
Abstract
The chemokine CCL2 is considered as one of the main effectors driving postischemic infiltration of monocytes into the brain parenchyma. New experimental data, however, suggest that CCL2 could also participate in blood–brain barrier (BBB) ‘opening’ during the transmigration of monocytes. The current study examines the role of CCL2 in regulating BBB permeability after ischemia in vitro. To address this issue, an in vitro BBB model (coculture of astrocytes and brain endothelial cells) was subjected to 5 h of oxygen glucose deprivation, followed by reoxgenation (in vitro ischemia/reperfusion (I/R)) for 0 to 48 h. During reperfusion, there was a biphasic enhancement of barrier permeability, with a 200-fold increase in barrier permeability to FITC-albumin at 6 h and a further period of disruption around 24 h. The latter coincided with increased secretion of CCL2 by both astrocytes and brain endothelial cells and increased levels of the CCL2 receptor, CCR2. Applying antisense oligonucleotide or neutralizing antibody to block CCL2 significantly decreased I/R-induced enhancement of BBB permeability (approximately twofold) and redistribution of tight-junction (TJ) proteins (occludin, zonula occluden-1, 2, claudin-5). Similarly, absence of CCR2 from endothelial cells caused stabilization of TJ complexes and decreased the permeability of brain endothelial barrier during in vitro I/R. These data suggest CCL2/CCR2 has an important role in regulating brain endothelial permeability and might be a potential novel therapeutic target for stroke.
Introduction
During central nervous system (CNS) ischemia/reperfusion (I/R) injury, blood–brain barrier (BBB) disruption can be: (a) permanent, involving endothelial cell swelling, astrocyte detachment and possible blood vessel rupture in the ischemic area and (b) transient, consisting of local brain endothelial hyperpermeability to macromolecules in the periinfarct area (Dirnagl et al, 1999; Petty and Wettstein, 2001; Sharp et al, 2000). This transient disruption or BBB ‘opening’, which mostly occurs during reperfusion into and around the damaged area, generally has a biphasic character (Kuroiwa et al, 1985). In the early phase (the first 2 to 3 h after ischemic onset), BBB ‘opening’ is associated with the generation of free radicals and activation of matrix metalloproteinases. In contrast, in the later phase (24 to 48 h), ‘opening’ is associated with the production of proinflammatory cytokines (interleukin (IL)-1β and tumor necrosis factor (TNF)-α) and regulators of leukocyte trafficking (such as intercellular adhesion molecule-1 (ICAM-1) and IL-8), as well as leukocyte migration (Abbott, 2000; Wojciak-Stothard et al, 1998; Dietrich, 2002; Matsumoto et al, 1997). At the morphologic level, enhanced BBB permeability is correlated with reorganization of the endothelial actin cytoskeleton and a redistribution of tight and adherence junction proteins from the plasma membrane to the cytoplasm (Bolton et al, 1998; Couraud, 1998).
Chemokines are proinflammatory mediators mainly involved in selectively directing leukocyte migration into the brain parenchyma. High levels of chemokines are found in the brain perivascular space in many CNS pathologic states accompanied by inflammation, and are mostly associated with formation of a chemoattractant gradient for leukocyte influx (Rollins, 1997; Murphy, 1994). One of the most common chemokines expressed in the CNS during inflammation is monocyte chemoattractant protein-1 (MCP-1, CCL2). CCL2 is a member of the CC subfamily of chemokines (Yoshie et al, 1997). There is strong evidence that CCL2 is involved in the recruitment of both monocytes/macrophages and activated lymphocytes into the brain in neuropathologic states (Menicken et al, 1999). Experiments on I/R brain injury have found CCL2 mRNA in the core of the infarct within the first 6 h after ischemia. In the ischemic penumbra, CCL2 production was significant within 12 to 48 h (peak of expression) and persists for up to 5 days (Kim et al, 1995; Wang et al, 1995; Che et al, 2001). The presence of CCL2 was highly correlated, both temporally and spatially, with upregulation of cytokines and adhesion molecules as well as an increase in leukocyte migration into the ischemic area (Kim, 1996; Stanimirovic and Satoh, 2000). Clinical studies have also found CCL2 in the colony-stimulating factor (CSF) of ischemic stroke patients 24 h after the onset of neurologic symptoms (Losy and Zaremba, 2001). Mice lacking CCL2 (CCL2–/–) have smaller infarct volumes 24 h after MCA occlusion, as well as a reduced inflammatory response compared with wild-type controls (CCL2 +/+) (Hughes et al, 2002).
In combination, these findings strongly suggest that the chemokine CCL2 plays a crucial role in development of the postischemic inflammatory response, regulating recruitment of monocytes/macrophages at the site of injury, and expression of adhesion molecules and cytokines. Recent studies also indicate that chemokines (including CCL2) can regulate endothelial cell permeability, further highlighting their potential contribution to BBB disruption and enhancement of leukocyte entry during inflammatory responses (Matsumoto et al, 1997; Schraufstatter et al, 2001; Stamatovic et al, 2003, 2005). Based on these findings, we hypothesized that CCL2 and its receptor CCR2, which is expressed on brain endothelial cells, have important roles in regulating BBB permeability during the postischemic inflammatory response and that both of them could be potential therapeutic targets for maintaining BBB integrity during I/R.
The experiments in this study, therefore, aimed at elucidating the role of CCL2/CCR2 in BBB ‘opening’ during I/R. The experiments use an in vitro model of the BBB (brain endothelial cell/astrocyte coculture) and an in vitro model of I/R (oxygen glucose deprivation plus reoxygenation).
Materials and methods
Brain Endothelial Cell Culture
Mouse
Astrocyte Cell Culture
Cerebral astrocyte cultures were prepared from 1-day-old mice according to the method of Hertz, with minor modifications (Hertz et al, 1982; Andjelkovic et al, 1999). The mice were either CD-1 strain, CCR2–/– or CCR2 +/+ mice pups on a 50:50 C57BL/6 ×129Sv background. Brain tissue was mechanically dissociated in Ca2+/Mg2+ -free HBSS, 0.035% sodium bicarbonate and 1 mmol/L sodium pyruvate, and then digested with 0.25% Trypsin-EDTA (Invitrogen Corp., Carlsbad, CA, USA) at 37°C for 30 mins, and 10 U/μ1 DNAse I (Sigma Chemical, St Louis, MO, USA). Trypsin digestion was stopped by washing cells with the Ca2+/Mg2+ -free HBSS supplemented with 10% fetal bovine serum (FBS), followed by low-speed centrifugation to remove debris. To obtain single-cell suspensions, the brain tissue was triturated with solution Ca2+/Mg2+ -free HBSS supplemented 10 U/μL DNAse I and 3.8% w/w MgSO4. Cells were then washed twice in HBSS, resuspended in complete astrocyte media (DMEM, 10% inactivated FBS, 1 × glutamine, 1 × A/A, (Invitrogen Corp, Carlsbad, CA, USA)) and seeded at a density of 2.5 × 107 per 75 cm2 tissue culture flask. Monolayers were grown under an atmosphere of 5% CO2/95% air at 37°C. At 2 weeks after initial plating, flasks of cells were shaken at 220r.p.m. for 2 h at 4°C. After this time, the supernatant, containing mainly microglia, was removed. Cultures were then shaken for an additional 18 h at 37°C to remove neurons. After this time, cultures were approximately 99% positive for GFAP by immunocytochemistry. First or second passages were used in the experiments.
In Vitro Model of the Blood-Brain Barrier. Coculture of Astrocytes and Mouse Brain Microvascular Endothelial Cells
For coculturing, the cells were plated on both sides of polyester membranes with 0.4 or 12 μm pores and an insert diameter of 12 or 24 mm (Transwell, Costar Corning Inc, Corning, NY, USA). The upper (endothelial) surface was first coated with collagen IV (BD Bioscience, San Jose, CA, USA). Mouse astrocytes (1 × 104) were then seeded on the underside on the insert. After 6 h, when cells were attached, inserts were turned over and placed in a 12- or 6-well culture plate. Mouse brain microvascular endothelial cells (densities of 2.5 × 10/cm2) were then seeded on the upper side of each insert (Pardridge, 1999; Kondo et al, 1996). Cocultures were maintained at 37°C under normoxic conditions of 95% air/5% CO2. At 4–7 days after initial coculturing with astrocytes, transendothelial electrical resistances (TEERs) were ~300 Ω cm2 and they were utilized for experiments.
Oxygen-Glucose Deprivation Model of Ischemia/Reperfusion Injury
Astrocyte/mBMEC cocultures were subjected to oxygen-glucose deprivation (OGD) injury. The OGD injury was initiated by placing cocultures in an anaerobic chamber (Coy Labarotory, Great Lake MI, USA) and in the presence of OGD media (DMEM glucose-free solution purged with an anaerobic gas mixture, 5% CO2/95% N and 10% H2, to remove residual oxygen) for 5 h at 37°C. This condition was denoted as the ischemic condition. After that, cocultures were removed from the chamber, the exposure medium exchanged with oxygenated DMEM, and the cells placed in an incubator at 37°C, with 5% C02-containing normal oxygen (21% O2) to mimic the reperfusion condition. The reperfusion period was 0 to 48 h. For every set of experiments, a cell viability assay was performed (Lactate Dehydrogenase (LDH) Assay, Cytotoxicity Detection Kit, Promega WI). Only cell cultures with the similar LDH release under control conditions were selected for further experiments.
Transendothelial Electrical Resistance
Electrical resistances of established astrocyte/mBMEC cocultures were measured with an ENDOHM apparatus (World Precision Instruments Inc, Sarasota, FL, USA). Transendothelial electrical resistance was measured at 6-h intervals during reperfusion. The resistance of a blank filter was subtracted from the measured values to calculate final TEER values (cm2) (Kondo et al, 1996). All experiments were performed in triplicate.
In Vitro Permeability Assay
The permeability of the cocultures was assessed with FITC-albumin at 6-h intervals after reperfusion (0 to 48 h). At those different time points, FITC-albumin (Sigma Chemical Co., St Louis, MO, USA) was added to the apical (donor) chamber containing 0.4 mL of DMEM (Invitrogen Corp. Carlsbad, CA, USA). At 2 h after FITC-albumin addition, the basal (receiving) chamber, containing 1.2 mL of DMEM, was sampled (Kazakoff et al, 1995). The permeability coefficient (PC; cm/min) of the monolayer during the time interval (T) was calculated as:
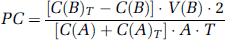
where C(B) and C(B) T are the concentrations (ng/mL) of FITC-albumin in the basal (receiving) chamber at the start and at the end of the time interval, respectively, and V(B) is the volume of the basal chamber (in mL). C(A) and C(A) T are, respectively, the concentrations of FITC-albumin in the apical (donor) chamber at the start and at the end of the time interval and (C(A) + C(A) T )/2 is the average concentration over the time interval. T is the duration of the time interval (2 h), while A is the area of the filter (in cm2). All samples were read on a fluorescent reader (Bio-Tek Instruments, Inc., Winooski, VT, USA; emission 485 and excitation 540 nm). The concentration of FITC-albumin in samples was calculated from a standard curve derived using known concentrations of tracer.
Semi-Quantitative Reverse Transcriptase-Polymerase Chain Reaction (PCR)
Total RNA was prepared using TRIZOL reagents (Invitrogen Corp., Carlsbad, CA, USA) according to the manufacturer's instructions. Aliquots (1 μg) of RNA were reverse transcribed (RT) with an Invitrogen cDNA synthesis kit (Invitrogen Corp., Carlsbad, CA, USA). A mix of equal amounts of mouse primers for β-actin (Biosource International Inc., Camarillo, CA, USA) and either CCL2 (5′-CTCACCTGCTGCTACTCATTC-3′ and 5′-GCATGAGGT GGTTGTGAAAAA-3′) or CCR2 (SuperArray, Frederick, MD, USA) was used to prime PCR. A total of 30 amplification cycles for CCL2 and 28 cycles for CCR2 were applied. The PCR cycles included 1 min denaturation at 94°C, 1 mins annealing at 55°C and 1 mins extension at 72°C, except for the first cycle with 2 mins denaturation and the last cycle with 10 mins elongation. The PCR products were resolved using electrophoresis in a 2% agarose gel in 1 × TBE buffer (Tris-HCl/EDTA/boric acid, pH 8). The gel was stained with ethidium bromide and photographed.
CCR2 Binding Assay
Quantitative immunofluorescence using a CCR2 receptor detection kit (R&D System Inc. Minneapolis, MN, USA) was used to show binding of CCL2 to CCR2 on mBMEC or astrocytes. Briefly, mBMEC or astrocytes in coculture were incubated with biotinylated recombinant mouse MCP (MCP-1/JE, CCL2) in binding buffer (Dulbecco PBS, supplemented with 1 mmol/L glucose, Invitrogen Corp. Carlsbad, CA, USA) for 2 h at 4°C. This was followed by avidin fluorescein (R&D System Inc. Minneapolis, MN, USA) for 1 h at room temperature. Quantitative immunofluorescence was used to quantify binding of CCR2 on the mBMEC. Briefly, 250 randomly selected areas from 10 different samples were used. Mean pixel intensity value was obtained using the ImageJ software program (NIH Image, Bethesda, MD, USA). Every value was subtracted from a previously defined background.
Monocyte Chemoattractant Protein-1 ELISA
Coculture supernatants were assayed for CCL2 levels by ELISA, using the mouse CCL2 Quantikine kit (R&D System Inc., Minneapolis, MN, USA). The sensitivity of detection using this assay is 32 pg/mL.
Immunofluorescence
Cocultured cells were fixed in 4% paraformaldehyde for 20 mins at 20°C. After fixation, samples were preincubated in blocking solution (5% bovine serum albumin, 0.05% Tween and PBS), and then incubated overnight at 4°C with the primary antibody. Rabbit anti-occludin, mouse anti-ZO-1, anti-claudin-5 (Zymed Laboratories, Carlsbad, CA, USA) and anti-ZO-2 (BD Bioscience, San Jose, CA, USA) were used. Reactions were visualized by fluorescein-conjugated anti-mouse or anti-rabbit antibodies (Vector Laboratories, Burlingame, CA, USA). All samples were viewed on a confocal microscope (LSM 510 Zeiss, Germany), objective × 40, 1.3 numerical aperture.
Triton-X Extraction of Membrane-Associated Proteins
Monolayers of mBMEC (from Transwell insert) were overlaid with extraction buffer for 20 mins at 4°C on a gently rocking platform. The extraction buffer contained 0.5% Triton-X 100, 10 mmol/L Tris-HCl, pH 7.4, 100 mmol/L NaCl, 300 mmol/L sucrose, plus proteinase inhibitor mixture containing phenylmethylsulfonyl fluoride, iodoacetamide, benzamidine (each 1 μg/mL), aprotinin, leupeptin, pepstatin A (each 20 μg/mL, Roche, Indianapolis, IN, USA). The soluble supernatant was collected and this fraction was defined as the Triton-X soluble fraction. The residue of cells with well-preserved nuclei and cytoskeleton fibers adherent to the culture inserts were gently washed twice with Tris buffer saline (TBS) supplemented with the protease inhibitors aprotinin, leupeptin (each 20 μg/mL; Indianapolis, IN, USA) and then lysed with the RIPA buffer (10 mmol/L Tris; 140 mmol/L NaCl, 1% Triton, 1% Na deoxycholate, 0.1% SDS, 0.5 mmol/L phenylmethylsulfonyl fluoride and 1 μg/mL aprotinin, 1 μg/mL leupeptin (Sigma Chemical Co., St Louis, MO, USA)). The extract was collected and this fraction was defined as the Triton-X 100 insoluble fraction (Fey et al, 1984).
Western Blotting
Equivalent amounts of each protein sample were electrophoretically separated on a 5%, 7.5% or a 12% SDS-polyacrylamide gel and transferred to Trans-Blot nitrocellulose membrane (BioRad, Hercules, CA, USA). The membranes were immunoblotted with rabbit anti-occludin, mouse anti-ZO-1, anti-claudin-5 (Zymed Laboratories, Carlsbad, CA, USA) and mouse anti-ZO-2 (BD Bioscience, San Jose, CA, USA) antibodies. Immunoblots were then exposed to secondary anti-mouse or anti-rabbit-HRP conjugated antibody (Vector Laboratories, Burlingame, CA, USA) and visualized using a chemiluminescent HRP substrate kit (Pierce, Rockford, IL, USA).
Inhibition of CCL2 and CCR2; Cell Treatment
Two methods were used to inhibit CCL2 activity. Anti-rabbit CCL2 neutralizing antibody (10 μg/mL) (R&D System Inc., Minneapolis, MN, USA) was added to the incubation media at the time of reperfusion and the effect of CCL2 inhibition was evaluated at 24 h. The CCL2 activity was also inhibited by transfection with antisense CCL2 oligonucleotide (Oligos Ets, Wilsonville, OR, USA). Briefly, mBMEC and/or astrocytes were exposed for 4 h at 37°C to serum-deprived media (Opti-MEM, Invitrogen Corp., Carlsbad, CA, USA) supplemented with Lipofectin 10 μg/mL (Invitrogen Corp., Carlsbad, CA, USA) and 1 μmol/L phosphorothioated CCL2 antisense oligonucleotide (5′-AAG CGT GAC AGA GAC CTG CAT AGT CGT GG-3′). Phosphorothioated CCL2 sense oligonucleotide (5′-CCA CCA CTA TGC AGG TCT CTG TCA CGT TT-3′) was used as a control. We also performed transfection with fluorescein-conjugated CCL2 antisense and sense oligonucleotides to control for uptake of oligonucleotides. The efficiency of CCL2 inhibition was evaluated by RT-PCR and Western blot. After 6 h of transfection, cocultures were incubated with growth media for 24 h before experimental use.
Migration Assay
Cocultures of astocytes and mBMEC were established on collagen-coated Transwell plates (Transwell; Costar Corning Inc, Corning, NY, USA) as described previously, except that the membrane used in these experiments had a pore size of 12 μm. Migration assays were performed under three conditions: (1) I/R cocultures were exposed to 5 h of ischemia followed by reperfusion. (2) Control cocultures were exposed to normal oxygen and glucose supply. (3) CCL2 treatment (positive control) cocultures were exposed to murine recombinant protein CCL2 (5 nmol/L, PreproTech Inc, Rocky Hill, NJ, USA) added to the lower part of Transwell dual chamber system. RAW 264.7 cells (ATCC, Manassas, VA, USA) were loaded with Calcein-AM (4 μmol/L conc., Molecular Probes Carlsbad, CA, USA) for 30 mins at 37°C in 5% CO2, followed by washing for 45 mins. A total of 10,000 cells were added to the upper chamber after either 0, 4, 10, 16, 22, 28, 34, 40 or 46 h of reperfusion. The cells were allowed to migrate for 2 h at 37°C. Transendothelial migration was stopped by removing the insert from the lower part of Transwell system. The total number of migrated cells in the lower chamber as well as adherent cells on the surface of the brain endothelial cells (cells still adherent after two washes with PBS buffer) was evaluated using a confocal microscope (LSM 510 Zeiss, Germany) and using the Image J software program (NIH Image, Bethesda, MD, USA).
Statistics
All data in this study are presented as mean ± s.d. Data were analyzed by ANOVA, followed by a Dunnett's posthoc test (for comparison to control). Statistical significance was accepted at P < 0.05.
Results
Functional and Morphologic Alterations in Blood–Brain Barrier Integrity During Reperfusion
Exposure of endothelial/astrocyte cocultures to 5 h OGD with reoxygenation (in vitro I/R) induced marked alterations in BBB integrity (Figure 1A). A dramatic decrease in TEER was found after 6 h of reperfusion (24 ± 3 versus 295 ± 7 Ω cm2 measured in control, P < 0.001). There was then a partial recovery of integrity by 12 h of reperfusion (176 ± 16 versus 292 ± 11 Ωcm2, P < 0.001), followed by a second phase of barrier disruption by 24 h (75 ± 6 versus 283 ± 10 Ωcm2, P < 0.001).
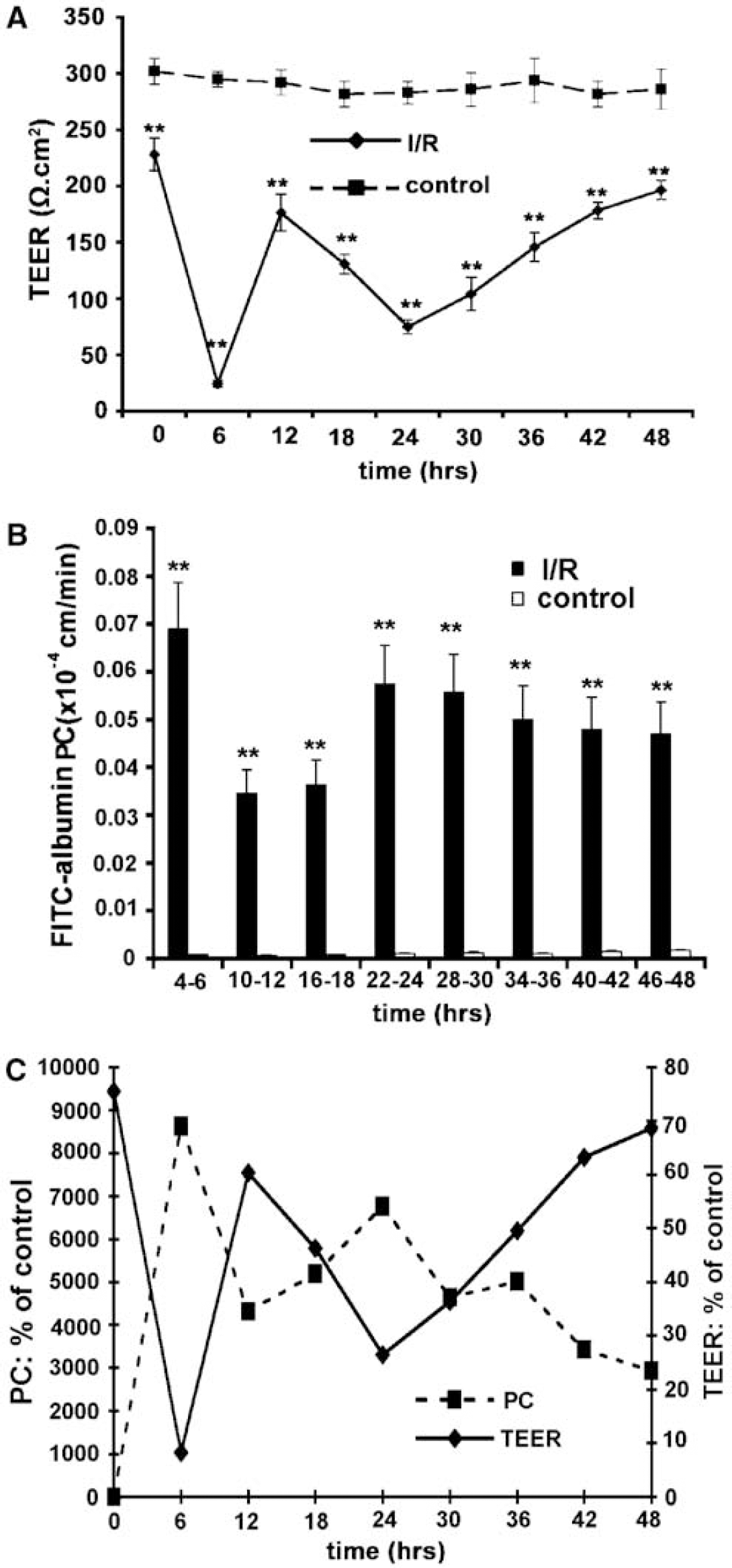
Effect of simulated in vitro I/R on brain endothelial permeability (in vitro model of the BBB). (
Similar changes were observed in FITC-albumin permeability studies. The permeability of the cocultures to FITC-albumin was significantly (~70-fold) increased between 0 and 6 h of reperfusion (Figure 1B). This was followed by a recovery of integrity between 6 and 18 h of reperfusion and then a second phase of disruption between 24 and 48 h (Figure 1B). For comparison, the % changes in FITC albumin permeability and TEER after reperfusion are shown in Figure 1C.
We further examined whether these functional alterations in BBB permeability in vitro were associated with morphologic and biochemical changes in endothelial tight-junction (TJ) structure. Figure 2A shows representative morphologic changes in the distribution of the TJ molecules occludin, ZO-1, ZO-2 and claudin-5 after 6, 12 and 24 h of reperfusion. Ischemia/reperfusion caused the loss and/or fragmentation of the staining for each of the junction proteins, particularly after 6h of reperfusion. These morphologic changes in TJ structure were confirmed by biochemical analysis. Triton-X 100 extraction of TJ molecules was used to examine possible alterations in the ‘functional’ status of these molecules. In cells not exposed to OGD, occludin, ZO-1, ZO-2 and claudin-5 were mostly extracted in the Triton-X 100 soluble fraction and also partial recovery could be seen after 12 h of reperfusion (Figure 2B). In contrast, after 6 and 24 h of reperfusion, the TJ proteins extracted in the Triton-X 100 insoluble fraction increased. This shift of TJ proteins from Triton-X 100-soluble to Triton-X 100-insoluble fraction reflects phosphorylation and association with the actin cytoskeleton (Tsukamoto and Nigam, 1997; Stamatovic et al, 2003).
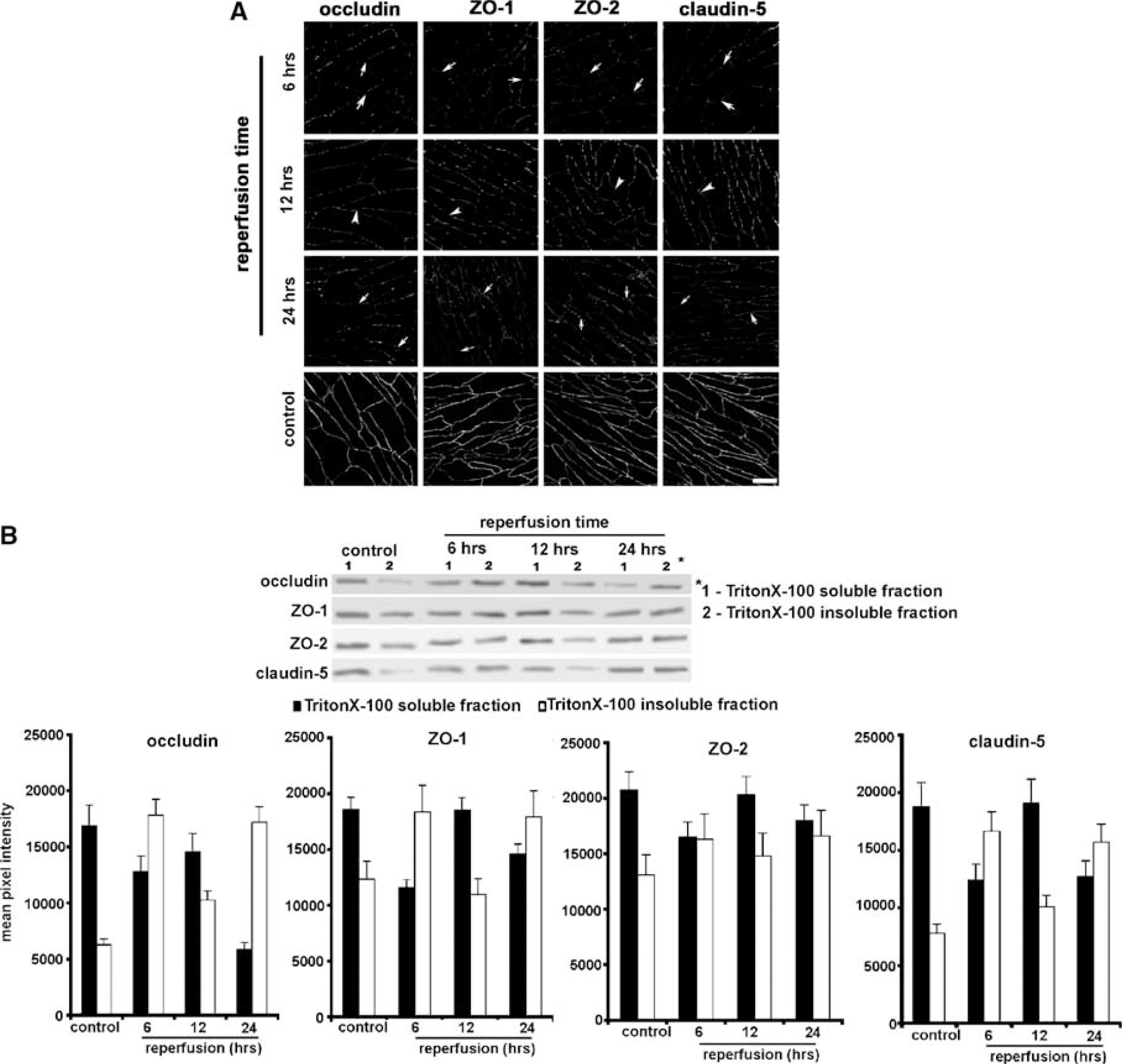
Effect of in vitro I/R on TJ protein distribution. Cocultures of mBMEC and astrocytes were subjected to 5 h of in vitro ischemia with reperfusion (0 to 48 h). (
Transmigration of Monocytes During In Vitro Ischemia/Reperfusion
In vitro, exposure of astrocyte/brain endothelial cell cocultures to OGD with reoxygenation resulted in a marked increase in transmigration of monocytic RAW 264.7 cells through the brain endothelial barrier (Figure 3). This increase was particularly pronounced after 24 to 48 h of reperfusion (up to 3000-fold increase compared with control).
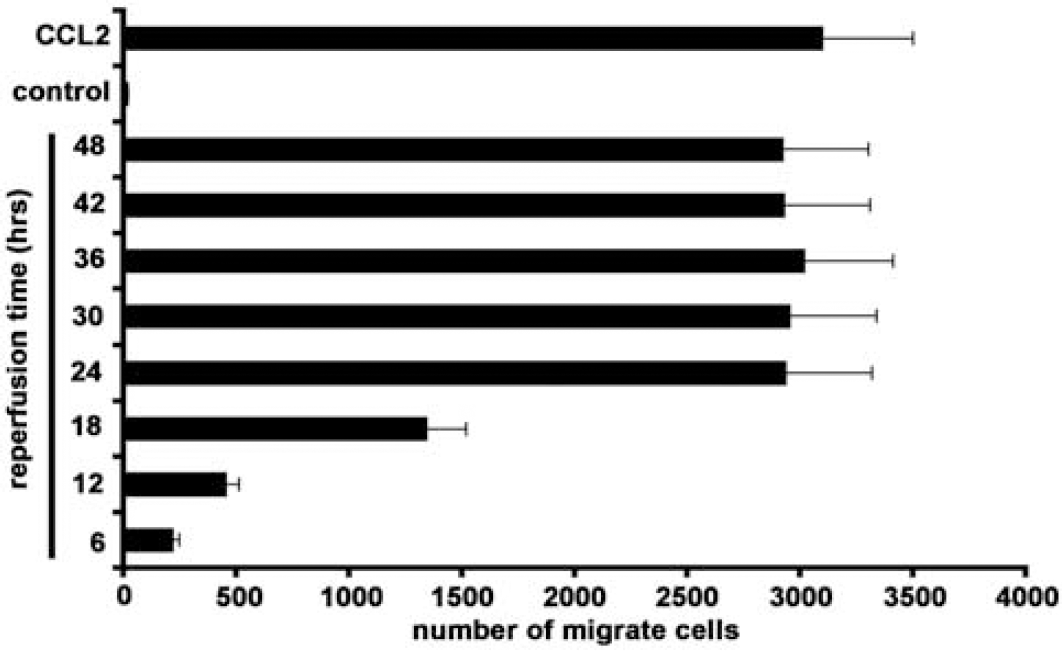
Effect of in vitro I/R on transendothelial migration of monocytic RAW 264.7 cells at the in vitro BBB. Cocultures of mBMEC and astrocytes were exposed to different durations of I/R or no ischemia (control). CCL2 indicates cocultures where murine recombinant protein CCL2 (5 nmol/L) was added in the lower chamber of Transwell system. For all experiments, monocytic RAW 264.7 cells were added to the upper chamber for 2 h and the number of migrated monocytes determined. Ischemia/reperfusion increased the number of migrating monocytes in a time-dependent fashion. The degree of migration at 24 to 48 h was similar to that found with exogenous CCL2. Values are means ± s.d.
In vitro Ischemia/Reperfusion Increases Expression of CCL2/CCR2 on Brain Endothelial Cells and Astrocytes
Exposure of mBMEC/astrocyte cocultures to in vitro I/R caused a marked increase in CCL2 mRNA levels during the reperfusion period (Figures 4A and 4B). Ischemia/reperfusion induced increases in CCL2 mRNA in both astrocytes and mBMEC, with a peak after 6 h of reperfusion (Figures 4A and 4B). The increase in CCL2 mRNA expression was associated with a progressive increase in protein levels in the coculture media during the reperfusion period (Figure 4C). Experiments with monoculture of either astrocytes or mBMEC indicated that CCL2 production by both cell types is enhanced after I/R (Figure 4C). Comparison of CCL2 production in those monocultures with the coculture experiments suggests that both cell types contribute significantly to enhanced CCL2 protein levels during a reperfusion. The increased production of CCL2 was temporally correlated with the peak in monocyte transmigration (Figure 3), suggesting that CCL2 could be the major reason for increased monocyte migration in the reperfusion period.
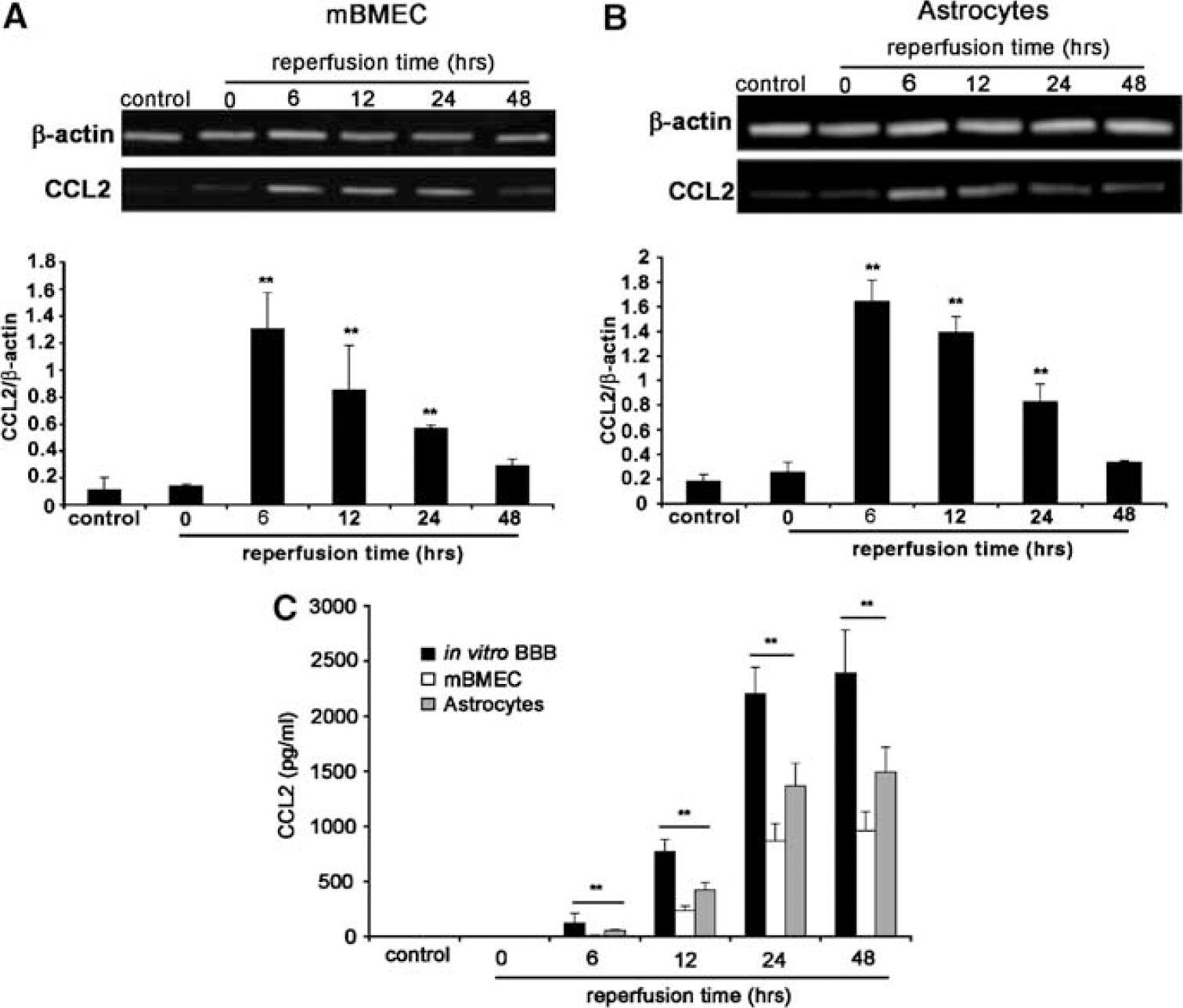
Effect of in vitro I/R on CCL2 mRNA and protein expression in brain endothelial cell/astrocyte cocultures or monocultures. Cocultures or monocultures of mBMEC or astrocytes were exposed to 5 h of ischemia and the indicated periods of reperfusion (0 to 48 h). Controls received no ischemia. In (
The increase in CCL2 expression during reperfusion was also accompanied by temporal changes in CCR2 expression, the CCL2 receptor (Figure 5). Whereas mBMEC showed a slight upregulation in CCR2 mRNA expression during reperfusion, there was a marked increase in astrocytes mRNA particularly between 6 and 24 h of reperfusion (Figures 5A and 5C). Furthermore, a CCL2-binding assay showed significant upregulation of binding on mBMEC and astrocytes in coculture after I/R, particularly between 24 and 48 h (Figures 5B and 5D). The higher CCL2 binding on both cell types was correlated with the peak in CCL2 protein levels in media. This suggests that CCL2 could be involved in regulating astrocyte and brain endothelial cell ‘activity’ during reperfusion.
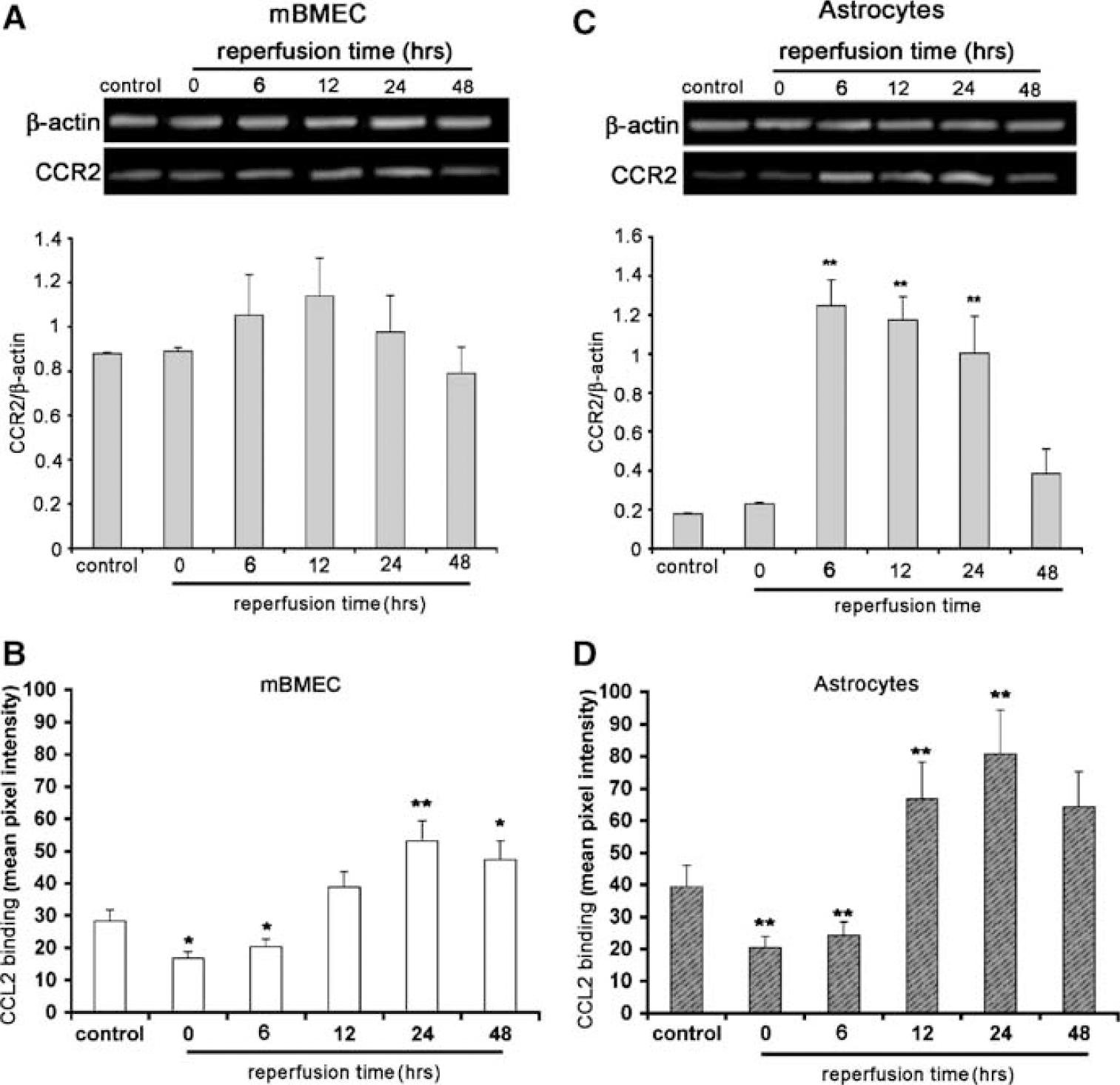
Temporal expression of CCR2 in brain endothelial cell/astrocyte cocultures after in vitro I/R. Cocultures were exposed to 5 h of ischemia and the indicated periods of reperfusion (0 to 48 h) to examine effects on CCR2 expression. In (
The Effect of CCL2 and CCR2 on Blood-Brain Barrier Permeability During In Vitro Ischemia/Reperfusion
To examine whether CCL2 production by astrocytes and brain endothelial cells participates in the ‘late opening’ of the BBB after in vitro I/R, we assessed BBB integrity under two conditions, where (a) CCL2 production/activity was blocked or (b) CCR2 was absent from the endothelial cells and/or astrocytes. In the first experiments, endothelial/astrocyte cocultures were exposed to phosphorothioated CCL2 antisense or sense oligonucleotide for 6 h, and 24 h later underwent in vitro I/R. In control (untreated) and sense oligonucleotide-treated cells, there was a marked decrease in TEER 24 h after I/R (Figure 6A). By contrast, antisense-treated cells had a significantly smaller reduction in TEER (P < 0.001; Figure 6A). Similarly, while the I/R-induced enhancement of barrier permeability to FITC-albumin was unaffected by sense oligonucleotide treatment, it was markedly reduced by phosphorothioated CCL2 antisense treatment (P < 0.001; Figure 6B). Antisense oligonucleotide treatment also resulted in a significant (~ two-thirds) reduction in I/R-induced monocyte transmigration across cocultures compared with control or sense oligonucleotide-treated cells (Figure 6C). There were also less morphologic changes in endothelial tight junctions after I/R in antisense-treated cultures than sense controls (Figure 6D). Thus, antisense-treated cultures had more continuous staining with rare fragmentation for TJ molecules (occludin, ZO-1, ZO-2, claudin-5), as monitored by immunocytochemistry. This morphologic alteration was confirmed by biochemical analysis of TJ proteins. Antisense treatment reduced the shift in occludin ZO-1, ZO-2 and claudin-5 from the Triton-X 100 soluble to the Triton-X 100 insoluble fraction during I/R (Figure 6E) that we found in control cultures (Figure 2B) and sense-treated cultures (Figure 6E).
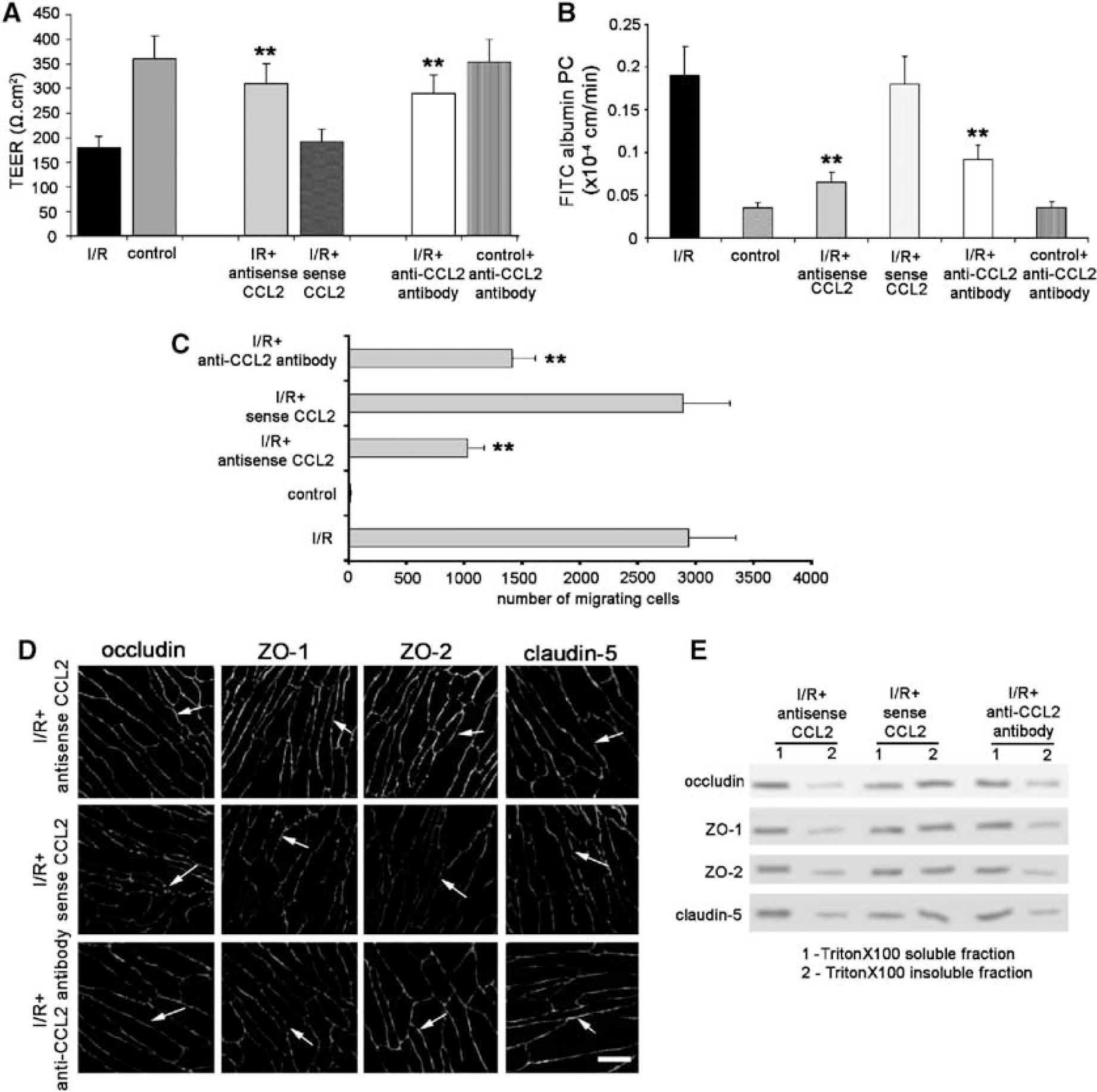
Effect of CCL2 inhibition on the response of brain endothelial cell/astrocyte cocultures to in vitro I/R. Cocultures were exposed to 5 h of ischemia followed by 24 h of reperfusion (I/R) or no ischemia (control). CCL2 was manipulated by two methods. CCL2 expression and production was inhibited by phosphorothioated CCL2 antisense oligonucleotide. Phosphorothioated CCL2 sense oligonucleotide was used as a control (see Materials and methods for details). Alternately, CCL2 activity was neutralized by anti-CCL2 antibody (10 μg/mL), applied at the beginning of reperfusion period. The cells were used to determine (
Similar effects were obtained if a CCL2 neutralizing antibody (concentration of 10 μg/mL) was added at the beginning of reperfusion. Thus, the antibody reduced I/R-induced hyperpermeability (Figures 6A and 6B), monocyte transmigration (Figure 6C) and structural changes in TJ proteins (Figures 6D and 6E).
To investigate the role of CCR2 receptors in regulating BBB permeability, experiments were performed where CCR2 was absent from endothelial cells, astrocytes or both cell types. Brain endothelial cells and astrocytes were prepared from mice of phenotypes CCR2 +/+ or CCR2–/–. Brain endothelial cells did not show any morphologic or functional alterations before I/R if CCR2 was absent. The absence of CCR2 also did not affect CCL2 release during in vitro I/R (data not shown). However, in cocultures where CCR2 was absent from both cell types, the degree of reduction in TEER and enhancement of FITC-albumin permeability induced by 24 h of reperfusion was less than with cocultures from CCR2 +/+ cells (Figures 7 A and 7B). Similarly, compared with cultures of CCR2 +/+ astrocytes and endothelial cells, CCR2–/– cocultures had reduced monocyte migration (Figure 7C), more continuous staining of TJ proteins with fewer areas of fragmentation (Figure 7D) and less shifting of TJ proteins from the Triton-X 100 soluble to the Triton-X 100 insoluble fraction during I/R (Figure 7E).
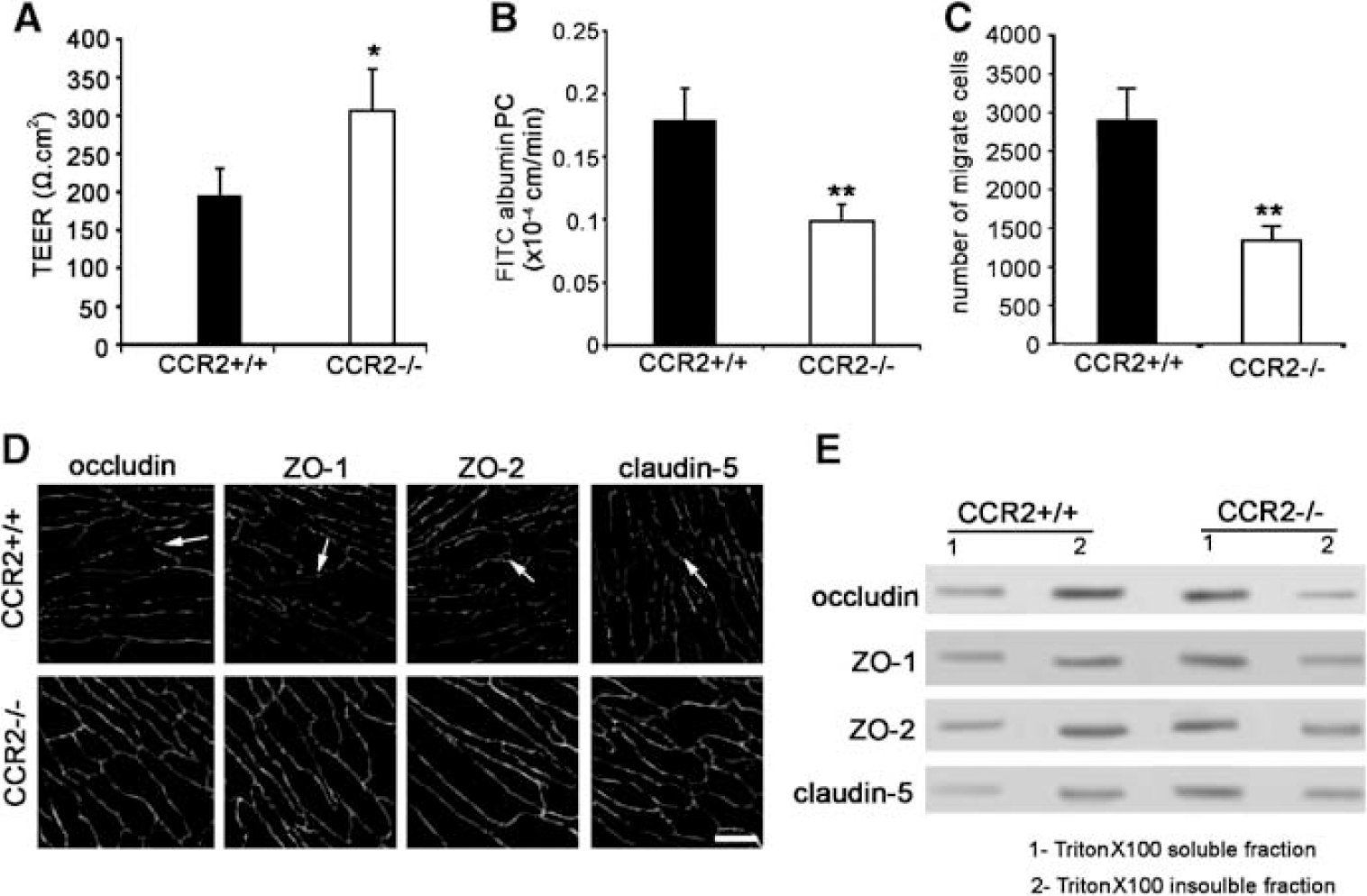
Absence of CCR2 receptor has a protective role on brain endothelial permeability during I/R condition. Mouse brain microvascular endothelial cells/astrocyte cocultures were prepared form CCR2–/– or CCR2+/+ mice. (
The reduced barrier injury found in CCR2–/– cocultures was mimicked by cocultures of CCR2–/– endothelial cells with CCR2 +/+ astrocytes, but not CCR2 +/+ endothelial cells with CCR2–/– astrocytes. Thus, for example, the PC for FITC-albumin was significantly decreased after 24 h of reperfusion if CCR2 was absent from brain endothelial cells (CCR2 +/+ 0.179 ± 0.025 versus CCR2–/– 0.098 ± 0.014 × 10−4cm/min) or from both endothelial cells and astrocytes (CCR2 +/+ 0.182 ± 0.03 versus CCR2–/– 0.084 ± 0.017 × 10−4 cm/min), but there was no alteration if CCR2 was absent only from astrocytes (CCR2 +/+ 0.189 ± 0.023 versus CCR2–/– 0.172 ± 0.034 × 10−4 cm/min). These effects of endothelial CCR2 deletion on FITC-albumin permeability were paralleled by changes in TEER. Similarly, morphologic and biochemical assays showed less alterations in TJ molecules and less monocyte migration during reperfusion if CCR2 was absent from brain endothelial cells. Taken together, these data indicate that CCL2 through CCR2 on brain endothelial cells plays an important role in regulating BBB permeability during I/R in vitro.
Discussion
The present study examined the effects of CCL2/CCR2 on BBB permeability during in vitro I/R. The major points of this study are as follows. (a) Chemokine CCL2 and its receptor CCR2 are significantly upregulated on brain endothelial cells and astrocytes during I/R injury. (b) The presence of CCL2 at the BBB during I/R injury is not only associated with chemotaxis of monocytes but it might also have a profound effect on the BBB, contributing to alterations in BBB integrity, (c) CCL2 antisense oligonucleotide or neutralizing antibody targeting CCL2, as well as absence of active CCR2 on brain endothelial cells, can help preserve BBB integrity after I/R.
Before discussing these results and their implications further, it is necessary to discuss caveats in the use of in vitro models of the BBB and I/R. Currently, cocultures of brain endothelial cells with astrocytes represent the best in vitro BBB model. Such cocultures do not, however, fully replicate the in vivo BBB. Thus, for example, the TEERs in the current experiments are a tenth of those recorded in vivo (Gaillard et al, 2001; Huber et al, 2001). However, in terms of the focus of this paper, it should be noted that we have found that application of exogenous CCL2 in vitro and in vivo has qualitatively similar effects on BBB disruption (Stamatovic et al, 2005).
Oxygen-glucose deprivation followed by reoxygenation is a commonly used model to investigate I/R-induced injury to different cell types. It should be noted, though, that it does not mimic all the elements of the complex process of in vivo I/R. In particular, it examines the effect of a cessation and restoration of nutrient (oxygen and glucose) delivery, but does not mimic the other effects of a reduction and restoration of blood flow (Nakagawa et al, 1990; Kastrup et al, 1999). In terms of the current experiments, qualitatively, the temporal changes in BBB permeability reported in this study are similar to those reported in vivo (Kuroiwa et al, 1985). Also, our observation showing that CCL2 expression appears to be highest during late reperfusion is substantiated with in vivo findings, providing additional support for the use of in vitro model of I/R injury. Finally, this model allows us to examine in great detail the cellular and molecular mechanisms involved in reperfusion injury. Such an examination is very difficult to perform in vivo.
There is limited evidence on the molecular and cellular events at the brain endothelial barrier during I/R period (Kondo et al, 1996; Utepbergenov et al, 1998; Abbruscato and Davis, 1999; Mark and Davis, 2002; Muruganandam et al, 2000). Those studies mostly focused on the effects of hypoxia on the brain endothelial barrier and did not evaluate how some of the events triggered by restoration of blood flow influences brain endothelial barrier and TJ complexes. However, reperfusion after ischemic brain injury triggers a cascade of events that ultimately result in secondary injury and development of BBB breakdown.
One of the classic hallmarks of reperfusion (secondary) injury in the brain is an inflammatory reaction (Stanimirovic and Satoh, 2000). Neutrophil influx occurs in the first 24 h after ischemia followed by monocyte migration from 24 h up to 5 days (Hallenbeck et al, 1986; Danton and Dietrich, 2003). The inflammatory response involves rolling, firm adhesion and transendothelial migration of leukocytes on the level of postcapillary venules, BBB dysfunction and increased production of proinflammatory mediators including reactive oxygen species, nitric oxide, cytokines, adhesion molecules and chemokines (Peters et al, 1998; Schuerer et al, 1994; Lindauer et al, 1996; Okada et al, 1994; Che et al, 2001; Yang et al, 1999). All of these mediators have been suggested as being involved in BBB ‘breakdown’ during inflammation, causing a dysfunction of TJ structure (abnormality in occludin and ZO-1), greater influx of blood-borne cells and further amplification of inflammation and brain parenchymal damage (Plumb et al, 2002; Gloor et al, 2001). Most of these observations imply that reperfusion-induced BBB disruption might be caused by different mediators with different time frames, although so far there is no solid evidence to show this. After reperfusion, there is an initial period of BBB disruption and, although there is then a recovery of endothelial permeability, there is a second phase of hyperpermeability (24 to 48 h) associated with morphologic and biochemical changes of TJ proteins. This ‘late’ opening of brain endothelial barrier coincides with high expression of proinflammatory mediators and temporally it occurs during enhanced monocyte migration into the postischemic brain and progression of the inflammatory response (Kastrup et al, 1999; Danton and Dietrich, 2003).
There are many controversies about the events at BBB during leukocyte trafficking. One line of evidence supports the hypothesis that leukocyte (particularly neutrophil) migration does not affect brain endothelial junction structure and, because of that, their migration occurs on the tricellular corners where three endothelial cells come together and where endothelial cell tight junctions are intrinsically discontinuous (Burns et al, 1997, 2000). In contrast, analysis of postmortem multiple sclerosis brains as well as studies on experimental allergic encephalomyelitis revealed that, during inflammation and leukocyte migration, the molecular organization of tight junction was altered with breakdown of occludin and ZO-1, and reorganization of actin cytoskeleton (Bolton et al, 1998; Couraud, 1998; Plumb et al, 2002; Kirk et al, 2003).
Proinflammatory cytokines such as IL-1α, IL-1β, TNF-α, IL-6 and granulocyte-macrophage (GM)-CSF have been found to induce alterations in TJ integrity, causing increased BBB permeability and BBB breakdown (Yang et al, 1999; Blamire et al, 2000; Paul et al, 2003). Recently, chemokines have also been reported to affect the BBB. For example, in vitro and in vivo data have clearly shown that CXCL8 (IL-8), through the CXCR2 receptor on the endothelial cells, increases the permeability of endothelial cell monolayers by inducing endothelial cell retraction and gap formation between endothelial cells, and it could also contribute to edema formation during I/R injury. During inflammatory response in the brain, alterations in endothelial TJ structure and BBB permeability can occur and chemokines may directly contribute to BBB ‘opening’ to facilitate leukocyte migration. The data presented in this study further extend our knowledge of the effects of chemokines on the BBB in pathologic conditions such as I/R injury. In particular, we have focused on the role of CCL2 in BBB ‘opening’ for two reasons. First, because CCL2 is one of the most abundant chemokines in brain tissue during the postischemic inflammation and lack of CCL2 dramatically reduces brain infarct size (Danton and Dietrich, 2003; Hughes et al, 2002; Stoll et al, 2002). Second, because recent studies have shown that brain endothelial cells express CCR2, the receptor for CCL2. Thus, CCL2 may directly influence endothelial cells and to induce BBB ‘opening’ (Bell et al, 1996; Stamatovic et al, 2003, 2005). To the best of our knowledge, this is the first study to provide evidence for the role of endogenous CCL2 in BBB ‘opening’ during I/R injury.
The main finding of this study was that CCL2, acting via its receptor on endothelial cells, may alter the endothelial TJ structure and increase brain endothelial barrier permeability during I/R. A possible scenario of CCL2 activity during I/R is that endogenous (de novo synthesized) CCL2, through the endothelial CCR2 receptor, induces signal transduction pathways that result in phosphorylation/dephosphorylation of TJ proteins. The change in phosphorylation state results in their redistribution and gap formation between endothelial cells that may enhance monocyte migration through the brain endothelial barrier. The results also suggest that the main source of CCL2 is likely to be endothelial cells and astrocytes because CCL2 was markedly upregulated on both of these cell types during I/R.
What are the important implications of our findings? First, is that regulating the CCL2 presence at the BBB during I/R could significantly modulate the inflammatory reaction by affecting not only leukocyte migration into parenchyma but also BBB integrity. Second, CCL2 inhibition or brain endothelial cell CCR2 receptor blockade act in the same manner to protect against brain endothelial barrier ‘opening’ and impair leukocyte trafficking. Third, these results also indicate that the CCL2/CCR2 axis has a more complex role in inflammation than previously thought. The cellular and molecular effects of CCL2/CCR2 suggest multiple potential effects, with regulation of BBB permeability being only one of them.
In summary, regulating CCR2 levels on brain endothelial cells and/or inhibiting synthesis or activity of CCL2 not only markedly reduce I/R-induced monocyte migration across the BBB in vitro but also reduce brain endothelial barrier disruption. By regulating these two processes at the BBB, CCL2 may enhance inflammatory injury during reperfusion in brain. Therefore, CCL2/CCR2 might be a promising therapeutic target for treating postischemic inflammation.
Footnotes
Acknowledgements
The authors thank David Oldfield (Department of Pediatrics), University of Michigan Medical School, for his technical assistance.