
Abstract
Ischemic depolarizing events, such as repetitive spontaneous periinfarct spreading depolarizations (PIDs), expand the infarct size after experimental middle cerebral artery (MCA) occlusion. This worsening may result from increased metabolic demand, exacerbating the mismatch between cerebral blood flow (CBF) and metabolism. Here, we present data showing that anoxic depolarization (AD) and PIDs caused vasoconstriction and abruptly reduced CBF in the ischemic cortex in a distal MCA occlusion model in mice. This reduction in CBF during AD increased the area of cortex with 20% or less residual CBF by 140%. With each subsequent PID, this area expanded by an additional 19%. Drugs that are known to inhibit cortical spreading depression (CSD), such as N-methyl-D-aspartate receptor antagonists MK-801 and 7-chlorokynurenic acid, and σ-1 receptor agonists dextromethorphan and carbetapentane, did not reduce the frequency of PIDs, but did diminish the severity of episodic hypoperfusions, and prevented the expansion of severely hypoperfused cortex, thus improving CBF during 90 mins of acute focal ischemia. In contrast, AMPA receptor antagonist NBQX, which does not inhibit CSD, did not impact the deterioration in CBF. When measured 24 h after distal MCA occlusion, infarct size was reduced by MK-801, but not by NBQX. Our results suggest that AD and PIDs expand the CBF deficit, and by so doing negatively impact lesion development in ischemic mouse brain. Mitigating the vasoconstrictive neurovascular coupling during intense ischemic depolarizations may provide a novel hemodynamic mechanism of neuroprotection by inhibitors of CSD.
Keywords
Introduction
The hemodynamic evolution of acute focal cerebral ischemia is poorly understood. Electrophysiologically, severely ischemic core (i.e., residual cerebral blood flow (CBF) ≤ 20%; Hoehn-Berlage et al, 1995; Hossmann, 1994; Sick et al, 1998) undergoes anoxic depolarization (AD), whereas in ischemic penumbra synaptic activity ceases, but residual CBF is sufficient to maintain membrane ionic gradients. With continuing ischemia, however, more penumbral tissue undergoes AD as the ischemic core expands over time. The expansion of depolarized core coincides with the occurrence of repetitive and spontaneous periinfarct spreading depolarizations (PIDs) (Hossmann, 1996). Anoxic depolarization and PIDs are associated with massive redistribution of ions across membranes, an increase in extracellular potassium ([K+]e) up to 60 to 80 mmol/L, and reduction in tissue adenosine triphosphate (ATP), oxygen and pH (Back et al, 1994; Busch et al, 1996; Hossmann, 1994; Nedergaard and Astrup, 1986; Somjen, 2001). Therefore, it has been presumed that ischemic depolarizing events increase the metabolic burden and exacerbate the energy deficit, thereby expanding the infarct (Back et al, 1996; Selman et al, 2004).
In addition to the expansion of depolarized core, there is also evidence for expansion of CBF deficit during the acute phase after middle cerebral artery occlusion (MCAO) (LaManna et al, 1985; McColl et al, 2004; Zaharchuk et al, 2000). The mechanism for this deterioration is not known, although endothelial swelling and microvascular plugging by activated leukocytes have been proposed (del Zoppo and Hallenbeck, 2000). In the normal brain from most species, cortical spreading depression (CSD) causes marked hyperemia. Under hypoxic or partially ischemic conditions, however, or in the presence of artificially elevated [K+]e and reduced nitric oxide concentration, CSD causes hypoperfusion rather than hyperemia (Dreier et al, 1998; Sonn and Mayevsky, 2000), suggesting that intense depolarizing events during focal ischemia (i.e., AD and PIDs) may variably impact CBF in penumbra, depending on the energy status of ischemic tissue and [K+]e levels. The survival of penumbra depends on the balance between CBF and metabolic demand; therefore, hemodynamic impact of AD and PIDs might be as important as their metabolic consequences in determining the tissue outcome.
Although the metabolic and electrophysiologic consequences of AD and PIDs have been extensively investigated, their impact on cerebral vasculature and CBF are less known. Using laser speckle flowmetry (LSF), a two-dimensional CBF imaging technique with high spatiotemporal resolution, we present evidence for a novel form of neurovascular coupling during focal ischemia. Our data suggest that vasoconstrictive neurovascular coupling during ischemic depolarizations contributes to the hemodynamic progression in acute focal cerebral ischemia, and that mitigating the adverse vascular effect of tissue depolarization might be a critical mechanism by which neuroprotective drugs reduce tissue injury.
Materials and methods
Drugs
MK-801 ((+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate; 0.5 or 1 mg/mL in saline), 7-chlorokynurenic acid (10 mg/mL in 0.1 N NaOH), carbetapentane (2.5 mg/mL in saline) and NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline, disodium salt, 24 mg/mL in saline) were purchased from Tocris (Ellisville, MO, USA), whereas dextromethorphan (2 mg/mL in saline) was purchased from Sigma (St Louis, MO, USA).
Surgical Preparation and Physiologic Monitoring
Mice (C57BL/6J, 25 to 35 g, n = 64) were anesthetized with isoflurane (2% induction, 1% maintenance), endotracheally intubated and ventilated (70% N2O, 30% O2; SAR 830/P, CWE, Ardmore, PA, USA). Femoral artery was cannulated for blood pressure (BP) and arterial blood gas measurements (ETH-400 transducer amplifier, ADInstruments, MA, USA). Blood pressure and heart rate were continuously recorded (PowerLab, ADInstruments, Colorado Springs, CO, USA). Mice were paralyzed (pancuronium 0.4 mg/kg/h intraperitoneal), placed on a stereotaxic frame, and scalp and periosteum were pulled aside. The adequacy of anesthesia was regularly checked by the absence of a BP response to tail pinch. Body temperature was kept at 37.0°C using a thermostatic heating pad (FHC, Brunswick, ME, USA). Arterial blood gases and pH were measured at least once every hour in 30 μL samples (Blood Gas Analyzer 248, CIBA/Corning, Corning, NY, USA). All physiological parameters were within previously reported normal limits (Table 1) (Dalkara et al, 1995). Institutional guidelines for animal care and use for research purposes were strictly followed.
Physiological parameters
Focal Cerebral Ischemia
After general surgical preparation as described above, mice were placed in a stereotaxic frame. The temporalis muscle was separated from the temporal bone and removed. A burr hole (2 mm diameter) was drilled under saline cooling in the temporal bone overlying the distal MCA just above the zygomatic arch. The dura was kept intact and a microvascular clip (Ohwa Tsusho, Tokyo, Japan) was used to occlude distal MCA (dMCAO).
Infarct Volume
In 15 mice, the MCA was exposed as described above, and permanently ligated using a 10-0 nylon suture. Direct infarct volume was determined 24 h later using triphenyltetrazolium chloride (TTC)-stained 1-mm-thick coronal brain sections. To avoid the morbidity associated with endotracheal and arterial cannulations, mice were allowed to breathe freely and physiological monitoring was not performed during the surgical procedure.
Laser Speckle Flowmetry
Laser speckle flowmetry was used to study the spatiotemporal characteristics of CBF changes during focal ischemia. The technique for LSF in mice has been described in detail elsewhere (Ayata et al, 2004a; Dunn et al, 2001). Briefly, a charge-coupled device (CCD) camera (Cohu, San Diego, CA, USA) was positioned above the head, and a laser diode (780 nm) was used to illuminate the intact skull. Raw speckle images were used to compute speckle contrast, a measure of speckle visibility inversely related to the velocity of the scattering particles, and therefore CBF. The speckle contrast is defined as the ratio of the standard deviation of pixel intensities to the mean pixel intensity in a small region of the image (Briers, 2001). In all, 10 consecutive raw speckle images were acquired at 15 Hz (an image set), processed by computing the speckle contrast using a sliding grid of 7 × 7 pixels, and averaged to improve the signal to noise ratio. Laser speckle flowmetry image sets were thus obtained every 7.5 secs. Speckle contrast images were converted to images of correlation time values, which represent the decay time of the light intensity autocorrelation function. The correlation time is inversely and linearly proportional to the mean blood velocity. Relative CBF images (percentage of baseline) were calculated by computing the ratio of a baseline image of correlation time values to the subsequent images.
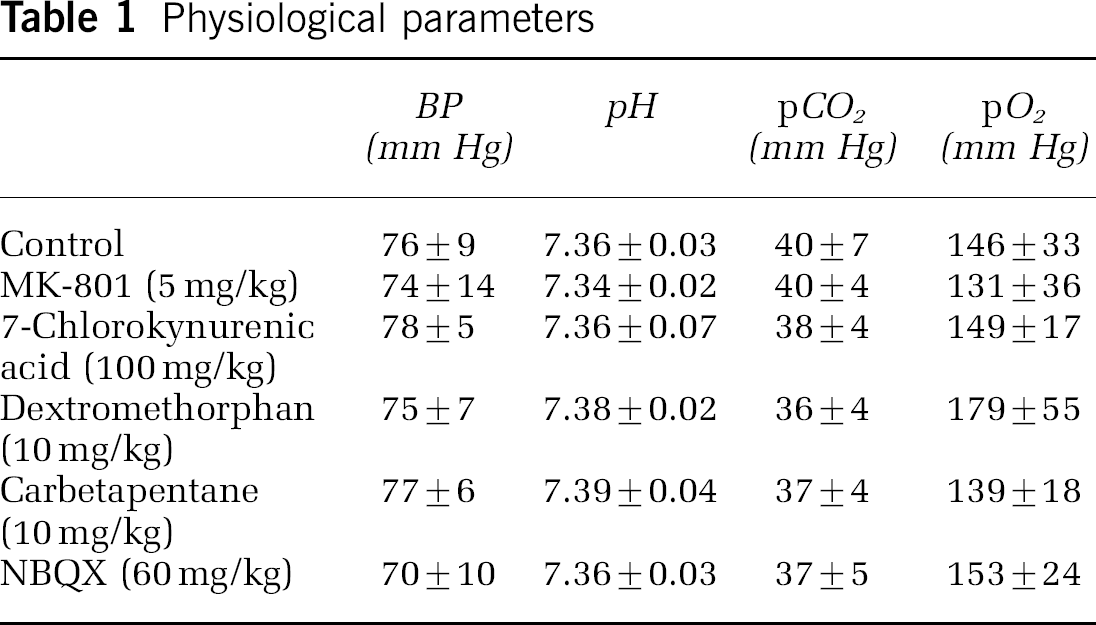
Laser speckle flowmetry imaging was started 1 min before dMCAO and continued for up to 90 mins. Based on the severity of CBF reduction during the first minute of dMCAO, three cortical regions of interest (ROI, 250 μm × 250 μm) were identified corresponding to the core (center of severe CBF reduction), hemodynamic penumbra (steep portion of CBF gradient between the core and nonischemic cortex) and nonischemic cortex (Figure 1, C, P and N, respectively). Relative CBF changes within these ROIs were recorded over time.
Hypoperfusion evoked by AD.
The amplitude of CBF changes during AD and their latency from the onset of dMCAO (0 min) were measured and averaged at 4 deflection points: the initial abrupt CBF decrease on dMCAO, the onset, the trough and the partial recovery of secondary hypoperfusion, as well as at 4 and 6 mins after dMCAO. The amplitude of CBF changes associated with PIDs and their latency from the onset of hypoperfusion were measured and averaged at 3 deflection points: the onset (0 min), trough and recovery of secondary hypoperfusion, as well as at 3 and 5 mins after the onset of hypoperfusion.
In addition, the severity of CBF deficit was assessed two-dimensionally by calculating the area of the severely hypoperfused cortex using a thresholding paradigm (pixels with residual CBF ≤ 20% of the preischemic baseline, expressed in mm2).
Closed Cranial Window
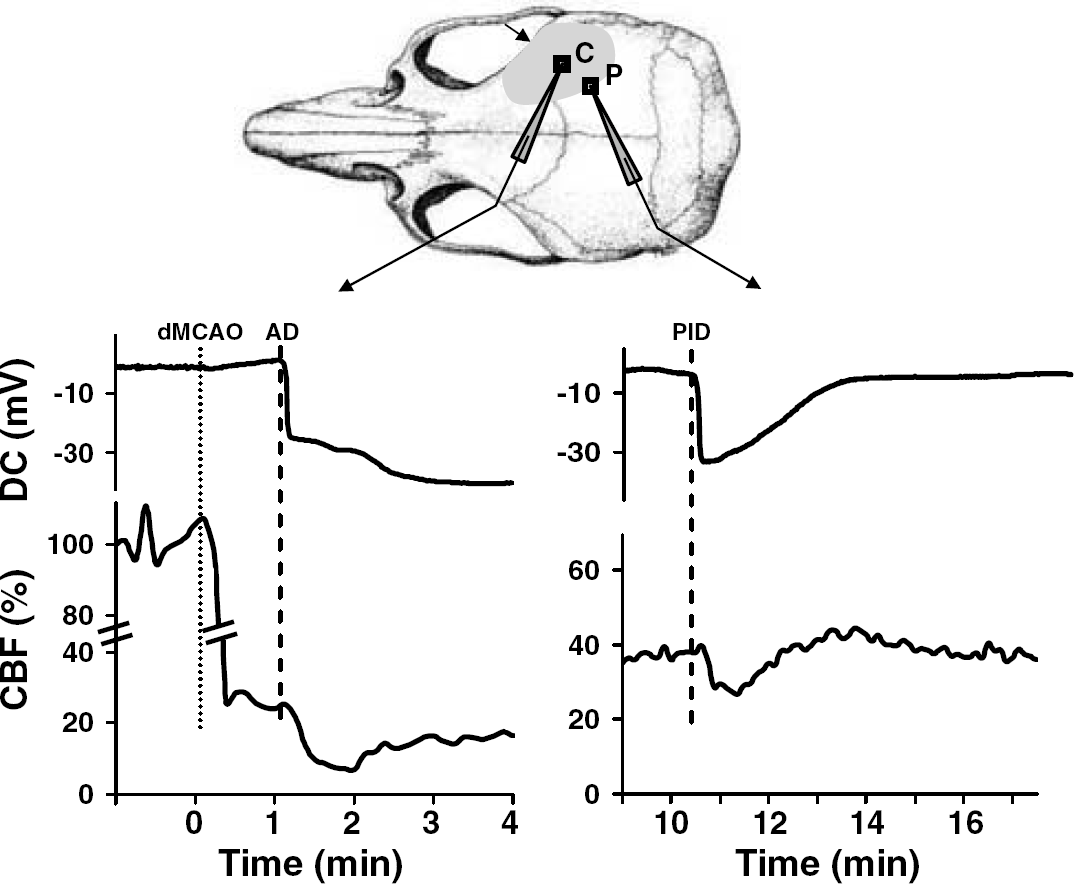
A closed cranial window was used in three experiments to directly visualize the pial arterioles for vessel diameter measurements during AD. The window was constructed as described previously (Ma et al, 1996), with modifications. Briefly, a circular window was constructed on the parietal bone using dental cement. After hardening of the cement, a burr hole of 3 mm diameter was drilled in the center of the window under saline cooling. The bone was removed with care to keep the dura intact. The window was filled with artificial cerebral spinal fluid, covered using a glass coverslip (12 mm in diameter, 150 μm thickness), and the circumference sealed with dental cement. The depth of the window was approximately 1 mm.
Electrophysiology
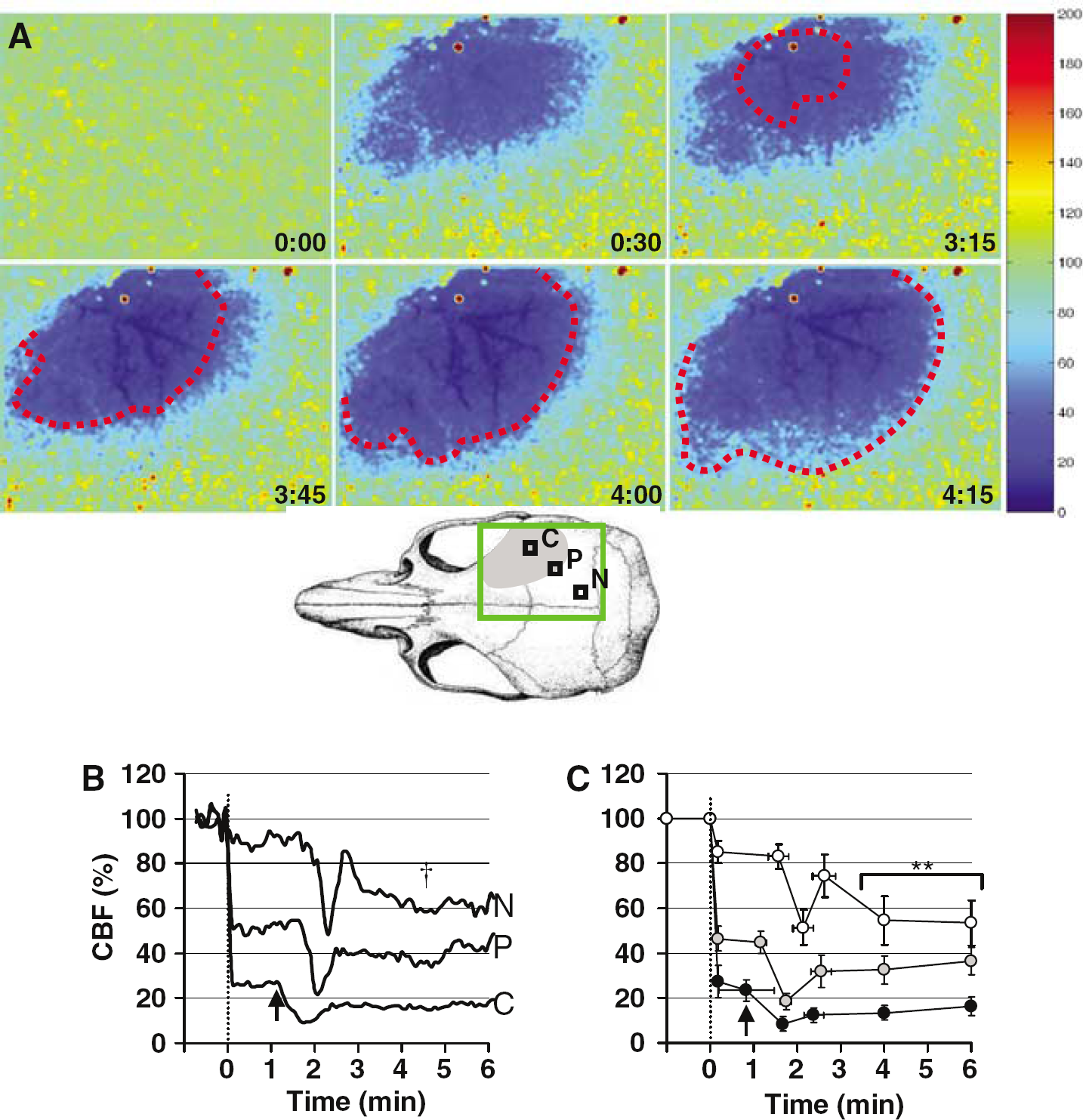
In a subgroup of mice (n = 5), the spatiotemporal relationship between cortical slow (DC) potential shifts and CBF changes was studied by electrophysiological recordings (Axoprobe 1A, Axon Instruments, Union City, CA, USA), simultaneously with LSF. For this, a small burr hole was drilled (< 0.5 mm diameter), positioned either over the core or penumbra. A glass micropipette was inserted through the dura to a cortical depth of 200 pm. A ground electrode (Ag/AgCl) was placed subcutaneously in the neck. The shank of glass microelectrode and its tip location were identified on speckle contrast images, and ROIs were positioned to place the microelectrode tip in the center of ROI. Therefore, CBF changes were recorded simultaneously with the DC shifts from the same cortical location.
Experimental Groups
Mice were treated with MK-801 (1 or 5 mg/kg, n = 5 and 9), NBQX (60 or 120 mg/kg, n = 5 and 4), dextromethorphan (10 mg/kg, n = 6), carbetapentane (10 mg/kg, n = 6), 7-chlorokynurenic acid (100 mg/kg, n= 6) or saline (0.1 mL, n = 15). All injections were made intraperitoneally 1 h prior to the onset of dMCAO. NBQX has a plasma half-life of 4 h when administered intraperitoneally at a dose of 30 mg/kg in mice (Dalgaard et al, 1994).
Statistics
One- or two-way analysis of variance (ANOVA) followed by Student-Newman–Keuls multiple comparisons test were used to compare physiological parameters and changes in CBF and the area of ischemic cortex, between the control and drug-treated groups. Paired t-test was used to test the effects of PIDs. Results were expressed as mean ± standard deviation. P < 0.05 was considered statistically significant.
Results
Distal MCAO abruptly decreased CBF over the dorsolateral cortex (Figure 1A, 0:30 mins; Figure 1B and 1C, dotted line). Cerebral blood flow was reduced to 26%75% of the baseline in ischemic core (Figure 1C, n = 15). Surrounding the core, there was a steep CBF gradient towards the nonischemic cortex (Figure 1A). This region, in which CBF was reduced to 46% ± 5%, was arbitrarily defined as hemodynamic penumbra (Figure 1A,‘P’). The nonischemic cortex also showed a small but consistent CBF reduction immediately after dMCAO (85% 75%, P < 0.01 versus preischemic baseline; Figure 1C). Within 2 mins after dMCAO, CBF in the ischemic core started to decrease acutely (Figures 1A to 1C). This abrupt secondary hypoperfusion was coincident with AD (Figure 2, left panel), and associated with profound vasoconstriction (n =3, Figure 3).
Episodic hypoperfusions were coincident with AD and PIDs. Electrophysiological recordings show that hypoperfusion in the core (left) and penumbra (right) was due to AD and PID (obtained from two different experiments). Both AD and PID were associated with abrupt hypoperfusion. The onset of depolarization preceded the onset of hypoperfusion by less than 10 secs. The upper drawing shows the location of microelectrode tips (C, core; P penumbra) in the center of ROIs (250 × 250 μm) within which CBF changes were measured. The arrow and shaded area indicate the location of dMCAO and CBF deficit, respectively.
Anoxic depolarization is associated with vasoconstriction. Images of a pial arteriole viewed through a closed cranial window during the first 90 secs of dMCAO. Within 45 secs after dMCAO, pial arteriolar diameter abruptly decreased, and at 60 secs the vessel completely collapsed. The vessel diameter incompletely recovered at 90 secs and remained less than pre-AD values throughout the imaging. Pial arterioles from the dorsal frontal cortex were imaged with the CCD camera, contrast was enhanced using a green-filtered light source, and images were rendered off-line for improved edge detection. Vessel diameter (μm) is indicated in the upper left corner of each image, and measured at the dashed line shown in the image taken at −15 secs.
Multiple spontaneous waves of hypoperfusion originated from the core and propagated into the penumbra during 90-min recordings (2.9 ± 1.8/h; Figure 4, arrowheads). Simultaneous electrophysiological and LSF recordings demonstrated that AD and PIDs (n = 2 and 15, respectively, in a total of 5 experiments) were associated with abrupt hypoperfusion (see Figure 2 for representative tracings). In all of these events, the onset of depolarization preceded the onset of hypoperfusion by less than 10 secs, but never followed it. Prolonged DC shifts during PIDs were associated with prolonged hypoperfusion. The majority of PIDs originated from the anterior ischemic regions, consistent with previous observations (Selman et al, 2004; Strong et al, 1996).
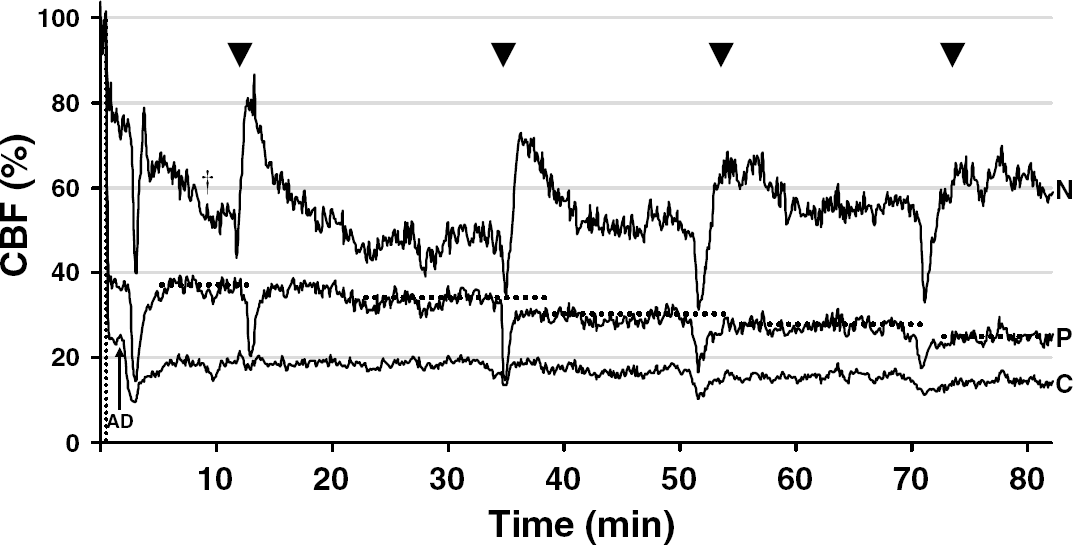
Cerebral blood flow in the core and penumbra worsened in association with AD and PIDs. Cerebral blood flow changes in the core, penumbra and nonischemic cortex after dMCAO (vertical dotted line) are shown from a representative experiment (C, core, P, penumbra, N, nonischemic cortex, as shown in Figure 1A). During the first 5 mins, AD (arrow) evoked a propagating hypoperfusion wave. Repetitive PIDs (arrowheads) were also associated with hypoperfusion. In the core and penumbra, each PID caused a permanent stepwise reduction in residual CBF (horizontal dotted lines). In the nonischemic cortex, episodic hypoperfusion was followed by hyperemia. t, Post-CSD oligemia.
In the nonischemic cortex, PIDs typically caused a brief hypoperfusion followed by longer-lasting hyperemia (Figure 4), similar to CSD in the normal mouse cortex (Ayata et al, 2004b). In the ischemic cortex, the hyperemic component was absent in both core and penumbra, and PIDs were mainly associated with prolonged hypoperfusion (19% and 32% CBF decrease in the core and penumbra, respectively). Cerebral blood flow appeared to be permanently reduced in the wake of each PID wave in both penumbra and core (Figure 4, horizontal dotted lines).
Anoxic depolarization more than doubled the cortical territory with residual CBF ≤ 20% (1.5 ± 0.8 mm2 before AD to 3.6 ± 1.3 mm2 after AD; P < 0.01, n = 15; see Figure 5 for representative images and tracings). Furthermore, each PID expanded this area, and did so in a stepwise fashion (0.3 ± 0.4 mm2). In the interval between the PIDs, the area with residual CBF < 20% decreased by 0.2 ± 0.3 mm2 (P < 0.01 PID versus no PID; Figure 5).
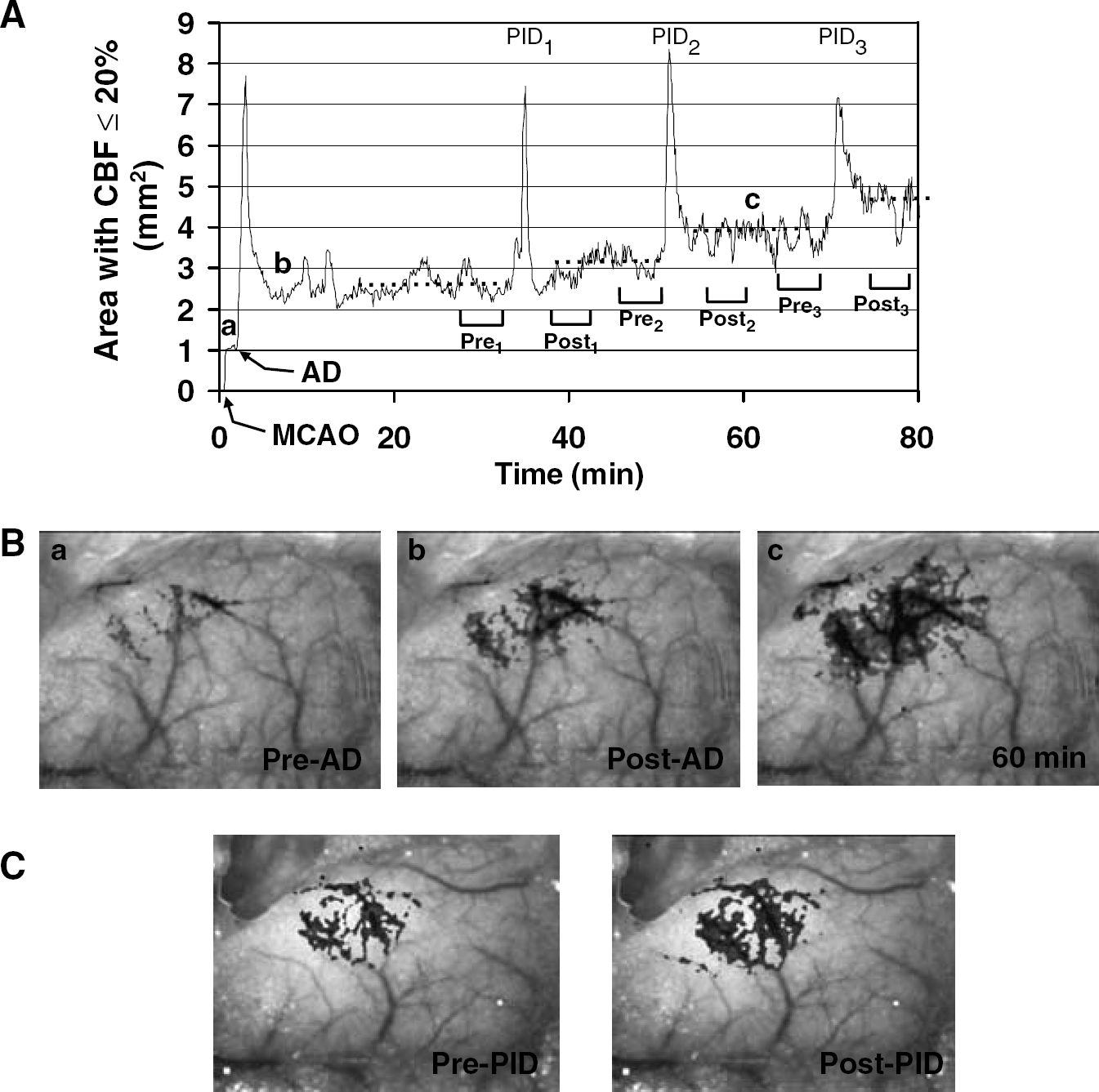
Expansion of severely hypoperfused cortex during AD and PIDs.
To determine whether neuroprotective agents ameliorate the unfavorable vasoconstrictive effect of ischemic depolarizing events and suppress the expansion of hypoperfused territory, we treated mice with N-methyl-D-aspartate (NMDA) receptor blockers MK-801 and 7-chlorokynurenic acid, σ-1 receptor agonists dextromethorphan and carbetapentane, or AMPA receptor blocker NBQX. MK-801 (5 mg/kg, n = 9) diminished the hypoperfusion associated with AD and PIDs in both core and penumbra (Figure 6). When analyzed two-dimensionally, MK-801 significantly attenuated the expansion of severe CBF deficit during AD (Figure 7, arrow), and completely blocked it during PIDs (0.1 ± 0.1 mm2 decrease in the territory with residual CBF < 20%, compared with 0.3 ± 0.4 mm2 increase in controls, P < 0.01). By so doing, MK-801 prevented the expansion of cortex with severe CBF deficit when examined over 90 mins (Figure 7). A lower dose (1 mg/kg, n = 5) seemed equally effective (Figure 8), although at this dose MK-801 did not prevent the spread of PIDs into the nonischemic cortex.
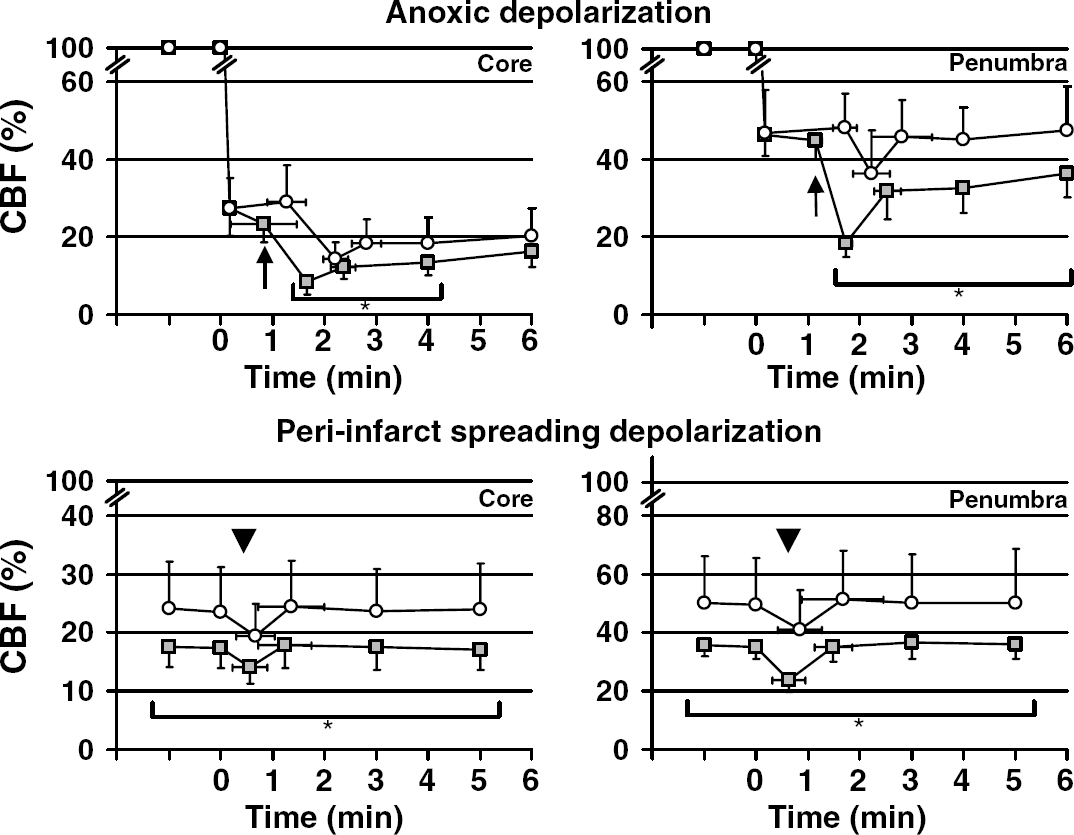
MK-801 ameliorated CBF worsening during AD and PIDs. The time course showing the impact of MK-801 on the abrupt secondary hypoperfusion associated with AD (upper panel, arrow) and PIDs (lower panel, arrowhead), in control (square) and MK-801 groups (5 mg/kg, circle). MK-801 administered 1 h before dMCAO did not significantly alter the onset of secondary hypoperfusion associated with AD in core, but delayed its spread into penumbra by 1 min (P < 0.01). MK-801 ameliorated the hypoperfusion in both core and penumbra (upper panels). Cerebral blood flow was 36% and 39% higher in the core and penumbra, respectively, in the MK-801 group compared with controls at 4 mins. This corresponded to 6% and 13% higher residual CBF in absolute terms. MK-801 did not reduce the frequency of repetitive episodic hypoperfusions (2.8 ± 1.2/h) compared with controls (2.9 ± 1.8/h), but completely prevented their spread into the nonischemic cortex; therefore, episodic hypoperfusion waves spread only within the penumbra in the MK-801 group. MK-801 ameliorated the hypoperfusion associated with PIDs in both the core (lower left) and penumbra (lower right). During PIDs, CBF in the penumbra decreased by 32% in control, and 17% in the MK-801 group (P < 0.05). Vertical and horizontal error bars indicate the standard deviations for the amplitude of CBF changes, and their latency from the onset of dMCAO (upper panel) or of hypoperfusion (lower panel), respectively. *P < 0.05, MK-801 versus controls.
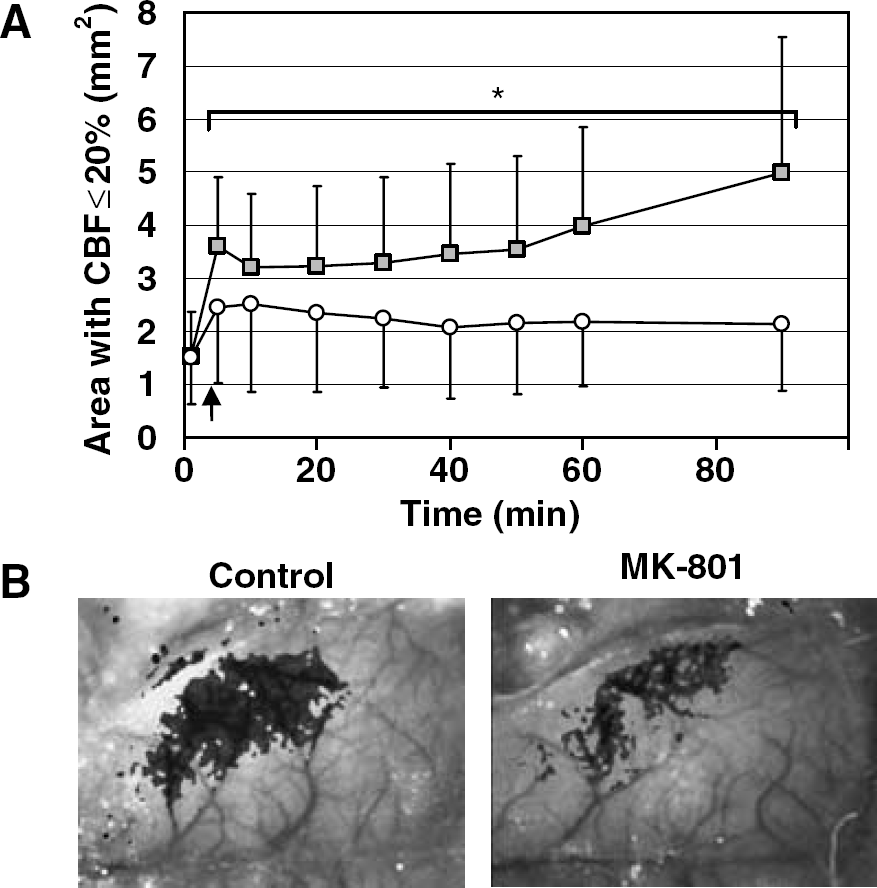
MK-801 prevented the expansion of severely hypoperfused cortex.
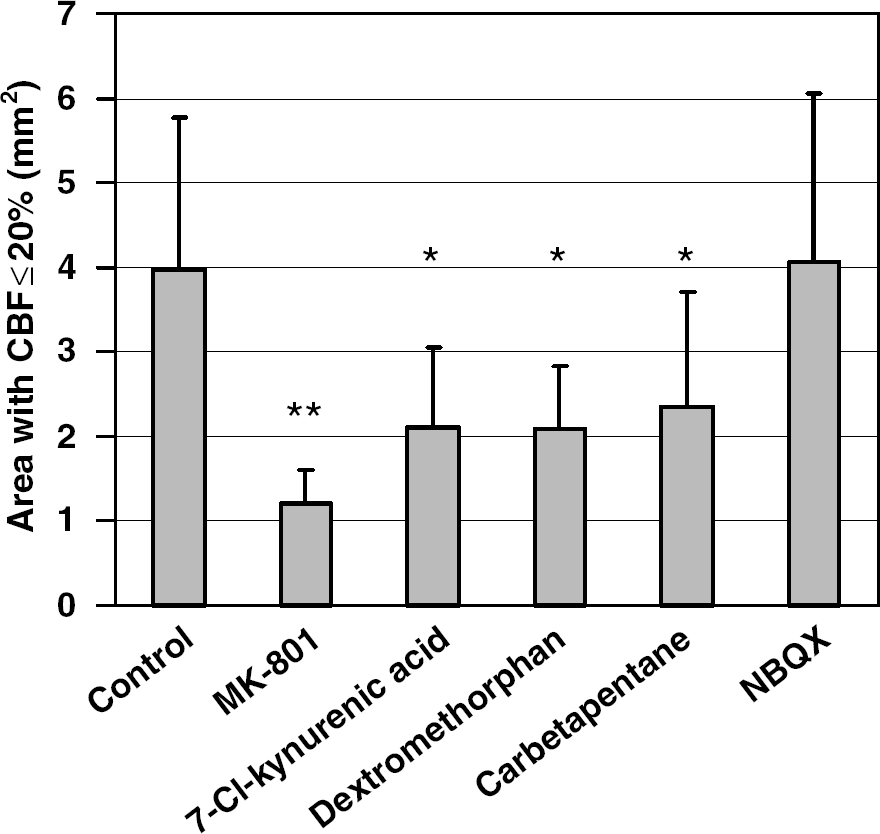
NMDA receptor inhibitors and σ-receptor agonists, but not AMPA receptor inhibitor, preserved CBF during focal cerebral ischemia. The area of severe CBF deficit (i.e., ≤ 20% residual CBF) was 70%, 47%, 48% and 41% smaller in MK-801 (1 mg/kg, n = 5), 7-chlorokynurenic acid (100mg/kg, n = 6), dextromethorphan (10 mg/kg, n = 6), and carbetapentane (10 mg/kg, n = 6), respectively, compared with controls (n = 15) 60 mins after dMCAO. NBQX (60 mg/kg) did not prevent the expansion of CBF deficit. Drugs were administered 1 h before dMCAO. *P < 0.05 and **P < 0.01 versus control.
Like MK-801, the NMDA receptor glycine site antagonist 7-chlorokynurenic acid (100 mg/kg, n = 6), the σ-1 receptor agonist and NMDA receptor antagonist dextromethorphan (10 mg/kg, n = 6), and the selective σ-1 receptor agonist carbetapentane (10 mg/kg, n = 6) also prevented the expansion of severely hypoperfused cortex (Figure 8). At 60 mins after dMCAO, the area of cortex with ≤ 20% residual CBF was approximately 50% smaller in 7-chlorokynurenic acid, dextromethorphan and carbetapentane groups compared with controls (P < 0.05). In contrast, AMPA receptor blocker NBQX (60 or 120 mg/kg, n = 5 and 4, respectively) did not preserve CBF (Figure 8). None of these drugs reduced the frequency of PIDs, or prevented their spread into the nonischemic cortex.
To determine whether the CBF preservation by these drugs corresponded to their neuroprotective efficacy, we administered MK-801 (5 mg/kg, n = 5) or NBQX (60 mg/kg, n = 5) 1 h before permanent dMCAO, and measured infarct size 24 h later. MK-801 decreased the infarct size by 70%, whereas NBQX did not (Figure 9).
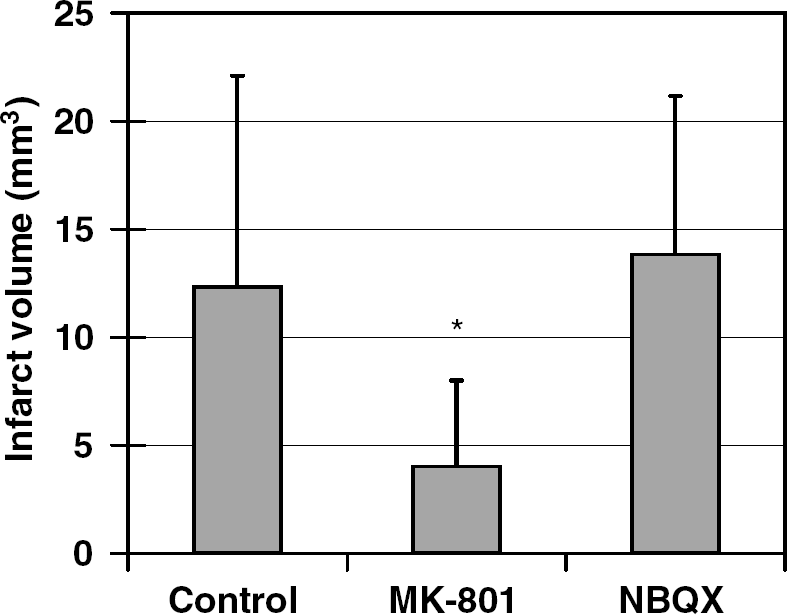
NMDA, but not AMPA receptor, inhibition reduced infarct size. MK-801 (5 mg/kg, n = 5) reduced infarct size by 70% compared with controls (n = 5), whereas NBQX (60 mg/kg, n = 5) was ineffective. Infarct size was measured 24 h after dMCAO using TTC staining in coronal sections. Owing to the small infarct size in this dMCAO model, only direct infarct volume was calculated. The large standard deviations are typical for this mouse dMCAO model. *P < 0.05 versus control and NBQX.
Discussion
In this study, we provide evidence for a vasoconstrictive form of neurovascular coupling in response to intense ischemic neuronal and astrocytic depolarization, as a novel hemodynamic mechanism by which focal ischemic depolarizations worsen outcome. Our data show that AD and PIDs cause vasoconstriction, worsen CBF and expand the area of severely hypoperfused cortex (i.e., CBF < 20%) in a stepwise fashion. By doing this, ischemic depolarizing events negatively impact tissue outcome by decreasing the blood supply, as well as increasing the energy demand. Our data also suggest that neuroprotective CSD inhibitors (i.e., MK-801, 7-chlorokynurenic acid, dextromethorphan and carbetapentane) prevent the CBF worsening, and that hemodynamic improvement is an important determinant of their neuroprotective efficacy.
Understanding the hemodynamic evolution of acute focal cerebral ischemia has so far been limited by poor spatial and/or temporal resolution of existing techniques. For example, indicator perfusion techniques lacked temporal resolution and, therefore, did not provide information about the dynamic CBF changes during the acute stage of focal ischemia (McColl et al, 2004; Selman et al, 1990). Similarly, episodic hypoperfusion was only occasionally observed during PIDs, probably owing to the lack of spatial resolution with laser Doppler flowmetry (Back et al, 1994; Iijima et al, 1992; Mies et al, 1994; Nallet et al, 2000; Ohta et al, 2001). In our study, we showed the adverse consequences of ischemic depolarizations on CBF using a novel real-time CBF imaging technique with high spatial and temporal resolution (12 mm/pixel, one image every 7.5 secs).
The CBF response to spreading depression in nonischemic mouse cortex is characterized by hypoperfusion, strikingly different from the large hyperemic response in rats (Ayata et al, 2004b). This raises the possibility that vasoconstrictive coupling during focal ischemic depolarizations is limited to mice; however, a review of the literature reveals both direct and indirect evidence that strongly argue against this possibility. For example, PIDs in cats were often associated with waves of hypoperfusion; the authors observed that persistent depolarization caused a secondary regional CBF decrease in the periinfarct zone (Ohta et al, 2001). Others also occasionally observed episodic hypoperfusion during PIDs in rat cortex (Back et al, 1994). Using reflectance spectroscopy, transient, spreading episodes of increased NADH fluorescence were recorded within penumbra in cats, suggesting that PIDs reduce oxygen availability, possibly due to reduced CBF (Strong et al, 1996). This was later confirmed in preliminary experiments using LSF, where PIDs were associated with propagating hypoperfusion in penumbra (Strong et al, 2003). Direct visualization of the ischemic cortex through a closed cranial window showed that during PIDs capillary erythrocyte velocities are transiently reduced in the penumbra, and flow direction reversed in rats (Pinard et al, 2002). Although authors attributed their findings to a steal phenomenon caused by hyperemia in the nonischemic cortex, our data show that the penumbral hypoperfusion associated with PIDs precede the delayed and longer-lasting hyperemia in nonischemic cortex (Figure 4), and thus suggest that hypoperfusion is caused by vasoconstriction rather than a steal towards the nonischemic cortex. This conclusion is further supported by data showing four-fold increase in cerebrovascular resistance due to arteriolar constriction during 10 mins of forebrain ischemia in rats (Siemkowicz, 1980). Altogether, these data suggest that vasoconstrictive neurovascular coupling takes place during ischemic neuronal and astrocytic depolarization, in species with either lissencephalic or gyrencephalic brains.
Vasoconstriction in association with AD was observed in studies using cerebral blood volume (CBV) as an indirect measure. Cerebral blood volume was greatly reduced specifically in the core (i.e., AD with restricted water diffusion) in both rats and cats using arterial spin labeling and steady-state or dynamic susceptibility contrast MRI (Caramia et al, 1998; Zaharchuk et al, 2000). The decrease in microvascular CBV was progressive, and was a good predictor of lesion expansion and final infarct size in rats (Zaharchuk et al, 2000). The same relationship between AD and severely reduced CBV was also observed during hyperacute stroke in humans, wherein reduced CBV was most closely correlated with restricted water diffusion as an indicator of AD (Sorensen et al, 1999).
Intense neuronal and astrocytic depolarization such as CSD is usually a potent stimulus to increase CBF in normal cortex. However, a hyperemic response to CSD depends on the physiological status of tissue. Under pathological conditions, such as hypoxia or partial ischemia, vasoconstriction and hypoperfusion become the predominant vascular response to CSD, rather than hyperemia (Sonn and Mayevsky, 2000). Furthermore, episodic vasoconstriction was shown during intense depolarizations in traumatized human brain (Mayevsky et al, 1996).
The mediators of vasoconstrictive neurovascular coupling during AD and PIDs are not known. [K+]e is implicated, because the onset of hypoperfusion and the DC shift, and hence the [K +]e surge up to 80 mmol/L (Nedergaard and Hansen, 1993; Somjen, 2001), are temporally coincident. [K +]e higher than 20 mmol/L directly depolarizes cerebrovascular smooth muscle and increases [Ca2+]i via voltagegated Ca2 + channels and intracellular stores, thus causing vasoconstriction (i.e., electromechanical coupling). Elevated [K +]e also depolarizes endothelial cells, reduces [Ca2 +]i and inhibits endothelium-dependent relaxations (Nilius and Droogmans, 2001; Seol et al, 2004). Furthermore, hypoperfusion during intense depolarization and vasoconstriction of isolated vessels in response to elevated [K +]e are both augmented by nitric oxide synthase inhibition (Ayata et al, 2004b; Dreier et al, 1995; Duckrow, 1993; Fabricius et al, 1995; Schuh-Hofer et al, 2001). In a series of elegant studies using cranial window in rats, Dreier et al (1998, 2000, 2001, 2002) have shown that the hyperemic response to CSD in nonischemic cortex transforms into a spreading hypoperfusion (i.e., ‘inverse coupling’) if [K +]e is artificially elevated in the superfusion solution and either nitric oxide synthesis inhibitors or scavengers are coadministered (Petzold et al, 2005). In this respect, vasoconstrictive neurovascular coupling in ischemic mouse brain might be analogous to inverse neurovascular coupling observed in rat cortex. The first of the two factors required for inverse coupling to occur in the nonischemic cortex (i.e., elevated [K +]e) mimics the core and penumbra conditions. Increased, rather than decreased, nitric oxide production was reported in ischemic tissue acutely after MCAO (Goyagi et al, 2001; Lin et al, 1996; Malinski et al, 1993). However, changes in nitric oxide production may depend on the severity of ischemia, since reduced nitric oxide production was observed when measurements were made within the severely ischemic cortex undergoing AD (Ohta et al, 1997). It is possible that endothelial nitric oxide production is selectively inhibited in ischemic tissue (Rikitake et al, 2005; Takemoto et al, 2002), and sets the stage for vasoconstriction during ischemic depolarizations. Although inverse coupling in nonischemic cortex, with artificially elevated [K +]e and reduced nitric oxide, has been shown to cause cortical spreading ischemia and neuronal damage (Dreier et al, 2000), the novel observation in our study is that a similar vasoconstrictive coupling mechanism is operational in focal cerebral ischemia and contributes to infarct expansion.
In addition to elevated [K +]e, there might be vasoconstrictor mediators (e.g., endothelin, neuropeptide Y, thromboxane A2) released during ischemic depolarizing events either from the depolarized tissue or from the vascular structures (Rubanyi and Vanhoutte, 1985). Recently, elevations in astrocytic [Ca2 +]i were shown to cause vasoconstriction via increased 20-HETE production (Mulligan and Mac Vicar, 2004). Astrocytic [Ca2 +]i rise occurs during intense neuronal and astrocytic depolarizations, and might be a vasoconstrictive mechanism operational in cerebral ischemia, along with microvascular compression by acute swelling of astrocytic end-feet (Simard and Nedergaard, 2004). Lastly, physical factors such as low luminal perfusion pressure and shear stress may also promote vasoconstriction during AD and PIDs.
The expansion of the area of severely ischemic cortex was markedly attenuated by neuroprotective drugs MK-801, 7-chlorokynurenic acid, dextromethorphan and carbetapentane, but not by NBQX. The CBF preservation by these neuroprotective agents appeared to be more prominent in penumbra than core (see Figure 6); however, the impact on the tissue outcome of smaller CBF increases in core might be comparable to larger increases in penumbra, since the core CBF may rise above thresholds for AD and ATP depletion (Hossmann, 1994; Mies et al, 1991). MK-801 is a noncompetitive NMDA receptor antagonist (blocks the channel pore) and 7-chlorokynurenic acid blocks the NMDA receptor glycine site; both drugs show high selectivity for NMDA receptor and inhibit CSD (Lauritzen and Hansen, 1992; Marrannes et al, 1988). As dextromethorphan is a Ca2+ channel and NMDA receptor blocker, in addition to being a σ-1 receptor agonist, we used carbetapentane, a selective σ-1 receptor agonist (Leander, 1989), and showed that it was as effective as dextromethorphan. Both σ-1 receptor agonists are potent inhibitors of CSD (Anderson and Andrew, 2002; Anderson et al, 2005); therefore, our data suggest that CBF preservation in focal ischemia is common to neuroprotective inhibitors of CSD, and not limited to NMDA receptor blockers. Both NMDA receptor blockers and σ-1 receptor agonists reduce infarct size in focal ischemia when administered before or up to 1 h after the onset of ischemia (Britton et al, 1997; Buchan et al, 1992; Chen et al, 1993; Gill et al, 1991; Ozyurt et al, 1988; Park et al, 1988; Shimizu-Sasamata et al, 1996; Steinberg et al, 1993). Although the neuroprotective mechanism is presumed to be inhibition of excitotoxicity, MK-801, dextromethorphan, and a competitive NMDA receptor antagonist CGS-19755 were shown to preserve CBF in focal ischemia (Buchan et al, 1992; Liu et al, 1997; Lo and Steinberg, 1991; Meyer et al, 1990; Ohta et al, 2001; Takizawa et al, 1991), despite the absence of their receptors on cerebral vessels (Beart et al, 1988). None of the drugs tested in our study has known direct vasoactive effects. Furthermore, they did not alter the initial CBF reduction on dMCAO. None of the drugs tested significantly reduced the frequency of PIDs, and only MK-801 at a dose of 5 mg/kg prevented their spread into the nonischemic cortex. Therefore, CBF preservation by CSD inhibitors was not due to blockade of the remote vasoconstrictive effects of PIDs on collateral blood supply. The mechanism of CBF preservation by these drugs may relate to attenuation of [K +]e increase during neuronal and astrocytic depolarization, although, when [K +]e is artificially increased and nitric oxide is scavenged in rat cortex using a cranial window (i.e., a model of inverse coupling), NMDA receptor blockade did not prevent the occurrence of spreading ischemia or attenuate the rise in [K +]e during these events (Petzold et al, 2005). NMDA receptor antagonists elevate the [K +]e threshold for neuronal and astrocytic depolarization both in vivo and in slices (Katayama et al, 1991; Petzold et al, 2005). Therefore, it is also possible that neuroprotective agents prevent the catastrophic [K+]e rise in moderately ischemic tissue, thereby reducing the [K +]e that cerebral vessels are exposed. As astrocytes harbor NMDA and σ-1 receptors (Klouz et al, 2003), the increase in astrocytic [Ca2 +]i and 20-HETE production might also be inhibited by NMDA receptor antagonists and σ-1 receptor agonists, thus attenuating the vasoconstriction (Mulligan and MacVicar, 2004).
Unlike NMDA receptors, AMPA/kainate receptors do not impact the initiation or propagation of CSD (Kruger et al, 1999; Lauritzen and Hansen, 1992; Nellgard and Wieloch, 1992). NBQX is a competitive AMPA/kainate glutamate receptor antagonist that is neuroprotective in some, but not all, focal ischemia studies (Buchan et al, 1991; DeGraba et al, 1994; Graham et al, 1996; Lo et al, 1997; Pitsikas et al, 2001; Smith and Meldrum, 1993). Unlike MK-801, and consistent with our data, AMPA/kainate antagonists do not appear to impact CBF (Buchan et al, 1991; Mies et al, 1994). In our study, NBQX did not preserve CBF nor reduce infarct size, suggesting that CBF preservation by neuroprotective agents might be an important mechanism of infarct reduction. Regardless of its mechanism, this secondary hemodynamic benefit conferred by CSD inhibitors is a novel mechanism that might be shared by other neuroprotective drugs. Although PIDs are known to occur for up to 24 h (Hartings et al, 2003), the therapeutic window for neuroprotective drugs is relatively brief (e.g., approximately 1 h for MK-801) (Hatfield et al, 1992; Lyden et al, 1995; Ma et al, 1998; Margaill et al, 1996). Therefore, attenuation of vasoconstrictive coupling, as a relevant neuroprotective mechanism in vivo, appears to be effective only during the acute phase of ischemia. In this study, we performed imaging for up to 90 mins and only tested the effects of pretreatment with MK-801. More work is needed to determine whether PIDs continue to worsen CBF after the first 90 mins, and whether delayed postischemic administration of MK-801 impacts CBF.
Although much effort has been directed towards developing neuroprotective agents in acute stroke, the success rate in human trials has been generally disappointing. In experimental animals, neuroprotectants reduce infarct volume concentrically. Often there is pan-necrosis within the ischemic lesion (neurons, glia and blood vessels), whereas, surrounding the lesion, cells are rescued and appear normal regardless of whether they express the receptors targeted by neuroprotectants. Despite the clear demonstration in vitro that neuroprotectants target specific receptors (and by so doing ameliorate cell death), the pattern of neuroprotection in vivo is one that might be expected from interventions that augment CBF. If the mechanism of action of neuroprotective agents (e.g. NMDA receptor antagonists) only involved occupancy of specific receptors, one might expect a pattern similar to that seen in culture models of oxygen-glucose deprivation, where surviving cells are interspersed among nonviable ones, a pattern similar to that seen in neuroprotection in culture models of oxygen-glucose deprivation. Our data suggest that neuroprotective agents such as MK-801 improve the ischemic CBF deficit by stabilizing neuronal and astrocytic membranes, and, therefore, interfering with the adverse vasoconstrictive effects of tissue depolarization. Such a secondary CBF-dependent benefit might be the critical determinant of a concentric pattern of infarct reduction by neuroprotective drugs primarily targeting neurons and glia.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.