetamine. At the doses chosen, D-amphetamine (0.5 or 2.5 mg/kg i.p.) and raclopride (6 mg/kg i.v.) have been reported to modify behavior and to alter the regional cerebral metabolic rate for glucose (rCMRglc) (Liao et al, 2001; Orzi et al, 1983; Porrino et al, 1984; Ross et al, 2002; Szumlinski and Szechtman, 2002; Trugman and James, 1993). In anesthetized baboons, 0.1 mg/kg i.v. amphetamine also increased rCMRglc and rCBF (McCulloch and Harper, 1977a). An abstract of part of this work has been published (Bhattacharjee et al, 2005a).
Materials and methods
Animals
The study was approved by the Animal Care and Use Committee of the National Institute of Child Health and Human Development, and was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication 86-23). Male CDF (Fischer-344) rats, 12 weeks old and weighing 250 to 300 g (Charles River Laboratories, Wilmington, MA, USA) were studied. They were acclimated for at least 7 days in an animal facility in which temperature, humidity, and light cycle (on 0600 to 1800) were controlled, with ad libitum access to food and water.
Chemicals, Drugs
[1-14C]AA in ethanol (50 mCi/mmol, 99% pure) was purchased from Moravek Biochemicals (Brea, CA, USA). D-Amphetamine sulfate, S(•)-Raclopride (+) tartrate, HEPES, fatty acid-free bovine serum albumin, and propylene glycol were purchased from Sigma Chemical Co. (St Louis, MO, USA). Sodium pentobarbital was purchased from Richmond Veterinary Supply Co. (Richmond, VA, USA). D-Amphetamine and raclopride were dissolved in 0.9% saline.
Arterial and Venous Catheter Placement
Polyethylene catheters (PE 50) (Becton Dickinson, Sparks, MD, USA) filled with heparin (50 IU) in 0.9% saline were surgically implanted into the right femoral artery and vein in a rat anaesthetized with halothane (1% to 2.5% v/v in O2). The incision was infiltrated with 1% lidocaine hydrochloride and closed with clips. The rat was loosely wrapped with its upper body free in a fast-setting plaster cast that was taped to a wooden block. It was allowed to recover from anaesthesia in a sound-dampened temperature-controlled box for 3 to 4h, to prevent any effect of anaesthesia on brain metabolism (Kimes et al, 1985; Sokoloff et al, 1977). Arterial blood pressure, heart rate and rectal temperature were measured before and 55 min after injection of saline, D-amphetamine, or raclopride.
Radiolabeled Arachidonic Acid Infusion
An unanesthetized rat was administered either 0.9% saline i.p. (n = 12), D-amphetamine, 2.5 mg/kg i.p. (n = 10) or S(•)-Raclopride (+) tartrate, 6 mg/kg i.v. 10 min before either saline (n = 7), or 2.5 mg/kg i.p. D-amphetamine (n = 9). Each injection was made in a volume of 1 ml/kg. Exactly 45 mins after saline or D-amphetamine, the rat was infused intravenously with 170 μCi/kg [1-14C]AA in 2 ml of 5 m mol/L HEPES buffer, pH 7.4, containing 50 mg/ml fatty acid free bovine serum albumin. An infusion pump (Harvard Apparatus, Model 22, Natick, MA, USA) was used to infuse the solution at a rate 400 μl/mins over a period of 5mins. Timed arterial aliquots (70 to 100 μl) were collected and centrifuged to obtain plasma, before and during tracer infusion to time of death at 20mins. At 20 min, the rat was killed with i.v. sodium pentobarbital (60 mg/kg), and its brain was removed and quickly frozen in 2-methylbutane at −40°C, then stored at −80°C for sectioning. In a subsequent experiment, rats were injected with 0.9% saline i.p. (n =8), or D-amphetamine sulfate, 0.5 mg/kg i.p. (n = 9), and 45 mins later, [1-14C]AA infusion was performed as noted above.
Radiolabeled Unesterified Arachidonic Acid in Plasma
The arterial plasma samples were extracted with 3 ml of chloroform/methanol (2:1 v/v) and 1.5 ml 0.1 mol/L KCl (Folch et al, 1957). A total of 100 μl of the lower organic phase of the solution was used to determine the labeled unesterified AA concentration by liquid scintillation counting (Liquid Scintillation Analyzer Model 2200CA, Packard Instruments, Downers Grove, IL, USA).
Autoradiography
Frozen brains were sectioned in the coronal plane in a cryostat (Hacker Instruments, Fairfield, NJ, USA) at −20°C. Sets of three adjacent 20-μm sections were collected at 100-μm intervals on 22 × 40 mm2 coverslips and dried on hot plate at 55°C for at least 5 mins. The sections, together with calibrated [14C]methylmethacrylate standards (Amersham, Arlington Heights, IL, USA), were exposed to autoradiographic film (EMC1, Eastman Kodak Company, Rochester, NY, USA) for 6 weeks, then the film was developed following the manufacturer's instructions. Radioactivity in each of 62 brain regions, identified from a rat atlas (Paxinos and Watson, 1987), was measured bilaterally in sextuplicate by quantitative densitometry using the public domain Image Analysis NIH Program (version 1.62) created by Wayne Rasband (NIH, Bethesda, MD, USA).
Regional brain incorporation coefficients, k* (ml/s/g brain), were calculated as (Bhattacharjee et al, 2005b; DeGeorge et al, 1991), k* is in units of ml/s/g wet wt (specific gravity of brain approximates 1); c*brain (20 min) is brain radioactivity at 20 min after the onset of infusion, in units of nCi/g; c*plasma is plasma AA radioactivity in units of nCi/ml; and t is time after onset of [1-14C]AA infusion.
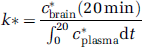
Statistics
Data are reported as means ± s.d. A one-way ANOVA with Fisher's LSD pair wise post hoc test was used to compare means between groups (rats administered saline, D-amphetamine (2.5 mg/kg), raclopride + saline, or raclopride + D-amphetamine (2.5mg/kg); all comparisons are shown versus control (saline) (SAS 9.0 Cary, NC, USA). An unpaired t-test was performed to compare means between the rats administered either saline or D-amphetamine (0.5 mg/kg). Statistical significance was taken as P < 0.05.
Results
Physiologic Parameters
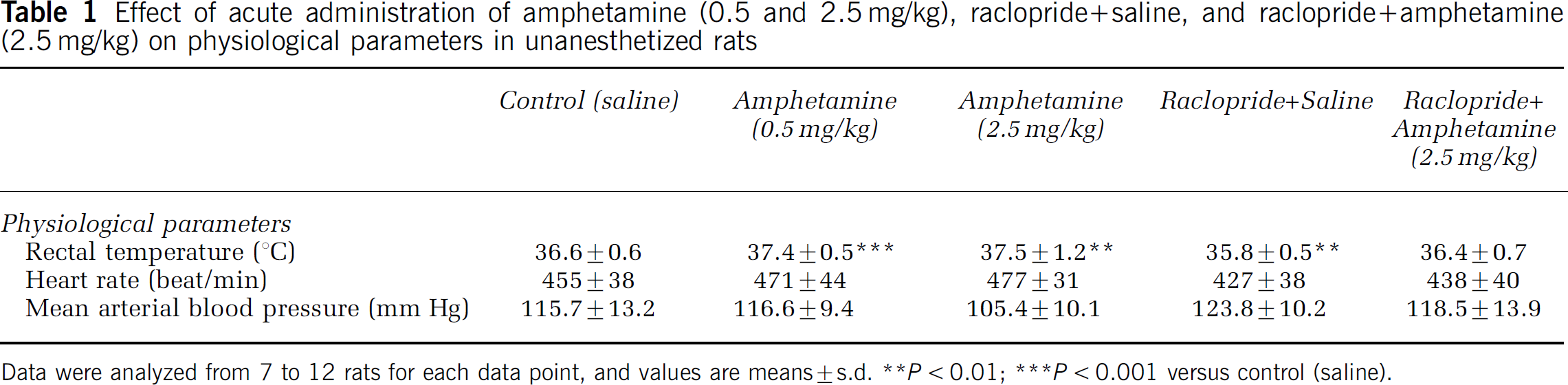
Table 1 presents mean values for body temperature, heart rate, and mean arterial blood pressure, 55 min after injection of saline or drug. No significant saline or drug effect was produced on heart rate or mean arterial blood pressure. However, D-amphetamine (0.5 or 2.5 mg/kg) significantly increased while raclopride significantly decreased body temperature, compared with saline, but raclopride followed by D-amphetamine had no effect on temperature. D-Amphetamine also produced hyperactivity and stereotyped sniffing, chewing, and licking, behaviors that we did not attempt to quantify, whereas prior administration of raclopride appeared to reduce these behavioral changes. Raclopride itself induced akinesia and catalepsy, which we also did not quantify.
Effect of acute administration of amphetamine (0.5 and 2.5 mg/kg), raclopride+saline, and raclopride+amphetamine (2.5 mg/kg) on physiological parameters in unanesthetized rats
Data were analyzed from 7 to 12 rats for each data point, and values are means ± s.d.
P < 0.01;
P < 0.001 versus control (saline).
The hyperthermia caused by D-amphetamine has been ascribed to induction of hyperactivity, decreased respiratory evaporative heat loss associated with cutaneous vasoconstriction (Lin et al, 1980), and to a thermoregulatory role of central DA (Ulus et al, 1975), while the hypothermia following raclopride has been ascribed to induction of akinesia and blocking of D-amphetamine's hyperthermic effects (Lin et al, 1980; Ross et al, 2002; Ulus et al, 1975).
Effects of D-Amphetamine on k*
Figure 1 presents color-coded images of k* in autoradiographs of coronal brain sections from a rat administered saline, 2.5 mg/kg i.p. D-amphetamine, 6 mg/kg i.v. raclopride followed by saline, or 2.5 mg/kg i.p. D-amphetamine. It illustrates that D-amphetamine compared with saline increased k* in many anterior brain regions. In contrast, raclopride, or raclopride followed by D-amphetamine, had no effect on k* compared with saline alone.
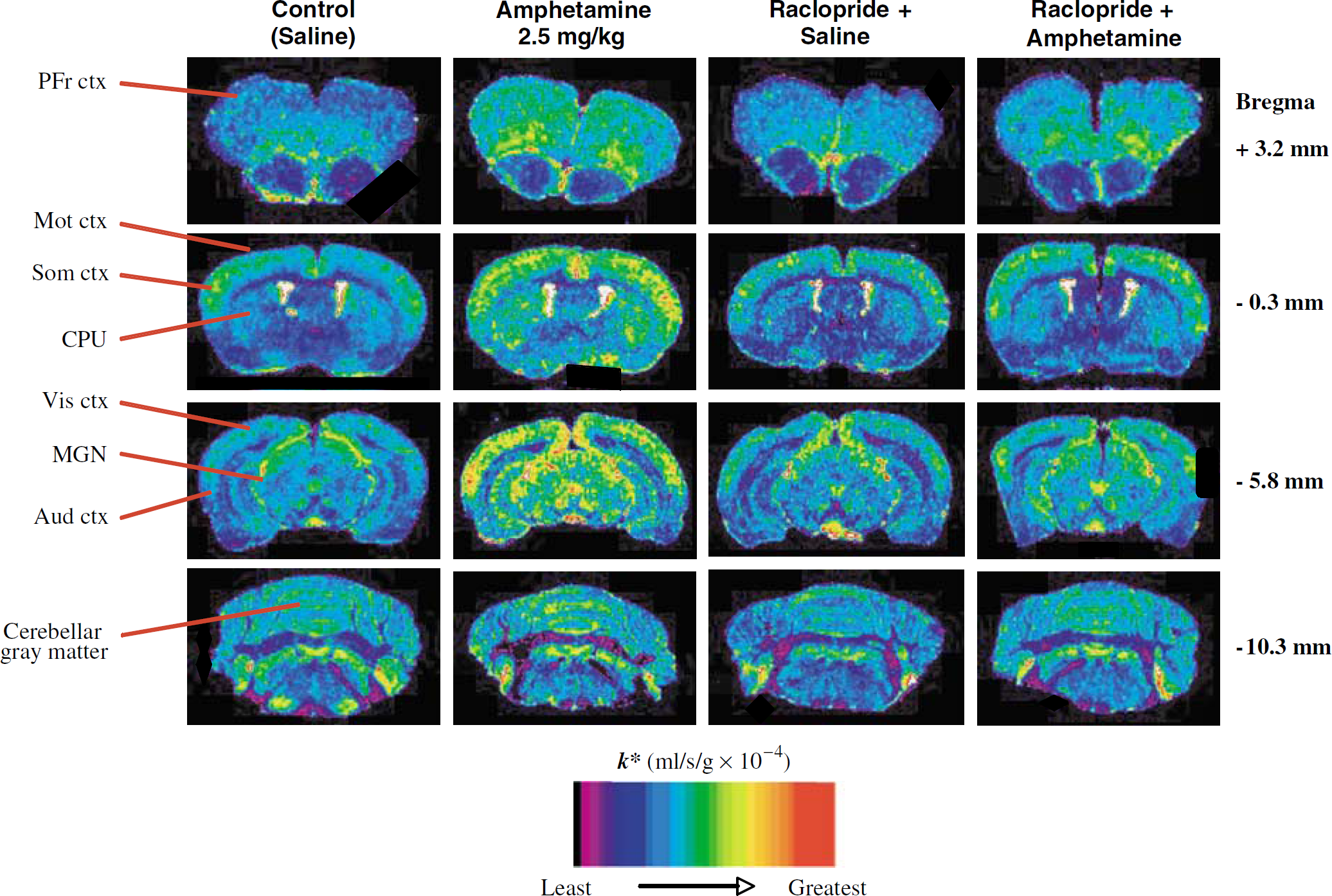
Coronal autoradiographs of effects of D-amphetamine (2.5 mg/kg i.p.) and of raclopride (6 mg/kg i.v.) before either saline or D-amphetamine, compared with i.p. saline, on regional incorporation coefficients k* for arachidonic acid in unanesthetized rats. D-Amphetamine increased k* in brain regions with high densities of D2 -like receptors. Raclopride blocked these effects and had no effect by itself. Values for k* are color-coded. Brain slices displayed are +3.2, then −0.3, −5.8, −10.3 from the bregma (Paxinos and Watson, 1987). Abbreviations: PFr ctx, prefrontal cortex; Mot ctx, motor cortex; Som ctx, somatosensory cortex; CPU, caudateputamen; Vis ctx, visual cortex; MGN, medial geniculate nucleus; Aud ctx, auditory cortex.
Mean values for k* in each of 62 brain regions, collated from autoradiographs, are presented in Table 2. D-Amphetamine 2.5 mg/kg compared with saline significantly increased k* in 27 of 62 brain regions examined, many of which are rich in D2-like receptors or of mRNA for such receptors (Hayakawa et al, 2001; Levant et al, 1992; Levey et al, 1993; Meador-Woodruff, 1994). In neocortical regions, D-amphetamine increased k* significantly in prefrontal (33% to 35%), frontal (31% to 45%), motor (35% to 45%), somatosensory (28% to 48%), and auditory neocortex (27% to 33%). In the pyramidal-extrapyramidal system, increments were produced in caudate-putamen (26% to 43%), globus pallidus (33%), and entopeduncular nucleus (52%). In limbic and associated regions, k* was increased in the anterior cingulate cortex (31%), accumbens nucleus (42%), Islands of Callaja (30%), ventral pallidum (43%), and anteroventral and anterome-
Effect of amphetamine and raclopride on regional unidirectional incorporation coefficients, k * for [1-14C]arachidonic acid in unanesthetized rats
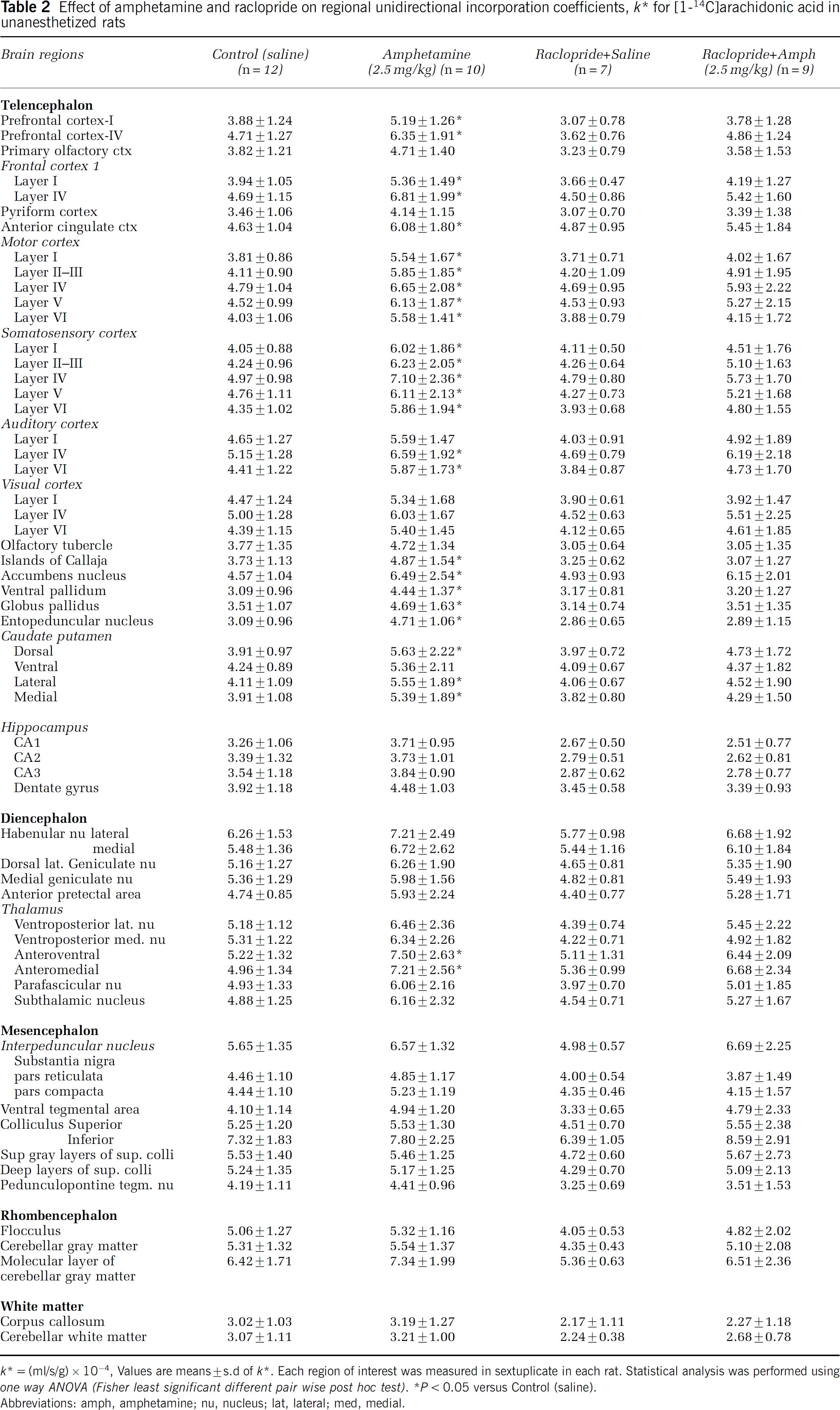
k* = (ml/s/g) × 10−4, Values are means ± s.d of k*. Each region of Interest was measured In sextuplicate In each rat. Statistical analysis was performed using one way ANOVA (Fisher least significant different pair wise post hoc test).
P < 0.05 versus Control (saline).
Abbreviations: amph, amphetamine; nu, nucleus; lat, lateral; med, medial.
dial thalamus (43% to 45%).
In a follow-up independent study, 0.5 mg/kg i.p. D-Amphetamine compared with saline increased k* in seven of the 62 brain regions examined (Table 3), including the nucleus accumbens (50%), layer IV of prefrontal (40%), frontal (37%), and motor (33%) cortex, and layers III-V of somatosensory cortex (34% to 38%).
Brain regions in which 0.5 mg/kg i.p. amphetamine significantly increased regional unidirectional incorporation coefficients, k* for AA in unanesthetized rats
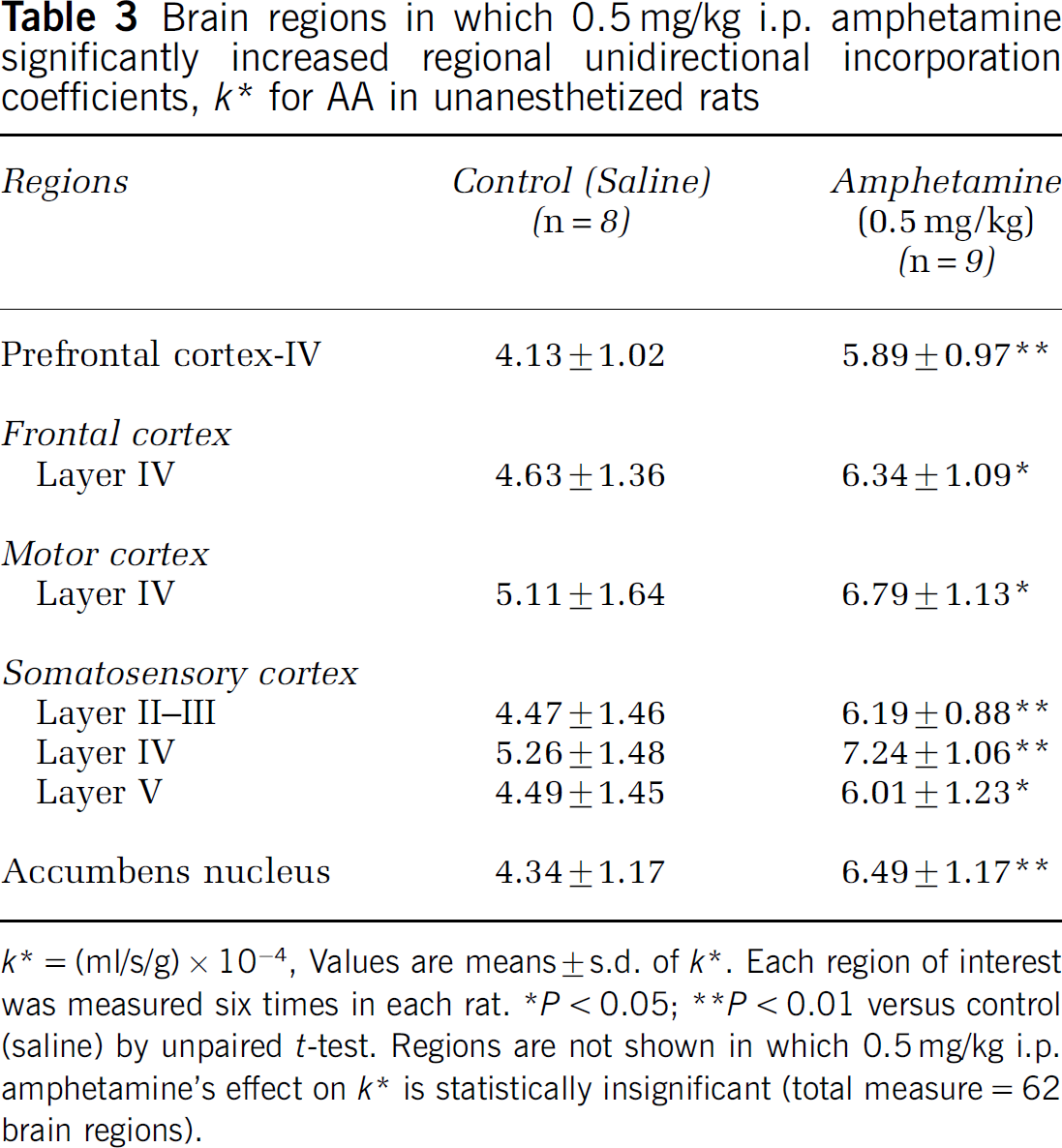
k* = (ml/s/g) × 10−4, Values are means ± s.d. of k*. Each region of Interest was measured six times In each rat.
P < 0.05;
1P < 0.01 versus control (saline) by unpaired t-test. Regions are not shown In which 0.5 mg/kg i.p. amphetamine's effect on k* is statistically insignificant (total measure = 62 brain regions).
Effects of Raclopride on k*
Rats were administered the D2-like receptor antagonist, raclopride, before either saline or 2.5 mg/kg i.p. D-amphetamine, to ascertain the extent to which D-amphetamine effects on k* were mediated by D2-like receptors. Figure 1 and Table 2 show that raclopride compared with saline did not significantly change k* for AA in any of the 62 brain regions examined. Furthermore, raclopride pretreatment prevented the significant increments in k* following D-amphetamine that occurred in rats administered 2.5 mg/kg D-amphetamine without pretreatment.
Discussion
Acute 2.5 mg/kg i.p. D-amphetamine compared with i.p. saline increased k* for AA in 27 of 62 brain regions examined in unanesthetized rats, whereas 0.5 mg/kg i.p. D-amphetamine increased k* in only seven of 62 regions, consistent with a dose effect. The regions affected by D-amphetamine are reported to have high densities of D2-like receptors or of mRNA for such receptors (Figure 1 and Table 2) (Cooper et al, 2003; Hayakawa et al, 2001; Levant et al, 1992; Levey et al, 1993; Meador-Woodruff, 1994). Affected at 2.5 mg/kg i.p. were neocortex, caudateputamen, globus pallidus, ventral pallidum, accumbens nucleus, and thalamic nuclei. Significant increments were absent in regions with few or no D2-like receptors, such as the hippocampus, habenular nucleus, subthalamic nucleus, substantia nigra, cerebellum, and ventral tegmental area. At 0.5 mg/kg i.p. D-amphetamine, k* was significantly elevated only in the nucleus accumbens (50%), layer IV of the prefrontal, frontal, and motor cortex, and layers III to V of the somatosensory cortex (Table 3).
Giving 6 mg/kg i.v. raclopride, a selective D2-like receptor antagonist, prior to i.p. saline or 2.5 mg/kg i.p. D-amphetamine did not change k* significantly in any of the 62 regions, confirming that the k* responses required activation via D2-like receptors. However, secondary activation via glutamatergic fibers derived from the thalamus also could have contributed to increased k* in layer IV of the prefrontal, frontal, motor cortex, and somatosensory cortex, the receptive fields for these fibers (Zilles, 1990). This is because ionotropic glutamatergic N-methyl-D-aspartate (NMDA) receptors can activate Ca2+-dependent cPLA2 by allowing Ca2+ into cells and thereby increase k* (Basselin et al, 2005a; Weichel et al, 1999). Targeting cortical layer IV, in any case, is consistent with the ability of D-amphetamine (5.0mg/kg i.p.) in rats to increase their expression of the immediate-early gene, c-fos, a marker of neural activity (LaHoste et al, 1996; Zilles, 1990).
As raclopride entirely blocked the increments in k* following 2.5 mg/kg D-amphetamine, our observations suggest that these increments were initiated by D2-like receptor activation of PLA2, and possibly by secondary activation of NMDA receptors in layer IV of the neocortex. Both D2-like and NMDA receptors can be coupled to cPLA2, via a G-protein or by allowing Ca2+ into the cell (Dennis, 1994; Nilsson et al, 1998; Vial and Piomelli, 1995; Weichel et al, 1999). Similarly, giving the D2-like receptor agonist quinpirole to unanesthetized rats increased k* for AA in many of the brain regions activated by 2.5 mg/ kg D-amphetamine, and the increases could be prevented by prior administration of the D2-like receptor antagonist, butaclamol (Basselin et al, 2005b; Bhattacharjee et al, 2005b; Hayakawa et al, 2001).
In addition to increasing the synaptic concentration of DA to activate D2-like receptors (see Introduction) (Butcher et al, 1988; Creese and Iversen, 1975; Fuller and Hemrick-Luecke, 1980; Houston et al, 2004; Jones et al, 1998; Kuczenski and Segal, 1989; Laruelle et al, 1997; Segal et al, 1980; Sharp et al, 1987), D-amphetamine can increase extracellular serotonin and epinephrine to indirectly stimulate serotonergic 5-HT2A/2C and β2-adrenergic receptors, respectively. These receptors, like D2-like receptors, can be coupled to PLA2 activation and AA release (Berg et al, 1998; Garcia and Kim, 1997; Kurrasch-Orbaugh et al, 2003; Pavoine et al, 2003; Qu et al, 2003). However, raclopride's ability to block k* increments following 2.5 mg/kg D-amphetamine (Table 2) indicates that the increments did not directly involve serotonin or epinephrine-initiated signaling.
Raclopride by itself did not change k* for AA significantly in any of the 62 brain regions analyzed (Table 2). This is consistent with a low abundance of DA synapses compared with a high (70%) abundance of glutamatergic synapses in rat brain (Fonnum, 1984; Raichle and Gusnard, 2002). Thus, baseline values of k* for AA in rat brain are decreased 20% to 50% following administration of MK-801, which blocks Ca2+-mediated activation of cPLA2 via the ionotropic NMDA receptor (Basselin et al, 2005a; Dumuis et al, 1988; Weichel et al, 1999).
Our studies are comparable to a dose finding study with D-amphetamine in unanesthetized rats (Porrino et al, 1984). In that study, 0.2 or 0.5 mg/kg i.v. D-amphetamine increased rCMRglc only in the nucleus accumbens, which has a very high density of D2-like receptors and is considered to regulate reward behavior and drug dependence (Cooper et al, 2003; Di Chiara et al, 2004; Salamone et al, 2005). D-Amphetamine at 1.0 mg/kg i.v. increased rCMRglc as well in prefrontal cortex and extrapyramidal regions having moderate-high densities of D2-like receptors, whereas 5.0 mg/kg i.v. D-amphetamine additionally increased rCMRglc in regions with few or no D2-like receptors (Porrino et al, 1984).
In this paper, we show that D-amphetamine modifies an aspect of brain metabolism other than rCMRglc or rCBF (McCulloch and Harper, 1977b; Norberg et al, 1978; Orzi et al, 1983; Porrino et al, 1984; Trugman and James, 1993; Wechsler et al, 1979), the receptor-initiated activation of PLA2 to release AA from membrane phospholipids and engender the brain AA metabolic cascade and the ATP-consuming recovery process (Purdon and Rapoport, 1998; Rapoport, 2001; Shimizu and Wolfe, 1990). However, whereas k* for AA is independent of changes in rCBF (Chang et al, 1997; DeGeorge et al, 1991; Robinson et al, 1992; Robinson and Rapoport, 1986), rCBF can be influenced by activation of the AA cascade, represented by k*. This is because some of the AA released by PLA2 activation will be converted by cyclooxygenase-2 (COX-2) to PGE2, a vasodilator (Li et al, 1997). When PGE2 formation is reduced in COX-2 knockout mice or following administration of a COX-2 inhibitor, k* responses to drug activation are absent (Basselin M and Rapoport SI, 2006, unpublished observation; Basselin et al, 2006; Rapoport et al, 2001) and rCBF responses to somatosensory stimulation are reduced (Li et al, 1997; Niwa et al, 2000, 2001). The k* response in the COX-2 knockout mouse is absent (Basselin et al, 2006) because it, and particularly the product of k* and plasma unesterified AA (actual rate of AA incorporation into brain phospholipid), represents the plasma AA that replaces the AA that is lost by metabolism in brain following activation via COX-2. COX-2 is closely coupled to cPLA2 activation (Naraba et al, 1998).
It now is possible to measure k* for AA in the human brain with positron emission tomography (PET) following the intravenous injection of [1-11C]AA (Esposito et al, 2003; Giovacchini et al, 2002, 2004; Rapoport et al, 2005). Thus, one might image D2-like receptor-mediated activation of AA metabolism in human brain diseases thought to have disturbed DA neurotransmission, such as Parkinson disease (Chalon et al, 1999; Hayakawa et al, 1998, 2001; Ross et al, 2001), cocaine abuse (Ross et al, 1996), attention deficit hyperactivity disorder and schizophrenia (Matochik et al, 1993; Wolkin et al, 1994). Not only might amphetamine be used in this regard, but so might apomorphine, a D1/D2-like agonist, considering prior imaging of rCMRglc or rCBF in animals and humans given one or the other drug (Cleghorn et al, 1991; Devous et al, 2001; Ernst et al, 1997; McCulloch and Harper, 1977a; McCulloch et al, 1982; Wolkin et al, 1994). In Parkinson disease, for example, based on behavioral and fatty acid animal studies, we would predict increased k* responses to apomorphine because of increased expression of D2-like receptors in the basal ganglia, but reduced responses to amphetamine, because of reduced pre-synaptic dopaminergic elements (Chalon et al, 1999; Guttman, 1992; Hayakawa et al, 2001; Metz and Whishaw, 2002). In attention deficit hyperactivity disorder, evidence of disturbed DAT expression and of the efficacy of psychostimulants that reverse the direction of the DAT so as to increase DA in the synaptic cleft (Biederman and Faraone, 2002; Lopez et al, 2004) predicts that the AA signal to apomorphine and D-amphetamine will be elevated at brain sites of D2-like receptors compared with controls.
In summary, we have imaged for the first time D2-like receptor initiated signal transduction involving PLA2 activation and AA release in unanesthetized rats given D-amphetamine. Increments in k* caused by D-amphetamine were found in brain regions having D2-like receptors, and were entirely prevented by pre-administration of the D2-like receptor antagonist, raclopride. At a low dose of 0.5 mg/kg i.p. D-amphetamine, comparable to what has been used in human studies when measuring rCBF or rCMRglc with PET (Devous et al, 2001; Wolkin et al, 1994), k* was increased in the nucleus accumbens and some layer IV of neocortical regions. This effect is consistent with a high D2-like receptor density in the nucleus accumbens, and with the reported low-dose activation of rCMRglc in this region.