
Abstract
We previously observed degranulated mast cells (MC) in association with perivascular brain edema formation during focal cerebral ischemia. Brain MC are typically located perivascularly and contain potent fast-acting vasoactive and proteolytic substances. We examined in a rat model of transient middle cerebral artery occlusion (MCAO) whether, in the early phase of ischemia, MC regulate microcirculation, the blood–brain barrier (BBB) permeability, and edema formation. First, animals received MC inhibitor (cromoglycate), MC-degranulating drug (compound 48/80), or saline. Thereafter, we performed transient MCAO in gene-manipulated MC-deficient rats and their wild-type (WT) littermates, calculating brain swelling, visualizing BBB leakage by intravenously administered Evans blue albumin, and determining neutrophil infiltration with light microscopy. Cerebral blood flow, monitored by laser-Doppler flowmetry in separate experiments, was similar among pharmacological treatments. Ischemic swelling resulted in increased hemispheric volume of 13.4% ± 1.0% in controls, 8.1% ± 0.4% (39% reduction) after cromoglycate, and 25.2% ± 2.0% (89% increase) after compound 48/80 (P < 0.05). Early ischemic BBB leakage was reduced by 51% after cromoglycate, and 50% enhanced by compound 48/80 (P < 0.05). The cromoglycate group showed 37% less postischemic neutrophil infiltration than did controls (P < 0.05). Furthermore, MC-deficient rats responded to focal ischemia with 58% less brain swelling (6.7% ± 1.2%) than did their WT littermates (15.8% ± 1.4%, P < 0.05). Blood-brain barrier damage was 47% lower in MC-deficient rats than in the WT (P < 0.05). Neutrophil infiltration after MCAO was decreased 47% in MC-deficient rats in comparison to WT (P < 0.05). Pharmacological MC inhibition thus appears to deserve further investigation regarding reduction of brain swelling and inflammation early after stroke.
Introduction
Stroke is one of the leading causes of death and disability worldwide, killing more than 150,000 individuals annually in the USA (Anderson et al, 1998). The leading cause of acute death after stroke is space-occupying brain swelling (Ayata and Ropper, 2002). Edema-based brain swelling occurs in approximately two-thirds of patients within 6 h of ischemic stroke onset, and in this period the extent of permanently lost tissue also becomes established (Kummer von et al, 2001). The two main detrimental effects of swelling are (1) mass effect with compression of normal brain structures (herniation), and (2) decreased blood flow in the ischemic penumbra. Both effects damage the penumbra's metabolism and facilitate its transformation into infarction. Blood-brain barrier (BBB) damage increases tissue water content, and the entry of serum proteins into brain tissue magnifies the vasogenic edema and swelling.
Mast cells (MC) reside in a variety of locations in the central nervous system (CNS) of different species, including humans (Hough, 1988), where they appear to be concentrated in the diencephalic parenchyma, thalamus, and cerebral cortex (Silver et al, 1996; Theoharides, 1995). Stationary MC found at the body surfaces form a first-line defense against toxic, immunologic, and physical challenges. In the skin, physical irritation leads to degranulation of local MC, after which the potent granule constituents cause an immediate phlogistic (vasodilation, permeability increase) dermal response. Indeed, MC contain metachromatic granules with potent preformed substances: vasoactive (histamine, bradykinin), anticoagulant (heparin), proteolytic (tryptase and chymase), and chemotactic factors. Mast cell-dependent hypersensitivity reactions are biphasic, the immediate degranulation phase occurring within minutes. The secondary phase follows several hours later and includes de novo synthesis of more vasoactive and chemotactic mediators (prostaglandins, platelet-activating factor, leukotrienes) and cytokines (tumor-necrosis factor a, interleukins (ILs)), with an ensuing influx of neutrophils and eosinophils. Mast cell degranulation has been documented to enhance vascular permeability and to regulate BBB permeability in some experiments (Wahl et al, 1988), but no data exist as to the role of MC in cerebral ischemia. Our observation that MC reside perivascularly in vessel-branching points suggests that they might be involved in regulation of blood flow or blood vessel permeability in the brain. This prompted us to study whether early pharmacological modulation of MC (both inhibition and aggravation) would influence acute BBB damage, brain swelling, and neutrophil infiltration in a transient middle cerebral artery occlusion (MCAO) model in rats. Furthermore, to support this hypothesis, we replicated the experiments in gene-manipulated MC-deficient rats (Niwa et al, 1991). We found that brain swelling was lowest in the MC-deficient rats (58% reduction in comparison to their wild type (WT)). Pharmacological MC blocking (sodium cromoglycate) led to a 39% decrease in brain swelling, and compound 48/80 (MC-degranulating agent) elevated it by 89%. Early ischemic BBB leakage and postischemic neutrophil infiltration were also significantly dependent on pharmacologic MC modulation, and were lower in MC-deficient rats than in the WT. Cerebral blood flow, monitored in separate experiments, was similar among pharmacological treatments.
Materials and methods
Animals
Adult male Wistar rats (Harlan Nederland, Horst, The Netherlands) and WsRcWs/Ws rats (Japan SLC, Inc., Tokyo, Japan), 290 to 340 g, were anesthetized by an intraperitoneal injection of ketamin hydrochloride (50 mg/kg, Ketalar, Parke-Davis, Detroit, MI, USA) and a subcutaneous injection of medetomidine hydrochloride (0.5 mg/kg, Domitor, Orion, Espoo, Finland). A polyethylene tube was inserted into the left femoral artery for blood pressure monitoring (Olli Blood Pressure Meter 533, Kone, Espoo, Finland) and for measurement of arterial pH, blood gases, and blood glucose (AVL OPTI, Roche, Basel, Switzerland), and another tube into the left femoral vein for drug and/or vehicle infusions. Rectal temperature was maintained at 37°C during the surgery with a heating blanket. We used the suture MCAO model with 60 mins of ischemia followed by 3 h of reperfusion, as described elsewhere (Takano et al, 1997a). The relevant Animal Research Committee had approved the study protocol.
Laser-Doppler Flowmetry
Cerebral blood flow (CBF) was measured on-line by the BF/F/0.5 bare-fibre flexible probe of the Oxy-Flow device (Oxford Optronix, Oxford, UK). The scalp was incised in the midline and the skull was exposed. The skull was thinned by a dental drill at an area ipsilateral to the ischemia between 1.0 to 2.5 mm posterior and 6.0 to 1.5 mm lateral from the bregma. The probe was attached, within the thinned area, to the skull surface at a place of representative baseline CBF signal, and fixed to that place. The CBF signal was then obtained from the same place throughout the entire experiment.
Pharmacologic Protocols
First, we examined the effect of pharmacological MC modulation (Table 1), by either cromoglycate (n = 14, a clinically used topically administered inhibitor of MC degranulation, given intracerebroventricularly because of minimal crossing of the BBB (Shapiro and Koning, 1985)), or by compound 48/80 (n = 11, a standard MC degranulating secretagogue (Dimitriadou et al, 1990)). All drugs have been purchased from Sigma-Aldrich (Steinheim, Germany). Saline infusion was applied to controls (n = 13). To determine the severity of ischemia and the degree of reperfusion in the pharmacologically treated animals, CBF was monitored by laser-Doppler flowmetry before and during MCAO and after reperfusion in three separate groups of rats: saline-treated (n = 4), sodium cromoglycate (n = 5), and compound 48/80 (n = 4) (Table 2). Furthermore, sham-operated animals (4 per group) underwent the same treatments (placebo, sodium cromoglycate, compound 48/80) and surgical procedures, except that the suture was advanced only 10 mm (not 17 mm) above the bifurcation and withdrawn after 10 secs (not 60 mins). CBF was also monitored during the sham surgery. In another set of experiments (Table 1), we induced transient MCAO in MC-deficient WsRcWs/Ws rats (Niwa et al, 1991), which carry a defective gene for c-kit (ligand for stem cell factor required for MC differentiation), and induced it in their WT (Table 1). All experiments were performed in a masked manner.
Drug protocols and corrected infarct volumes in rats undergoing middle cerebral artery occlusion (MCAO)
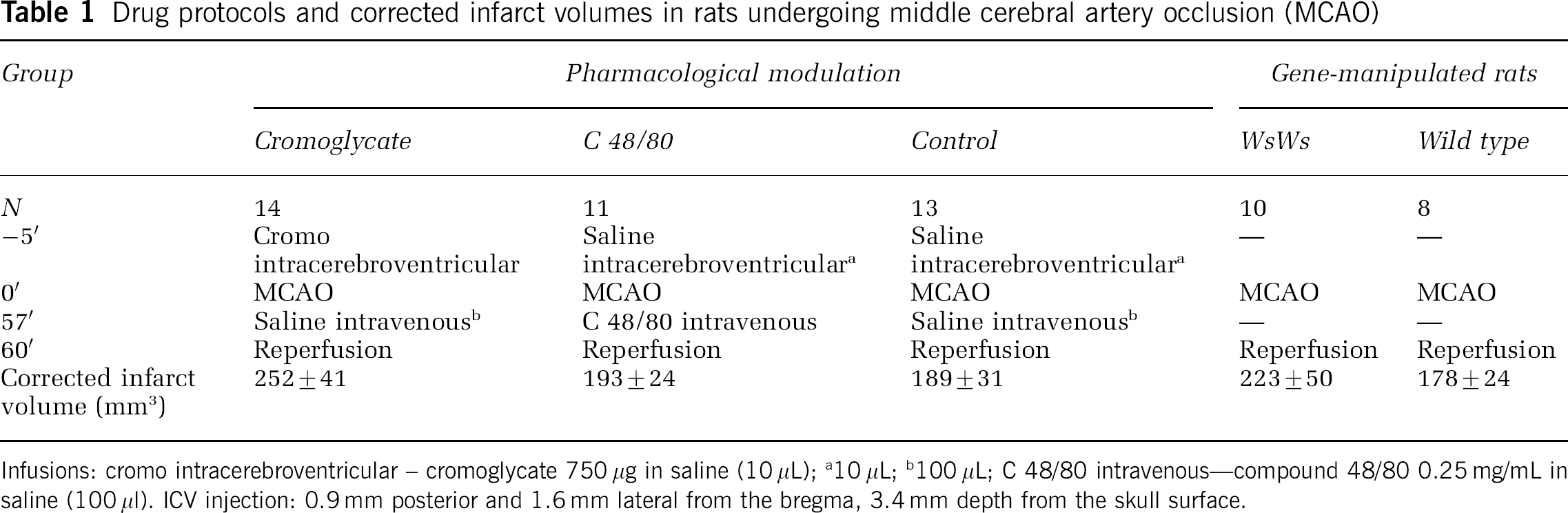
Infusions: cromo intracerebroventricular – cromoglycate 750 μg in saline (10 μL)
10 μL
100 μL
C 48/80 intravenous—compound 48/80 0.25 mg/mL in saline (100 μl). ICV injection: 0.9 mm posterior and 1.6 mm lateral from the bregma, 3.4 mm depth from the skull surface.
Corrected infarct volumes, brain swelling, and cerebral blood flow (CBF) in three separate groups of rats undergoing middle cerebral artery occlusion (MCAO)
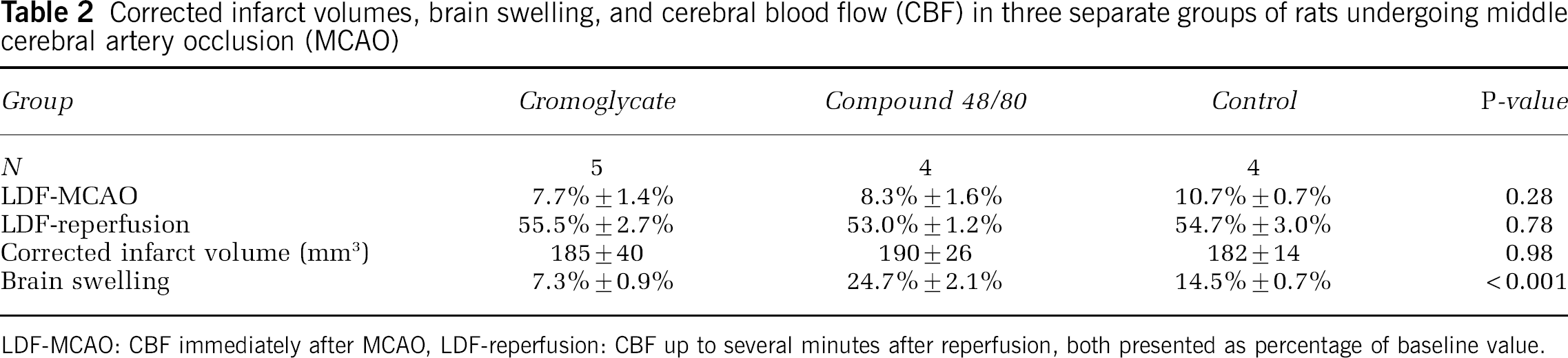
LDF-MCAO: CBF immediately after MCAO, LDF-reperfusion: CBF up to several minutes after reperfusion, both presented as percentage of baseline value.
Tissue Handling
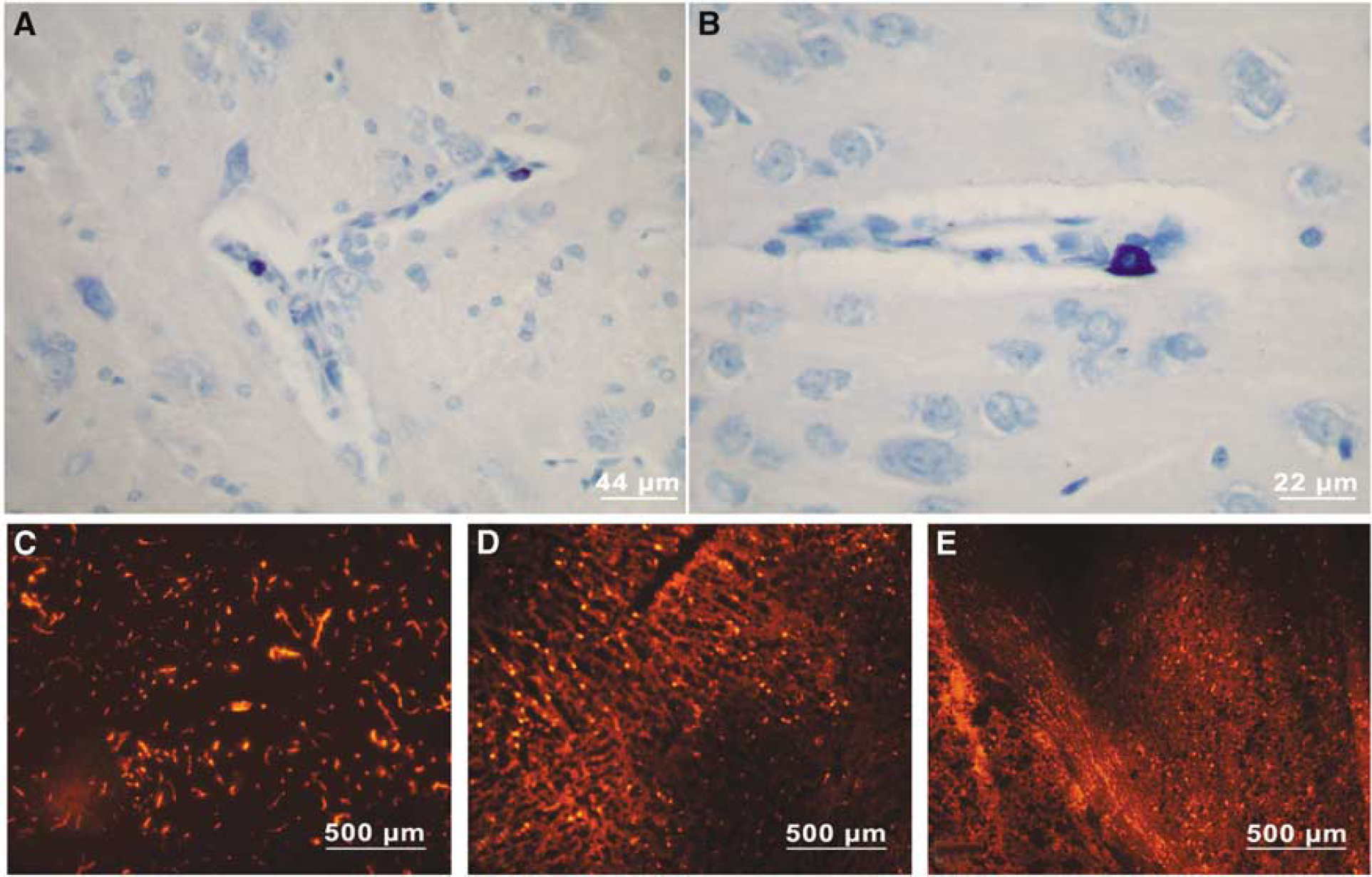
At 4 h after MCAO, the rats were reanesthetized with an overdose of pentobarbital (60 mg/kg, Mebunat, Orion). After cardiac perfusion with ice-cold saline (Takano et al, 1997a), the brains were quickly removed and dissected coronally into six 2-mm slices. Each third slice was cut into two 1-mm portions (rostral and caudal). The rostral part was embedded in Tissue-Tek (Sakura Finetek Inc., Tokyo, Japan), snap-frozen in liquid nitrogen, and kept thereafter at −80°C until 15-μm sections were cut. Then, 5-μm sections were cut from the caudal site of all slices and stained with Toluidine blue, a standard metachromatic histopathologic technique for detection of the heparin-containing granules present exclusively in MC (Figure 1A and 1B), and chloracetate esterase (Leder) (Leder, 1964) staining; these detect polymorphonuclear neutrophils. The remaining slices were then incubated for 15 mins in 2,3,5 triphenyltetrazolium chloride (TTC) at 37°C, and immersion-fixed subsequently in 10% formaldehyde.
(
Histopathologic Examinations
Light microscopy was performed by an experienced hematopathologist (M-LK-L) without prior knowledge of animal grouping with an Olympus BH-2 (Olympus, Tokyo, Japan) and photographed with a Nikon Eclipse E600 microscope connected to a Nikon Coolpix 995 digital camera (Nikon, Tokyo, Japan). Intravascular and emigrated neutrophils were counted in systematically placed target areas in 5-μm chloracetate esterase-stained cross sections through the area of maximal infarction with a morphometric 0.1 mm2 grid. Neutrophil number was counted in a total area of 10 mm2 in the temporoparietal infarction core characterized by pyknotic neuronal change, and in a 6-mm2 area in the adjacent parasagittal edematous infarct penumbra, as well as in the deep thalamic and basal ganglia.
Blood-Brain Barrier Damage
To visualize BBB leakage (Lindsberg et al, 1997), animals received a 2% solution of Evans blue (EB) albumin (fluorescent dye, Sigma, 20 mg/mL dissolved in 1% albumin) into the femoral vein (0.3 mL/100 g) 20 mins before cardiac perfusion.
In the first experiment (Table 1), the 15-μm brain sections were examined for distribution of characteristic red fluorescence of EB in the brain parenchyma by epifluorescent microscope Axioplan 2 (Carl Zeiss, Hallbergmoos, Germany) and a fluorescence filter specific for EB (Chroma, Rockingham, VT, USA; excitation at λ = 620 nm, emission at λ = 680 nm). For evaluation of BBB damage, images of 5 prespecified regions of interest (ROI) (three from the cortex and two from the basal ganglia) from the infarcted area, as well as a reference image from the healthy hemisphere, were captured with an AxioCAM HR digital camera (Carl Zeiss). For reliability of digital imaging fluorescence microscopy in measurement of tracer concentration in sectioned tissue, see Weinberg et al (1994). We quantified EBA-fluorescent pixels with Image J analyzing software (NIH, Bethesda, MD, USA). The level of autofluorescence was based on that obtained from the healthy hemisphere. For each animal, we calculated the difference between the fluorescence signal in five selected ROI (represented by the characteristic red fluorescence) and the level of autofluorescence. Finally, the corrected signals from all five ROI were averaged. After the acquisition of an improved fluorescence scanner Typhoon 9400 (Amersham Biosciences, Buckinghamshire, UK), a slightly revised technique was adopted. With this scanner, the difference in average fluorescence signal intensity between the entire infarcted area and the intact hemisphere could be measured by image analyzer software ImageQuant (Amersham). Briefly, the area with the lack of TTC staining signified the infarcted region, and the fluorescence signal was measured in the outlined TTC-based area.
Calculation of Volumes of Brain Infarction and Swelling
For each animal, all six TTC-stained brain slices mounted on a scale were photographed with a digital camera (Sony, Tokyo, Japan). Infarction and corrected infarction volumes were calculated as described (Takano et al, 1997b). Percentage of brain swelling was derived from volumetric growth of the ischemic hemisphere in comparison to the intact one as percentage of brain swelling = [(right hemisphere's volume/left hemisphere's volume)–1] × 100.
Statistics
Data are presented as mean ± s.e. Statistical analyses were performed with unpaired t-test for comparisons of two groups, and for multiple groups with one-way ANOVA in continuous variables followed by the Holm-Sidak post hoc test. Kruskal–Wallis ANOVA served for nonparametric variables. A two-tailed value of P < 0.05 was considered significant.
Results
Physiologic Variables and Infarct Volumes
No significant differences appeared in physiologic parameters (MABP, temperature, pH, paCO2, paO2, glucose) among the study groups (data not shown). A transient nonsignificant 7% reduction of MABP in the compound 48/80-treated group occurred at 100 min after MCAO when compared with control figures. Corrected infarct volumes within each experiment were similar (Tables 1 and 2), except that no infarctions occurred in sham-operated animals.
Laser-Doppler Flowmetry
No significant differences in CBF appeared among the study groups during any monitored time points (Table 2). CBF stayed similar during the sham surgery, and no difference existed among the three pharmacologically treated sham groups (data not shown).
Histopathologic Findings
Already in the early postischemic phase, 4 h after MCAO, ischemic neuronal necrosis appeared in hematoxylin-eosin-stained tissue sections. The sections were stained with toluidine blue for detection of heparin in MC granules (MC being the only cell type containing heparin (Khalil et al, 2003)). Mast cells were frequently visible in the vicinity of small cerebral cortical and thalamic penetrating vessels (Figures 1A and 1B), often in widened Virchow-Robin spaces, as described earlier (Dimitriadou et al, 1990). In ischemic areas, extracellular granules on the abluminal surfaces of blood vessels were accompanied by perivascular edematous changes. Histopathologic evaluation suggested milder ischemic edematous changes in MC-deficient WsRcWs/Ws rats than in their WT.
Blood-Brain Barrier Integrity
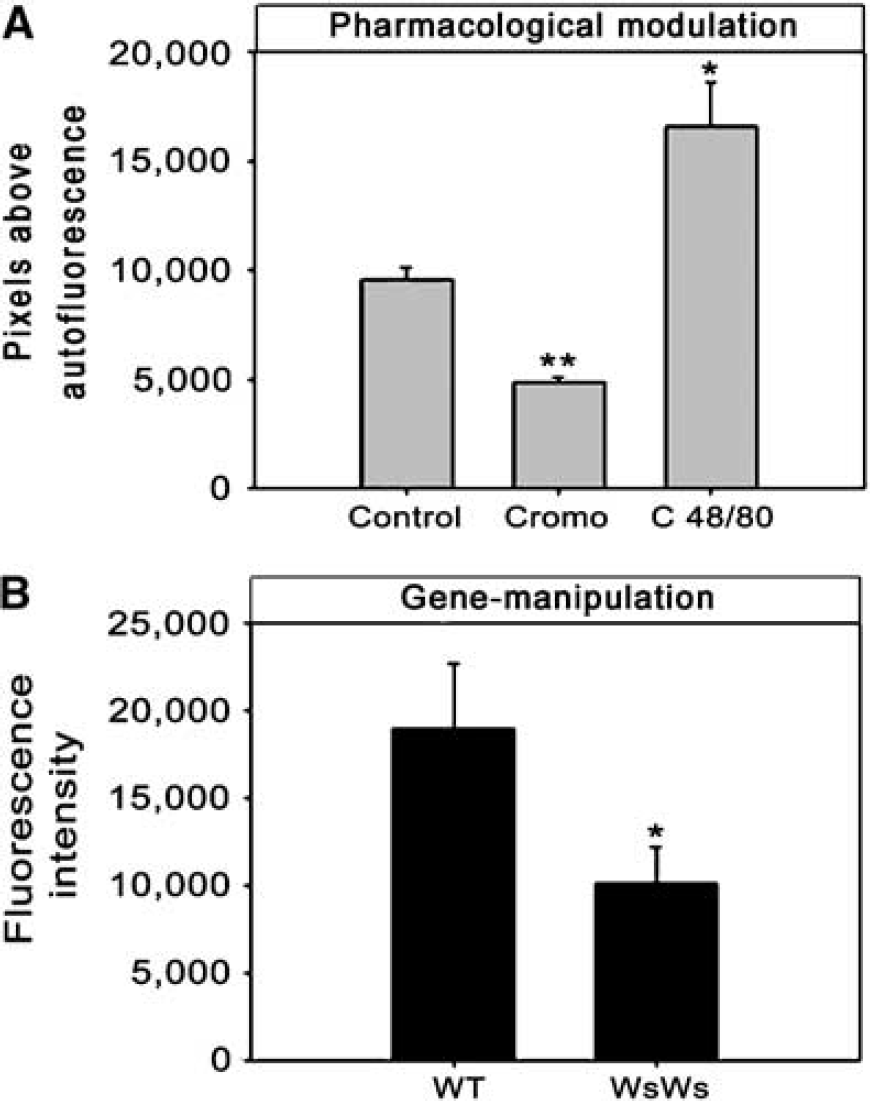
Visualization of EBA fluorescence in pharmacologically modulated (Figures 1C to 1E) and in MC-deficient animals suggested that the albumin extravasation was differentially regulated. When compared with controls, the number of fluorescence pixels indicating magnitude of extravasation was significantly elevated by compound 48/80 and reduced by cromoglycate (P < 0.05; Figure 2A). WsRcWs/Ws rats had significantly reduced fluorescence signal intensities when compared with their WT (P < 0.05; Figure 2b). Furthermore, when fluorescence intensity was measured in the three separate groups of animals, in which laser-Doppler flowmetry was also performed, we again observed a significant difference among the groups (P < 0.001): 19,464 ± 1034 (saline-treated), 8912 ± 600 (sodium cromoglycate), and 26,214 ± 2375 (compound 48/80). In the sham-operated animals, however, no significant changes occurred in amount of fluorescence between the left and right hemispheres (P = 0.96): 965 ± 492 (saline-treated), 804 ± 1363 (sodium cromoglycate), and 1163 ± 72 (compound 48/80).
EBA-fluorescent signal representing the magnitude of extravasation and BBB leakage. (
Brain Swelling
Pharmacologically induced MC degranulation led to an 89% increase in ischemic brain swelling (P < 0.05), whereas MC stabilizing reduced it by 39% (P < 0.05), compared with control values (Figure 3, left). Brain swelling caused by ischemic edema correlated with the extravasation of EBA (Figure 4). WsRcWs/Ws rats responded to transient MCAO with 58% less ischemic brain swelling than did their WT (P < 0.05; Figure 3, right). Accordingly, in the three separate groups of animals having laser-Doppler assessment of CBF, ischemic brain swelling showed a 50% reduction after sodium cromoglycate, and a 71% increase with compound 48/80 (P < 0.001). No differences (< 0.3%) appeared between the volumes of the right and left hemispheres in the sham-operated animals (P = 0.71).
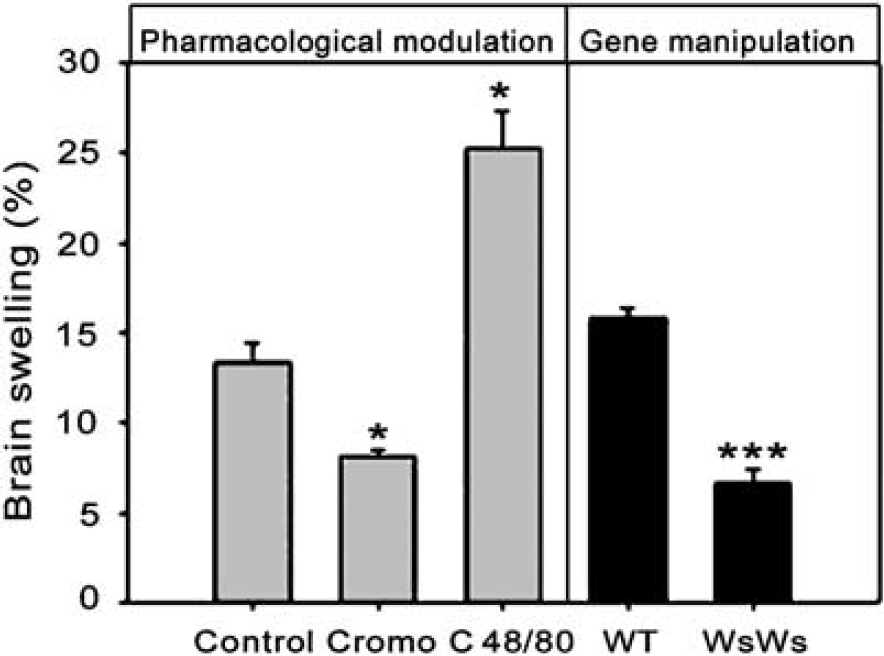
Brain swelling in pharmacologically modulated groups, and gene-manipulated rats. Kruskal-Wallis ANOVA showed a highly significant effect of pharmacological modulation (P < 0.001); post-hoc tests: *P < 0.05 (Cromo = cromoglycate, C 48/80 = compound 48/80, WT = wild-type littermates, WsWs = WsRcWs/Ws). No brain swelling occurred in any sham-operated groups.
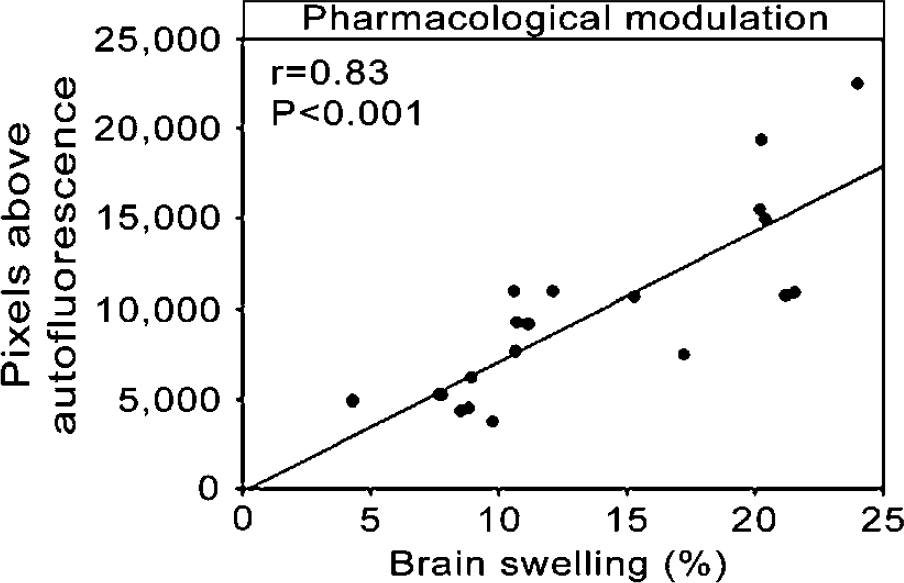
Correlation between brain swelling and BBB permeability, suggesting that swelling of the infarcted hemisphere derives principally from fluid shifts from inside the intravascular compartment.
Neutrophil Response
Even at this early postischemic time point, neutrophil density was up to 2.5-fold higher in the infarcted than in the noninfarcted hemisphere and in the sham-operated rats (P < 0.05). The lowest neutrophil counts appeared in the MC-deficient rats, which had 47% of the neutrophil count found in the infarcted hemisphere of WT (P < 0.05; Figure 5B). Cromoglycate significantly reduced the density of neutrophils in the infarcted hemisphere, by 37% (Figure 5A). Interestingly, cromoglycate reduced not only the number of emigrated neutrophils but also of those still detectable within the intravascular space (Figure 5A, right). This difference was most pronounced in the infarct core and basal ganglia (Figure 5A and 5B, left). Treatment with compound 48/80 was associated with a clear trend toward enhanced neutrophil response (Figure 5A), which was borderline significant in the area of basal ganglia (P = 0.06).
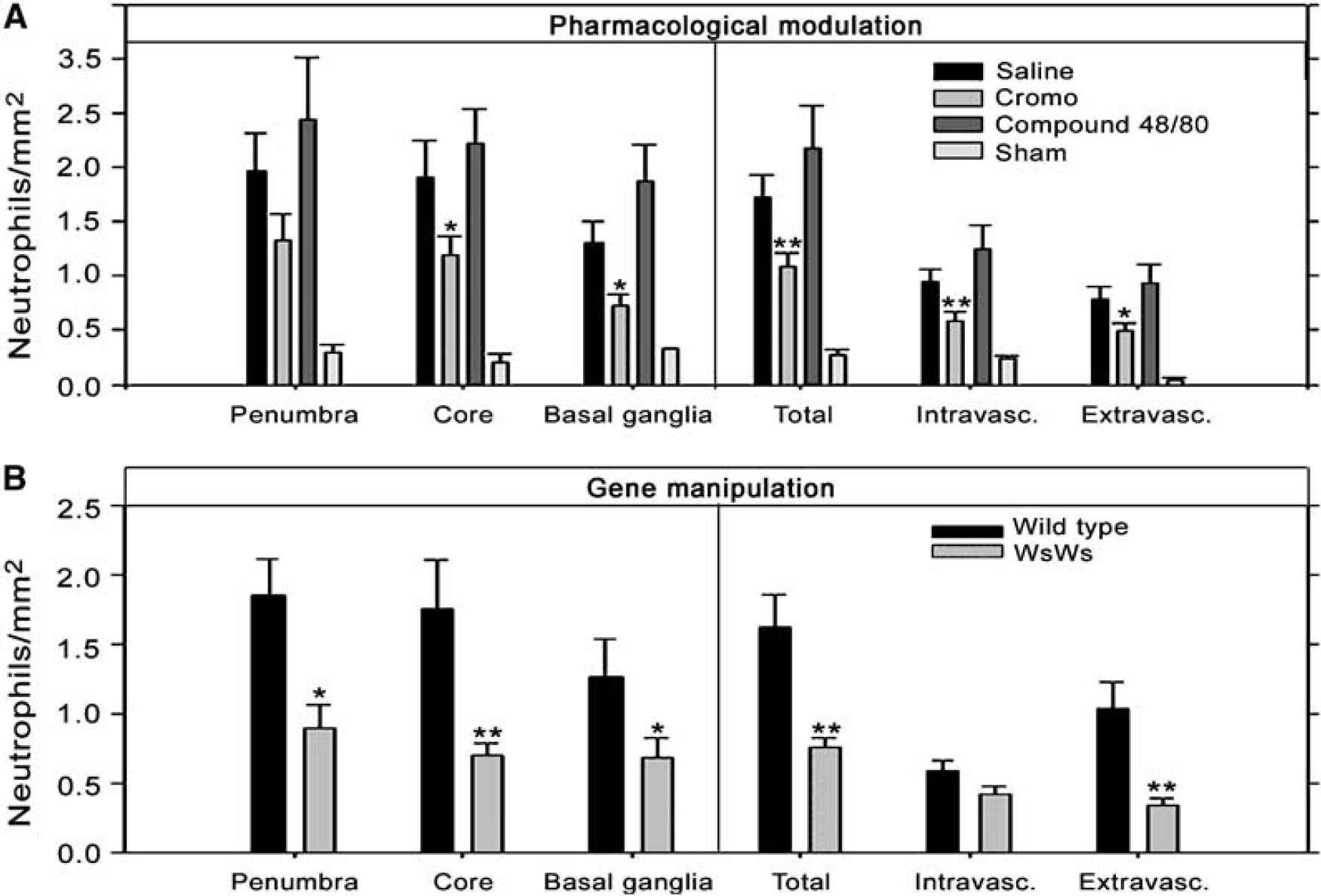
Neutrophil counts in sham-operated (saline-treated), and pharmacologically modulated (
In sham-operated rats, the pharmacological treatments as such did not cause any changes in neutrophil counts that would be comparable to the ipsilaterally elevated levels found in the rat brains subjected to MCAO (data not shown).
Discussion
Mast cells are stationary cellular mediators of immediate hypersensitivity and show local phlogistic reactions to mechanical, toxic, and allergic stimuli. We show that cerebral MC participated in regulation of early postischemic BBB failure, brain swelling, and inflammation. Both MC degranulation state and absence of MC in the gene-manipulated rats influenced early ischemic BBB permeability, brain swelling, and neutrophil infiltration. Accordingly, pharmacological MC degranulation led to an 89% increase in acute ischemic swelling, whereas blocking of MC degranulation by sodium cromoglycate reduced it by 39%, compared with control values (Figure 3, left). Similar effects were replicated in the LDF-monitored animals (Table 2). Moreover, MC deficiency in the WsRcWs/Ws rats resulted in even greater reduction (58%) in ischemic swelling than in the WT (Figure 3, right), supporting the specificity of the MC-targeted interventions.
Whereas cerebral as well as meningeal perivascular cells form a heterogeneous cell population, we and others have observed a subpopulation of mononuclear perivascular cells with morphologic and histochemic characteristics of MC positioned abluminally to the basal lamina and BBB (Figure 1A and 1B) (Dimitriadou et al, 1990; Letourneau et al, 2003). Mast cell as a regulator of BBB has thus far not been widely recognized. We found that the specific MC stabilizer cromoglycate robustly reduced acute postischemic BBB-damage (Figures 1C–1E and 2A). Mast cell-deficient WsRcWs/Ws rats showed an even more profound reduction in acute postischemic BBB leakage (Figure 2B). These experiments imply, in early BBB failure after ischemia, the strong involvement of this multi-active and potent cell type, with an armamentarium of bioactive granule mediators known to target vascular basal lamina. The mechanism of MC activation in ischemia, akin to the MC-mediated phlogistic dermal reaction, is unclear. However, mixing of serum with CSF in vitro, as well as plasma extravasation in BBB damage in stroke and in subarachnoid hemorrhage, yields complement proteins C3a and C5a, which belong among the most potent stimulators of MC (Lindsberg et al, 1996).
Ischemic BBB damage (as well as MC-mediated hypersensitivity reactions) has long been held to be biphasic, the first phase already beginning within minutes (Kuroiwa et al, 1985). The integrity of BBB is largely determined by basal lamina, the three main constituents of which are the matrix proteins laminin, fibronectin, and collagen type IV. The integrin-labeled basal lamina loses its integrity very early after ischemia onset (Tagaya et al, 2001). Secretory granules of cerebral MC contain chymase (Dimitriadou et al, 1990), a potent protease which cleaves fibronectin and also activates procollagenases (Saarinen et al, 1994; Tetlow et al, 1998), even in the presence of the tissue inhibitor of metalloproteinase-1 (Frank et al, 2001). Mast cells also release the gelatinases A (matrix metalloproteinase (MMP)-2) and B (MMP-9) (Fang et al, 1999), which degrade collagen type IV. Thus, MC-derived chymase and gelatinases may participate in this early phase of postischemic basal lamina degradation. We focused on the early ischemic brain swelling, which is already present in two-thirds of patients during the first 3 to 6 h (Kummer von et al, 2001), is dependent on early BBB failure, and is a proven major determinant of stroke mortality in general (Ayata and Ropper, 2002). Further studies are warranted to determine the full therapeutic window for MC blocking in acute stroke.
Mast cell granule constituents, especially histamine, heparin, and bradykinin, show significant microcirculatory effects, and modulation of their liberation during ischemia may influence the extent of ischemic injury. Infarct volumes were not influenced by the MC interventions (Table 1), as expected at this early postischemic time point and by the fact that we tested no drug with any direct neuroprotective property. Experiments performed after sham surgery and pharmacological MC modulation suggest that the changes observed were related to the ischemic response rather than to the pharmacological effects alone. Furthermore, the pharmacological manipulations seemed to cause no significant influences in postischemic CBF that could have mediated the effects observed (Table 2).
Reperfusion injury includes the release of free radicals and inflammatory mediators promoting leukocyte infiltration. After reperfusion, neutrophils start to accumulate within hours (Barone et al, 1992; Zhang et al, 1994). Importantly, we found this to depend on the presence of MC (Figure 5B), and to be reduced by cromoglycate (Figure 5A). That clinical trials based on inhibition of neutrophils have had no success (Investigators, 2001; Krams et al, 2003) might be explained by the fact that leukocytes arrive too late to influence damage propagation (Emerich et al, 2002). The extremely potent proinflammatory MC, already resident at the outset of ischemia, thus may offer an alternative target for early antichemotactic intervention before involvement of any circulating inflammatory cells. Indeed, MC liberate potent chemotactic substances platelet-activating factor (PAF), tumor necrosis factor α, IL-4, IL-5, IL-8, neutrophil, eosinophil, and macrophage chemotactic factors). Since cromoglycate reduced both the number of transmigrated neutrophils and of those still within the intravascular space (Figure 5b, right), MC blocking not only reduced passive neutrophil trafficking through BBB breaches, but probably also attenuated the perivascular chemotactic gradient up which neutrophils migrate.
A role for MC in ischemic stroke is supported by the reduced albumin extravasation and brain swelling in MC-deficient rats. That drugs targeting MC also could pharmacologically modulate elements of reperfusion injury suggests a potential therapeutic approach in a number of highly prevalent cerebral insults, in which extensive tissue injury is followed by florid acute extravasation, hazardous brain swelling, and inflammatory cell infiltration.