
Abstract
Hypoxic/ischemic and traumatic injury to central nervous system myelinated axons is heavily dependent on accumulation of Ca ions in the axoplasm, itself promoted by Na influx from the extracellular space. Given the high density of nodal Na channels, we hypothesized that nodes of Ranvier might be particularly vulnerable to Ca overload and subsequent damage, as this is the expected locus of maximal Na influx. Adult rat optic nerves were exposed to in vitro anoxia and analyzed immunohistochemically for the presence of spectrin breakdown. Cleavage of spectrin became detectable between 15 and 30 mins of anoxia, and increased homogeneously along the lengths of fibers; localized breakdown was not observed at nodes of Ranvier at any time point analyzed. Spectrin breakdown was also found in glial processes surrounding axons. Confocal imaging of axoplasmic Ca also revealed a gradual and nonlocalized increase as anoxia progressed, without evidence of Ca ‘hot-spots’ anywhere along the axons at any time between 0 and 30 mins of anoxic exposure in vitro. Calculations of Ca diffusion rates indicated that even if Ca entered or was released focally in axons, this ion would diffuse rapidly into the internodes and likely produce diffuse injury by activating Ca-dependent proteases. Western blot analysis for voltage-gated Na channel protein revealed that key functional proteins such as these are also degraded by anoxia/ischemia. Thus, proteolysis of structural and functional proteins will conspire to irreversibly injure central axons and render them nonfunctional, eventually leading to transection, degradation, and Wallerian degeneration.
Introduction
Disorders of white matter may cause serious disability if vital tracts are disrupted. Examples include neurotrauma, with both traumatic brain and spinal cord injuries invariably damaging white matter tracts, stroke which almost always affects myelinated fibers in the brain, and inflammatory demyelination such as multiple sclerosis. Much work has been performed on the pathophysiology of ischemic gray matter damage, and in the last decade significant insight has also been gained into the mechanisms of white matter damage during ischemic and traumatic injury. A complex interplay of voltage-gated and transmitter-activated ion channels, transporters, mitochondrial failure, nitric oxide (NO) production, and free radical generation is set in motion, resulting in tissue destruction, with mechanisms sharing significant overlap between ischemic and traumatic insults (for reviews, see Goldberg and Ransom, 2003; Maxwell et al, 1997; Park et al, 2004; Stys, 2004). As with gray matter injury, a final common step appears to be cellular overload of Ca, an ion that is normally very tightly regulated in all mammalian cells. Excessive cytosolic Ca will trigger a variety of deleterious Ca-dependent biochemical pathways, with important effectors being the Ca-activated proteases, the calpains.
The calpains are cysteine proteases that require Ca for activation, and function normally in growth, synaptic plasticity, and normal cellular remodeling (Croall and DeMartino, 1991; Melloni and Pontremoli, 1989; Ray and Banik, 2003; Suzuki et al, 1995). Given the ubiquitous nature of this protease, and its ability to degrade a large variety of key cellular proteins, such as spectrin, neurofilament, ion channels and receptors, myelin proteins, and many others (Buki et al, 1999; Deshpande et al, 1995; Jiang and Stys, 2000; McCracken et al, 2001; Schaecher et al, 2001; Schumacher et al, 2000; Shields et al, 1999), it has also been implicated in destruction of central nervous system (CNS) tissue during pathological conditions such as ischemia and trauma, in both gray and white matter regions (Buki et al, 1999; McCracken et al, 1999; Roberts-Lewis et al, 1994; Saatman et al, 1996). Unlike neurons, myelinated axons are unique in that their surface is covered almost entirely by the myelin sheath, with only ≈ 1% of their surface area exposed to the extracellular space at nodes of Ranvier. Given that Ca influx appears to be a key step in axonal injury, and that Ca-dependent calpain activation is an important consequence (Jiang and Stys, 2000), we hypothesized that nodes would be particularly vulnerable to calpain-mediated injury during anoxia. We performed a series of experiments in an in vitro model of anoxic white matter to examine the spatiotemporal profile of calpain-mediated cytoskeletal damage to see if nodes are initially or preferentially damaged. We also examined whether nodal Na channels, critical for supporting action potential conduction and present at very high densities at nodes of Ranvier, are damaged during anoxia.
Materials and methods
Tissue Preparation
Optic nerves were dissected from adult Long-Evans male rats (200 to 250 g; Charles River) as previously described (Stys et al, 1991). Nerves were equilibrated in 95% O2/5% CO2 for 60 mins in an interface chamber maintained at 37°C and were perfused at 2 mL/min with artificial cerebrospinal fluid (aCSF) containing the following (in mmol/L): NaCl 126, KCl 3.0, NaHCO3 26, MgSO4 2.0, NaH2PO4 1.25, CaCl2 2.0, and glucose 10 (pH 7.4). Anoxia was induced by switching to 95% N2/5% CO2 for 15 to 60 mins.
Immunostaining for Spectrin Breakdown Products (SBP), Neurofilament NF-160, and Na Channels
We used qualitative confocal immunofluorescence to examine which optic nerve elements were damaged by anoxia. Antiserum against SBPs (SBP 38-2 – a generous gift from Dr Robert Siman, University of Pennsylvania) was used to examine Ca-dependent calpain-mediated degradation of the structural protein spectrin in optic nerves. We did not attempt to precisely delineate the subcellular distributions of spectrin breakdown patterns, but instead used this marker to distinguish whether nodal regions were initially and preferentially damaged, which was our initial hypothesis. After surgical preparation, optic nerve dissection, incubation in aCSF, and exposure to normoxia or anoxia as described above, samples were fixed in 4% paraformaldehyde for 3 h, and then cryoprotected in 20% sucrose phosphate buffer at 4°C overnight. Tissues were then rinsed twice for 10 mins in 0.05 mol/L Tris, rinsed for 20 mins in methanol, rinsed again three times for 10 mins in Tris 0.05 mol/L. The sample was then blocked with 10% normal goat serum in 0.3% Tris-triton X-100, and incubated overnight in primary antiserum diluted in Tris-triton/normal goat serum at concentrations of 1:1000 for NF-160 (Sigma, St Louis), 1:5000 for SBP 38-2, and 1:500 for Na channels (Sigma). After 3 quick rinses in 0.05 mol/L Tris buffer, tissues were incubated for 2 h in goat anti-mouse Alexa-488 (Molecular Probes, Eugene, OR) at a concentration of 1:100 (against NF), goat anti-rabbit Alexa-594 at 1:100 (against SBP), and anti-mouse Alexa-488 at 1:250 (against Na channels) in 0.05 mol/L Tris/0.9% NaCl. Tissues were then quickly rinsed in 0.05 mol/L Tris three times, and mounted on microscope slides in Prolong Antifade Reagent (Molecular Probes). Images were collected on a Bio-Rad 1024 confocal laser-scanning microscope with an × 60 oil immersion objective.
Na Channel Western Blots
Frozen optic nerves (n = 4, each group) were homogenized in 200 μL of lysis buffer (M-PER, Pierce, Rockford, IL, USA) with protease inhibitor mixtures (pepstatin A, 1 μg/mL; leupeptin, 1 μg/mL; aprotinin, 1 μg/mL; Pefabloc SC, 0.2 mmol/L; benzemidine, 0.1 mg/mL; calpain inhibitors I and II, 8 μg/mL each; all from Sigma) on ice for 30 mins. The homogenates were centrifuged at 15,000g for 30 mins at 4°C. The supernatants were frozen at −80°C until use. The samples (15 μg/lane) were boiled for 5 mins in SDS sample buffer (glycerol 1 mL, β-mercaptoethanol 0.5 mL, 10% SDS 3 mL, 1.0 mol/L Tris-HCl 1.25 mL, pH 6.7, and bromophenol blue 2 mg) and run on 7% Tris acetate NuPAGE gel (Invitrogen, Carlsbad, CA, USA) at 30 V for 1.5 h. The polypeptides were electrotransferred to immunobilon membranes (Millipore) at 150 V for 1.5 h. Nonspecific binding was blocked using 5% non-fat milk in PBST (9.1 mmol/L dibasic sodium phosphate, 1.7 mmol/L monobasic sodium phosphate, 150 mmol/L NaCl, and 0.1% Tween 20) for 30 mins at room temperature (RT). The membranes were incubated with polyclonal rabbit anti-pan Na channel antibody (#06-811, Upstate, Lake Placid, NY; 1:80) specific for the intracellular III–IV loop of the Na channel α-subunit (amino acids (a.a.) 1491 to 1508 of full length a.a. 1–2005; P04775) overnight at 4°C, then incubated with HRP-conjugated secondary antibody (1:20,000, Vector Laboratories, Burlingame, CA, USA) for 1 h each at RT. The membranes were rinsed three times with TTBS (100 mmol/L Tris, 0.9% NaCl, and 0.1% Tween 20) for 5 mins at RT, incubated with WestDura Chemiluminescent Substrate (Pierce) for 5 mins, and then exposed to X-ray film (Biomax MS-1, Kodak, Rochester, NY, USA). The film was scanned and analyzed using ImageTrak software (written by PKS).
Ca Imaging in Live Optic Nerve
Adult Long-Evans male rats were anesthetized with 80% CO2/20% O2 and decapitated. Optic nerves were dissected out, immersed in Ca2+ -free aCSF at 4°C. Nerves were placed in an interface perfusion chamber and loaded for 1.5 h with fluorescent dyes through a suction pipette applied to the distal end. For Ca2+ imaging nerves were placed in a custom-built perfusion chamber, mounted on an upright Nikon Cl confocal laser-scanning microscope. Imaging was performed at 36°C with an ×60 immersion objective, itself maintained at the same temperature to avoid cooling of the sample. Images were analyzed using ImageTrak: axonal fluorescence changes were normalized to average basal levels and reported as a ratio of signal collected from Ca2 +-sensitive and -insensitive fluorophores plotted against time.
Oregon Green 488 BAPTA-1 Dextran (Kd ≈ 450 nmol/L) was used as a Ca2+ indicator. For visualization of axonal profiles, nerves were coloaded with red Alexa Fluor 594 Dextran. The red Ca2+-independent fluorescence was used to outline regions of interest (ROIs) from which green Ca2+-dependent fluorescence was measured. The loading buffer contained low Na+ concentration (NaCl was replaced by 126 mmol/L of N-methyl-D-glucamine) and low Ca2+ concentration (CaCl2 was omitted) to mimic intraaxonal concentrations. Dye concentrations in the loading pipette were: 75 μmol/L for Oregon Green 488 BAPTA-1 Dextran and 100 μmol/L Alexa Fluor 594 Dextran. Fluorescence dyes were purchased from Molecular Probes.
Results
Lack of Spectrin Breakdown in Control Nerves
Calpain-mediated spectrin breakdown is a reliable index of calpain activity (Siman et al, 1989). Optic nerves were dissected from freshly killed rats, placed in an in vitro perfusion chamber at 37°C, and allowed to equilibrate under normoxic conditions for ≈ 60 mins. Immunohistochemistry was then performed for SBP to examine if any elements of the optic nerve were damaged by excision and equilibration. Figures 1A to 1D shows representative confocal micrographs of rat optic nerve double stained with antineurofilament antibody to outline axon cylinders and SBP. Normoxic conditions did not induce significant axonal damage, showing no evidence of accumulation of spectrin cleavage products either immediately after dissection or after 60 mins of normoxic incubation. Figures 1E and 1F shows double staining with the same two antisera, but with images taken from the cut end of normoxic nerve: this traumatized region showed abundant spectrin breakdown as expected, serving as a positive control for this antibody. Oligodendrocytes labeled with anti-CNPase antibody (Trapp et al, 1988) and SBP exhibited very little cytoskeletal damage under normoxic conditions (Figures 1G and 1H).
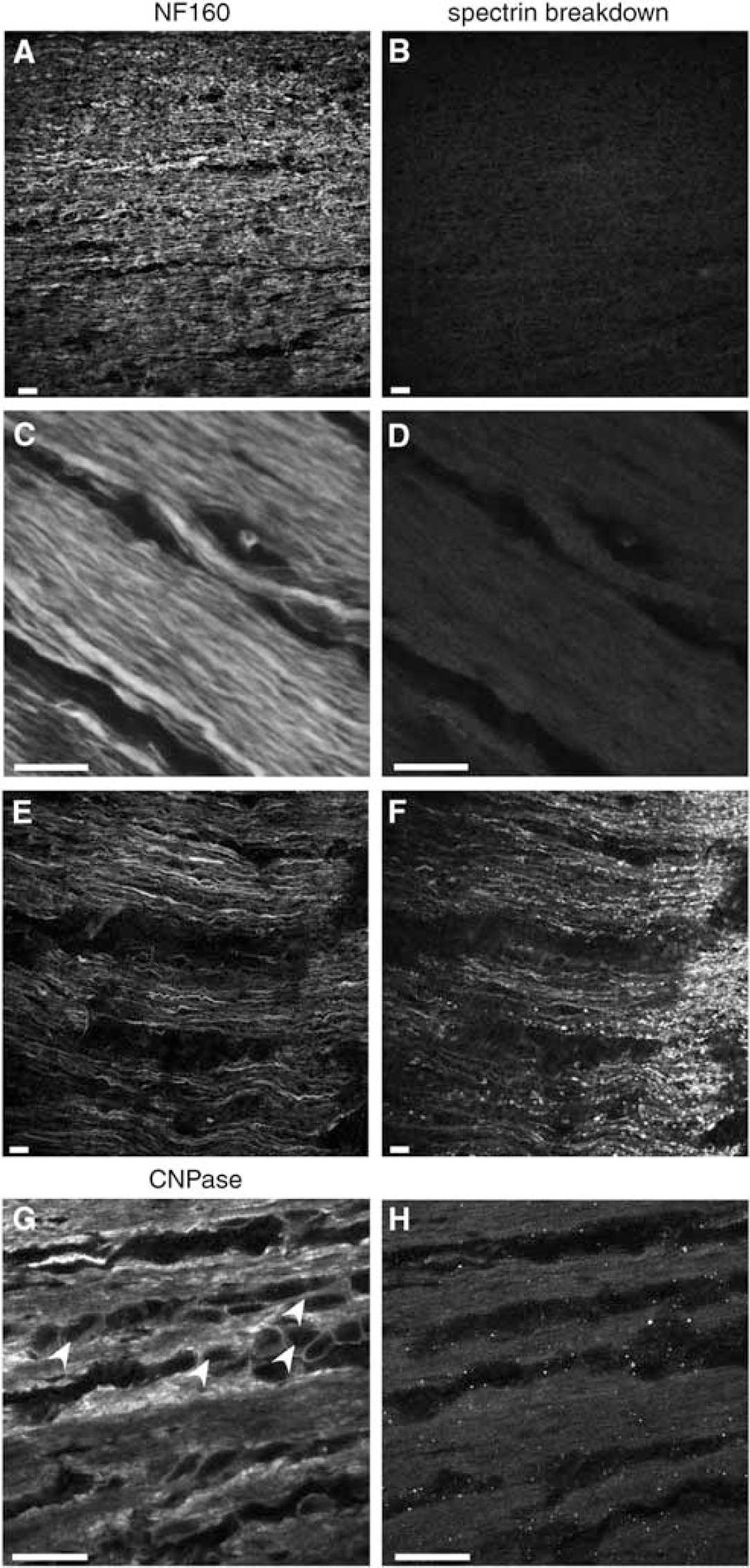
Confocal microscopic images of fixed normoxic control optic nerves stained immunohistochemically with standard markers (neurofilament and CNPase to identify axon cylinders and oligodendrocytes, respectively) and for spectrin breakdown. (
Figure 2 shows control optic nerve as above, but double labeled for Na channels and SBP. Numerous punctate regions reflect the high density of Na channels at nodes of Ranvier. Once again, control optic nerves did not exhibit spectrin breakdown, either diffusely or more specifically at the nodes of Ranvier.
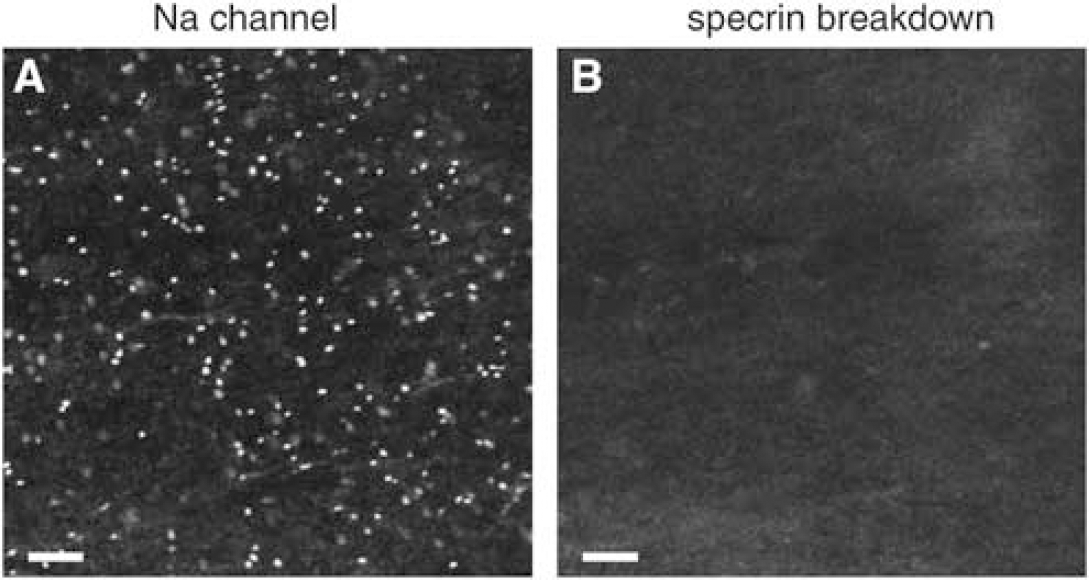
Confocal micrographs of control normoxic optic nerve labeled with anti-pan Na channel antibody (
Spectrin Breakdown in Anoxic Optic Nerves
Optic nerves exposed to 15, 30, or 60 mins of anoxia at 37°C in the in vitro perfusion chamber were processed for immunohistochemistry using the same markers as for control nerves above. Compared with normoxic controls, no significant spectrin breakdown was observed after 15 mins of anoxia (Figures 3A and 3B). However, significant spectrin cleavage was seen after 30 mins of anoxia (Figures 3C and 3D). The pattern exhibited a roughly linear appearance which followed the direction of the fibers, but was otherwise diffuse, with no indication of any focal onset of spectrin proteolysis. Given the expected ionic disturbances that may begin at nodal regions, we hypothesized that spectrin breakdown may start in this location; however, nodal labeling with Na channel antibody did not suggest preferential colocalization of spectrin breakdown at nodes of Ranvier (Figures 3E and 3F).
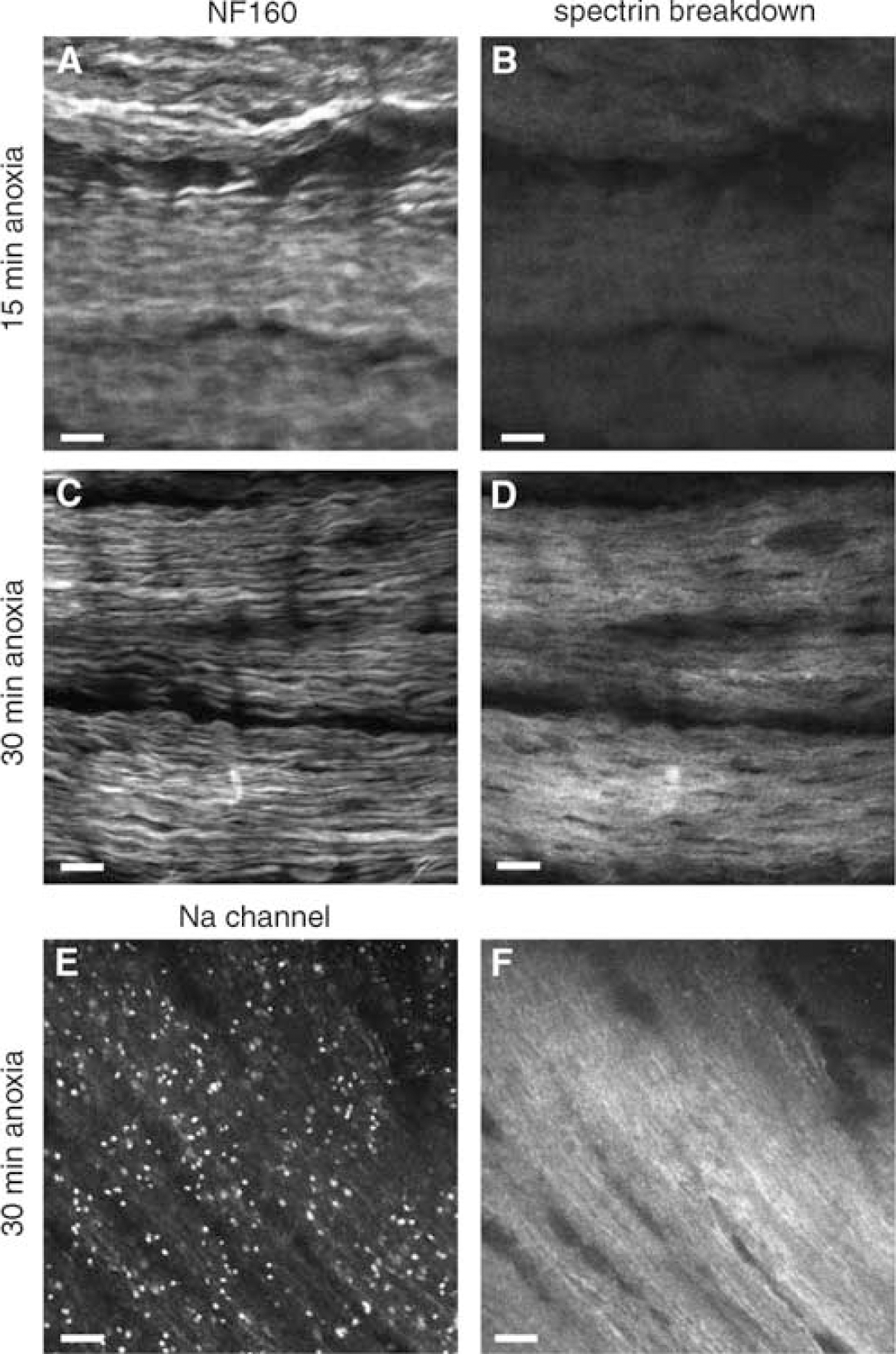
Confocal micrographs of optic nerves harvested after 15 or 30 mins of in vitro anoxia. (
After 60 mins of anoxia, substantial spectrin breakdown was seen as expected, but neurofilament profiles appeared more disrupted, indicating increasing damage to axon cylinders (Figure 4). Many axons (particularly larger diameter fibers) displayed strong SBP signal colocalized with much weaker and disrupted neurofilament staining (arrowheads in Figures 4B and 4C indicate identical regions of weak neurofilament colocalized with strong SBP). Interestingly, much of the SBP signal was found to lie outside axon cylinders, often immediately adjacent to these profiles (Figure 4E to 4G), even while oligodendroglial cell bodies did not exhibit significant spectrin breakdown even after 60 mins of anoxia (Figures 5A and 5B). Most of the SBP signal was associated either with distal processes of these cells and/or axon cylinders (Figures 4D to 4G and 5B). Even after 60 mins of anoxia, double-labeling with Na channel antibody and SBP did not indicate a preferential focus of spectrin cleavage at nodes of Ranvier (Figures 5C and 5D).
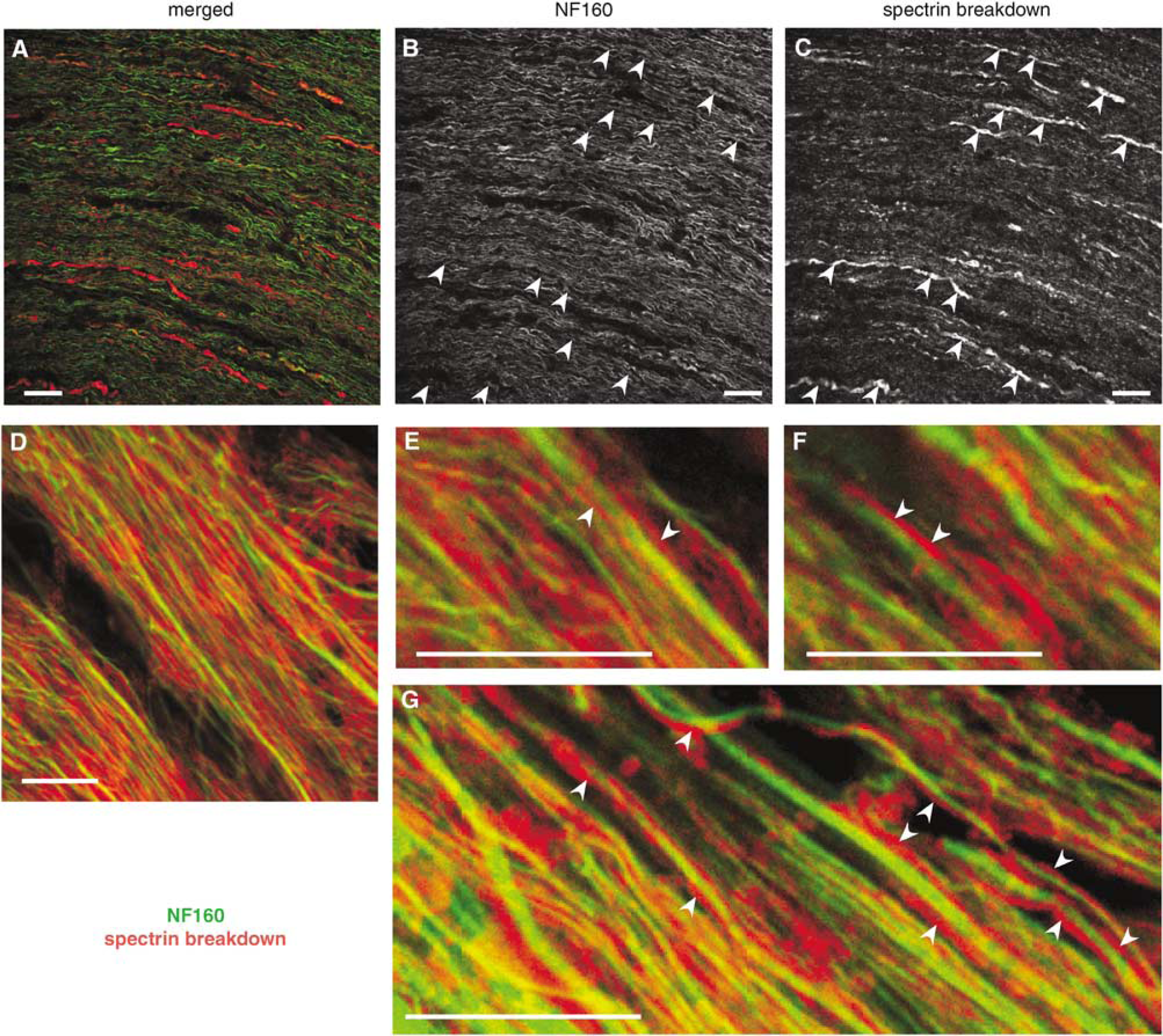
Confocal micrographs of optic nerve exposed to 60 mins of anoxia. At this time point, substantial spectrin breakdown was seen both within axon cylinders and in glial processes surrounding axons. Areas of more intense staining for spectrin breakdown (red channel panel
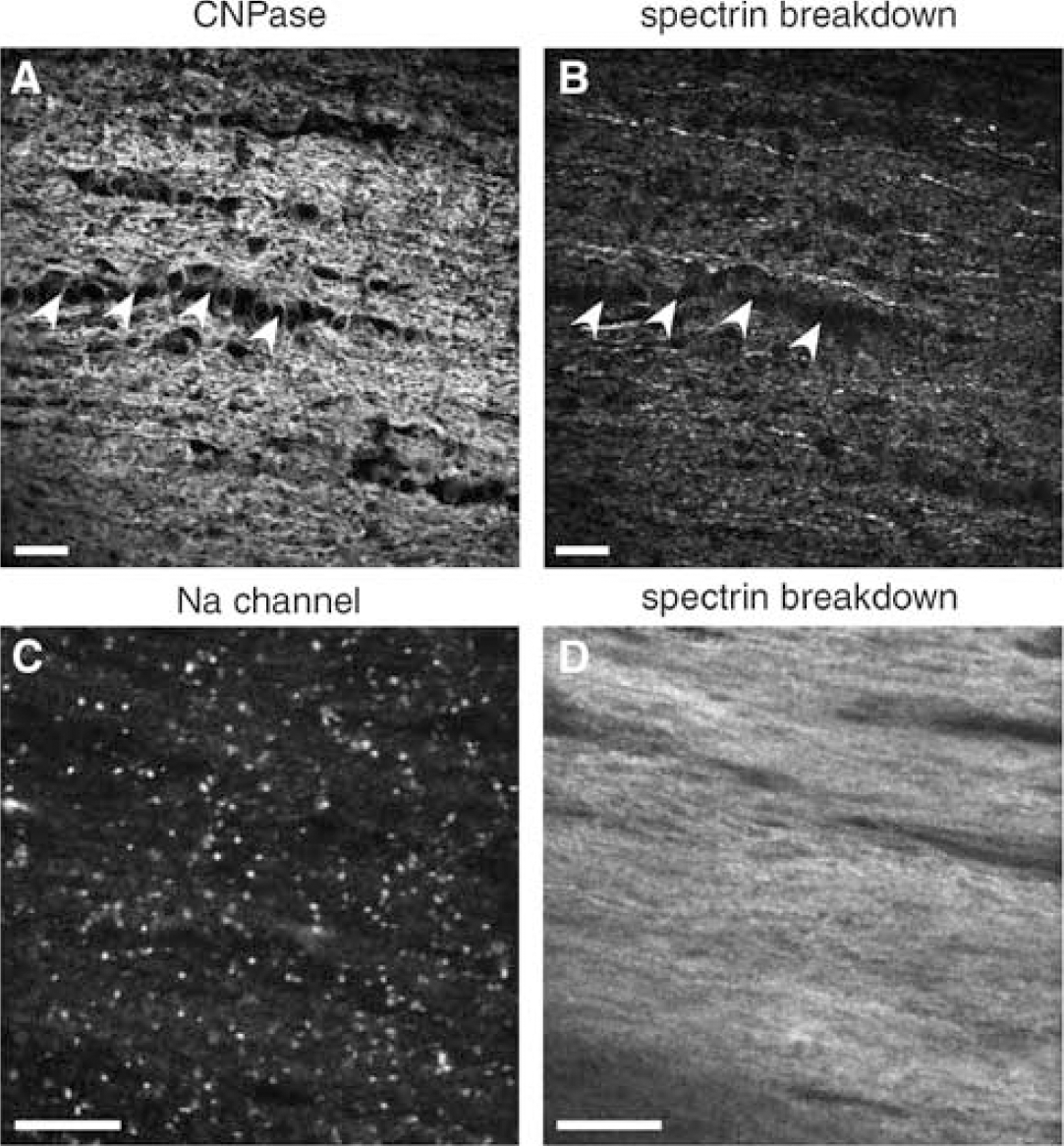
Confocal micrographs of fixed immunostained optic nerve exposed to 60 mins of anoxia. (
Spatiotemporal Changes in Axoplasmic [Ca] During Anoxia
While spectrin breakdown is a useful marker of Ca-dependent subcellular injury, because enzymatic action may require time to generate immunodetectable products, the spatial profiles of these breakdown products may not necessarily reflect initial spatial gradients of Ca accumulation. In an effort to confirm the occurrence of localized Ca ‘hot-spots’ in the early stages of anoxia, we imaged axoplasmic [Ca] directly in living optic nerve axons using fluorescent Ca reporters and confocal microscopy. Figure 6 shows representative confocal micrographs of optic nerve axons loaded with a red Ca-insensitive dye (Texas Red dextran), which conveniently outlines axon cylinders in isolation (myelin and glial cell bodies do not take up the large dextran conjugates), and a dextran-conjugated Ca reporter (Oregon Green 488 BAPTA-1 dextran). There was a slight rundown of the red reference channel beginning at 10 to 15 mins (typically 10% to 20% at the end of 30 mins), with a concomitant rise in Ca-sensitive emission indicating that axoplasmic free [Ca] began to rise within minutes of anoxia onset, as shown by the graph in Figure 6. After 30 mins of anoxia, Ca fluorescence increased approximately 2.5-fold over baseline. Careful spatial analysis of Ca-sensitive fluorescence patterns along the lengths of the fibers did not disclose any observable Ca ‘hot-spots’ the anoxia-induced Ca increases appeared to occur homogeneously along the length of the fibers, and continued in a gradual manner for the 30 mins duration of these experiments. Although we could not identify nodes of Ranvier in these live imaging experiments, there is no doubt that many nodes were included in each image judging by the density of nodes revealed by Na channel immunostaining (compare with Figure 5C, for example).
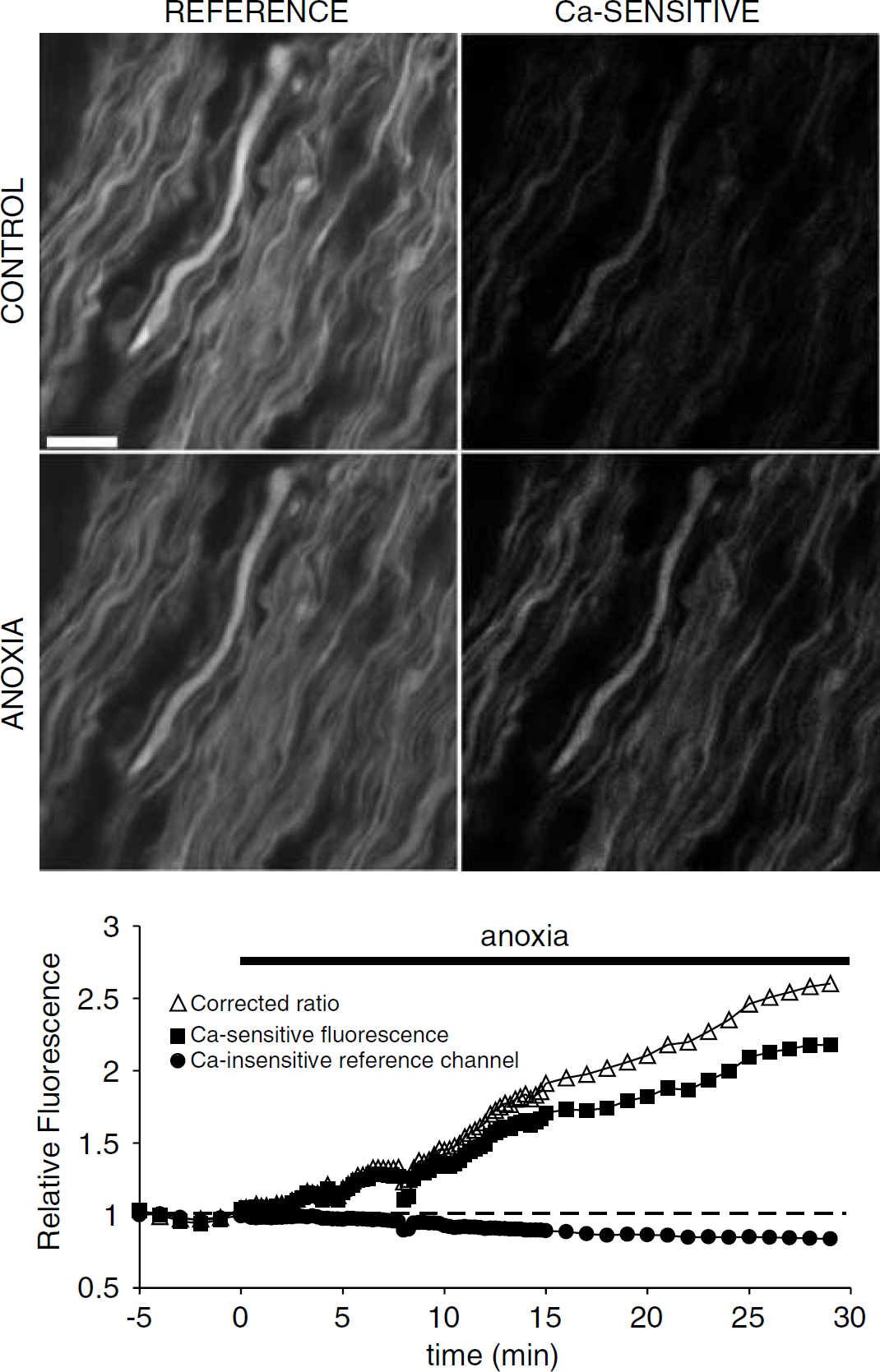
Confocal micrographs of live optic nerve loaded with a Ca-insensitive dextran-conjugated reference dye (left panels) designed to outline axon cylinders, and the Ca reporter Oregon Green 488 BAPTA-1 (right panels). The integrity of the axon cylinders remained relatively intact after 30 mins of in vitro chemical anoxia, as shown by the preservation of the profiles and modest dye loss. Mean axoplasmic [Ca] concentration began to rise within minutes of anoxia onset and continued throughout the 30 mins challenge. Detailed analysis of time series images failed to reveal focal Ca accumulations at any time point, indicating a rather homogeneous increase in Ca levels. Scale bar 10 μm.
Na Channel Proteolysis During Anoxia/Ischemia
Although spectrin is a key component of the cellular cytoskeleton and its breakdown products are a useful marker of structural injury, many other key functional proteins might also be damaged by anoxia/ischemia; one key target might be voltage-gated Na channels which are essential for generating and propagating axonal action potentials. Indeed, proteolysis of these channels has been shown to occur as a consequence of traumatic stretch injury to axons (Iwata et al, 2004). We performed Western blots probed using a panvoltage-gated Na channel antibody (Figure 7). Optic nerves were subjected to either 1 h of in vitro anoxia, oxygen-glucose deprivation (‘OGD’ to simulate a more severe ischemic injury) or normoxic incubation at 37°C. After anoxia, the integrated density of the Na channel bands decreased to 75% of control, and OGD further reduced the signal to 69% of normoxic control nerves. These results suggest that anoxia and ischemia induce a gradual degradation of the intact Na channel proteins.
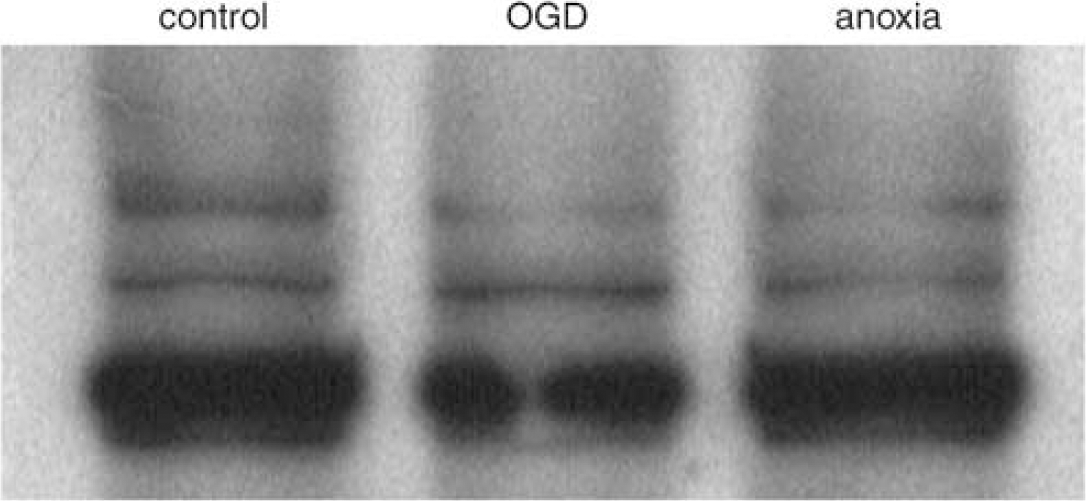
Immunoblot probed for the alpha subunit of the voltage-gated Na channel using a pan-specific antibody. Samples were obtained from optic nerves exposed to either 60 mins of OGD or anoxia in vitro. Anoxia reduced the integrated density of the Na channel bands to 75% of control, and OGD to 69% of normoxic control nerves, indicating proteolysis of Na channel subunits during anoxic/ischemic injury.
Discussion
Central axons suffer damage in a number of disorders, such as traumatic injury to the brain and spinal cord; acute stroke, neonatal periventricular leukomalacia and more chronic subcortical ischemia; inflammatory demyelinating diseases such as multiple sclerosis; and the encephalopathy associated with HIV infection (Back, 2001; Maxwell et al, 1997; Raja et al, 1997; Schwartz and Fehlings, 2002; Trapp et al, 1998). As distinct as the initial inciting events might be, these conditions result in overlapping white matter neuropathology, which includes demyelination, axonal damage, and finally irreversible loss of brain and spinal fibers. Indeed, it is thought that loss of central axons is a key predictor of clinical outcome (Medana and Esiri, 2003). Neuropathological examination reveals axonal damage that is surprisingly focal, at least early in the course before Wallerian degeneration results in complete disappearance of the fiber: axonal spheroids and swellings are characteristic findings in brains of patients with multiple sclerosis, neurotrauma victims, and those with ischemic injury (Povlishock and Christman, 1995; Trapp et al, 1998; Yam et al, 1998). Such swellings display strong staining for beta-amyloid precursor protein (suggesting focal failure of axonal transport) (Stone et al, 2004; Yam et al, 1998) and spectrin breakdown (Buki et al, 1999), indicating a Ca-mediated disruption of the axonal cytoskeleton. Thus, there might be selective regions of the axon that are vulnerable to spheroid formation and ultimately transection compared with other areas. The unanswered question is whether the initial locus of injury is random or whether it is governed by intrinsic structural/functional inhomogeneities of myelinated axons.
The most striking feature of myelinated axons is the highly specialized node of Ranvier (for reviews, see Poliak and Peles, 2003; Scherer and Arroyo, 2002). This interruption of the ensheathing myelin occupies only ≈ 1% of the axon's surface area, yet contains sufficient channel densities to rapidly conduct inward action currents to support saltatory conduction. Nodes contain high densities of Na channels, and thus would be expected to experience significant axoplasmic fluctuations of [Na] during normal action potential propagation, and also during pathological conditions such as anoxia/ischemia. Because much of the pathological accumulation of axonal Ca is driven both by depolarization and a rise in axoplasmic [Na] (for a recent review, see Stys 2004), we hypothesized that Ca deregulation and accumulation would begin at nodes of Ranvier, leading to preferential damage to the cytoskeleton and possibly to Na channels themselves, in this region.
Examining the spatio-temporal distributions of Ca ions as a function of injury in small-diameter central axons is very difficult. We adopted two complementary strategies: the first was to immunohistochemically assay the distribution of calpain-cleaved breakdown products of spectrin, a ubiquitous cytoskeletal structural protein. The calpains are a family of cysteine proteases whose enzymatic activity is stimulated by ambient free Ca ions (Croall and DeMartino, 1991; Melloni and Pontremoli, 1989); therefore, locally elevated Ca levels should activate calpain, and in turn result in preferential accumulation of breakdown products in regions of high [Ca]. Rat optic nerves exposed to 0, 15, 30, and 60 mins of in vitro anoxia failed to display preferential ‘hot-spots’ of spectrin cleavage: the pattern of spectrin breakdown evolved from virtually undetectable signal in normoxic control specimens and those exposed to 15 mins of anoxia (Figures 1 and 3B), to a fairly homogeneous increase in SBPs that began between 15 and 30 mins of anoxia (Figures 3D and 3F). Careful examination of sections double-labeled with Na channel antibodies which clearly identified nodes of Ranvier (Figures 2A, 3E and 5C) failed to reveal any preferential accumulation of SBPs in this area. The cut (and therefore traumatized) ends of control nerves, and nerves exposed to longer periods of anoxia (30 and 60 mins), exhibited clear and strong spectrin breakdown staining, indicating that the antibody was reliable and could easily detect these products in damaged regions.
After 60 mins of anoxia, many axons displayed fragmentation of their cylinders, as shown by a more mottled appearance of neurofilament staining, which often overlapped with stronger spectrin breakdown signal (Figures 4A to 4C). This was particularly evident in large-diameter fibers. Neurofilament breakdown is expected, as this structural protein is also a substrate for calpain (Banik et al, 1997; Schumacher et al, 2000) (as are many others such as tau, tubulin, ankyrin (Rami et al, 1997)), and cleavage of these proteins can be blocked by calpain inhibitors (Stys and Jiang, 2002). Interestingly, little spectrin breakdown was observed in the cell bodies of oligodendroglia, even after 60 mins of anoxia. Although these cells are known to be vulnerable to anoxic/ischemic injury (Dewar et al, 2003), our relatively limited 60 mins exposure to anoxia (compared with ischemia) might have produced only a minor degree of structural injury to these cells which can be detected by our antibody. In contrast, glial processes running parallel to axons (probably oligodendrocytic, e.g. Figures 4D to 4G) displayed significant spectrin degradation products, indicating that these distal glial extensions appear more vulnerable to anoxia than the parent cell bodies, possibly due to preferential localization of m-calpain to these regions (Ray et al, 2002).
Analyzing Ca-dependent enzymatic activity is a reliable, albeit indirect, method of inferring fluctuations in cytosolic Ca levels. It was also possible that Ca does accumulate focally at nodes, but because it may take time for the enzyme to be activated and to degrade spectrin, the resulting injury (as evidenced by SBP staining) might have been spatially ‘smeared’, and focal Ca rises missed. For this reason, we complemented the immunohistochemical studies with direct measurements of axoplasmic Ca using a high-affinity Ca indicator and confocal microscopy. Images were initially acquired every 15 secs after the start of anoxia in case any focal Ca elevations were brief, and could be missed with longer sampling intervals. Axoplasmic [Ca] began to rise within minutes of anoxia onset, and continued for the duration of the experiment (30 mins). No focal Ca elevations were observed in any axons at any time point: Ca levels rose gradually and homogeneously. Although we did not identify nodes of Ranvier in the live axon-imaging experiments, the high density of nodes found in fixed tissue implies that we had many nodes in each imaging field, so the absence of potential nodal Ca ‘hot-spots’ was not because we might have missed nodal regions in our sampling of random areas. As our Ca measurements could not be calibrated (due to the overlying myelin sheath which would prevent clamping axoplasmic [Ca] with ionophores), we cannot provide absolute [Ca]'s. However, based on previous experiments from our lab (Nikolaeva and Stys, 2002) and the Ca levels required to activate μ-calpain (Croall and DeMartino, 1991; Melloni and Pontremoli, 1989), we estimate that axoplasmic [Ca] rose into the micromolar range.
The absence of focal Ca accumulation implies either that (1) Ca entry across the axolemma (and potentially release from internal stores (Ouardouz et al, 2003)) occurred continuously or that (2) Ca influx/release was focal, but diffusion of the ion along the fibers was rapid enough to preclude focal accumulations. Numerical simulations of Ca diffusion along a cylindrical volume mimicking an axon are shown in Figure 8, using standard diffusion equations for a semi-infinite medium (Crank, 1975; David et al, 1997). This model assumes a large localized source of Ca (a completely open axon or highly permeable locus of entry) whose concentration approaches that of the extracellular space. The surface plot shows the profile of [Ca] concentration from this point source as a function of distance (we assume ions would diffuse similarly in each direction from this point source; therefore only one half of a model axon was simulated) and time. The diffusion coefficient of Ca in axoplasm varies with the state of the fiber, and is estimated at 1.0 ×10−7 cm2/s for Myxicola axoplasm in intact axons (al-Baldawi and Abercrombie, 1995), rising substantially to 5.3 ×10−6 cm2/s in metabolically poisoned fibers whose Ca sequestering and buffering systems were compromised (Donahue and Abercrombie, 1987). We used the latter value in our simulations, reasoning that anoxic axons would also have impaired Ca handling. Under these conditions, Figure 8 shows a remarkably rapid rate of Ca diffusion along the length of the axon. For a fairly large 2-μm-diameter optic nerve fiber, the Ca concentration profile rises fairly uniformly along the internodal length (the predicted internodal length for such a fiber would be ≈ 200 μm; so the furthest location from any node, the initially hypothesized point source of Ca, would be no greater than 100 μm). This model assumes an immediate local rise of [Ca] to 2 mmol/L at time 0. In reality, a more restricted permeability to Ca at a point source such as a node would increase focal intraaxonal Ca more slowly, yielding an even more homogenous spatial increase. These simulations support the notion that even if Ca accumulated focally in injured axons (be it from the influx at the nodes or focal release from cisternae of axoplasmic reticulum (Ouardouz et al, 2003)), this ion would rapidly diffuse into the internodes and not result in focal accumulations. In the absence of focal Ca accumulations in anoxic axons, it is interesting to speculate why focal structural damage (in the form of axonal spheroids) is often observed in anoxic/ischemic or traumatized axons. One possibility might be that the source of Ca is not focal, but that the vulnerability of axons to a damaging increase in axoplasmic [Ca] is inhomogeneous, with certain areas being intrinsically more susceptible than others.
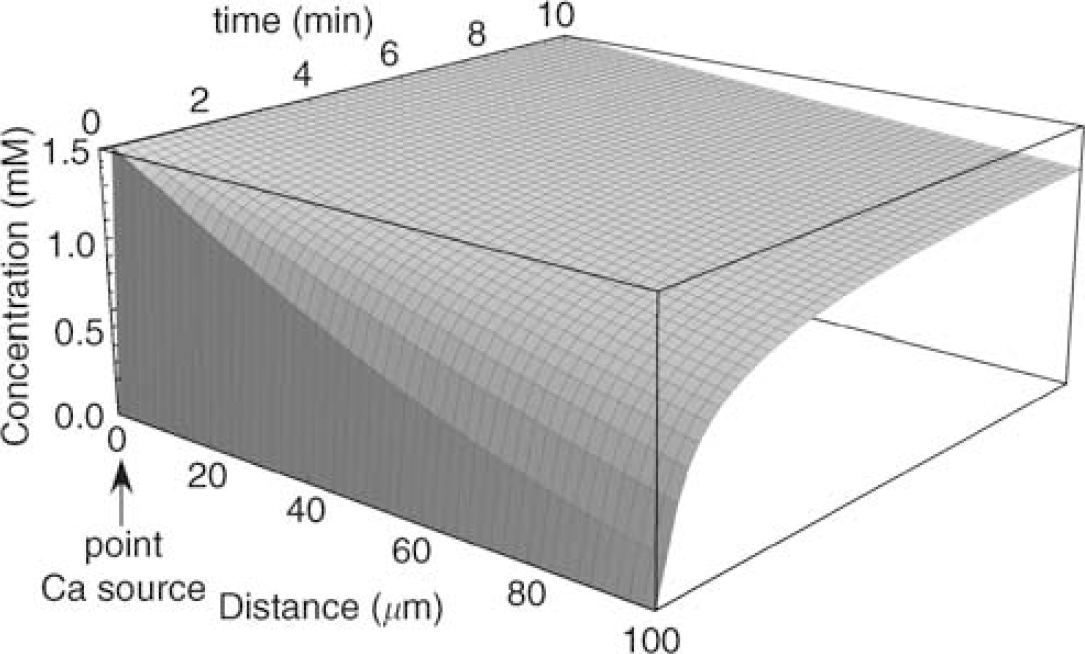
Surface plot showing calculated free [Ca] as a function of distance and time in an ideal cylinder simulating an axon. A point source of Ca is assumed at 0 μm, set at 2 mmol/L Ca. Note that the concentration of Ca along the vertical axis is in millimolar. Assuming that Ca influx into an anoxic fiber occurred at a single locus (e.g. node of Ranvier), this simulation shows that Ca diffusion would raise axonal Ca to very high levels within minutes, even many tens of microns away from the source. Therefore, the absence of focal injury (e.g. reflected by localized spectrin breakdown, for example) does not necessarily rule out a localized point of Ca entry into anoxic fibers. Similarly, the relatively rapid diffusion of this ion would quickly ‘smear’ the spatial profile and explain why real-time imaging of axoplasmic Ca failed to discern ‘hot-spots‘.
Although our results and those from previous studies (Buki et al, 1999; Saatman et al, 2003; Schumacher et al, 2000; Stys and Jiang, 2002) clearly indicate extensive injury to structural proteins in injured axons, we also wanted to examine whether key functional proteins were damaged by anoxia/ischemia as well. Voltage-gated Na channels are damaged by proteases in a Ca-dependent manner after mechanical stretch injury to axons, with the suggestion raised of an upregulation of the persistent component of Na currents resulting from this proteolytic cleavage of the alpha subunit (Iwata et al, 2004). Our results from anoxic and ischemic optic axons parallel these observations; while our results are only qualitative, we found a consistent reduction in the amount of immunodetectable Na channel protein after exposure to in vitro anoxia or simulated ischemia. Degradation of this key protein likely contributes to a significant degree to the overall functional injury caused by anoxia/ischemia in optic nerve.
In summary, the present results indicate that anoxia produces a sustained increase in axoplasmic [Ca], and degradation of the spectrin cytoskeleton in axons and distal glial processes beginning between 15 and 30 mins of anoxia. Contrary to our initial hypothesis, structural injury does not begin at nor is it preferentially localized to nodes of Ranvier, where exaggerated ionic dysregulation might be expected to occur. While we cannot exclude a focal source of Ca (either influx at nodal regions and/or focal release from internal stores), it appears that diffusion of this ion along the axoplasm in energy-depleted fibers is rapid enough to raise [Ca] fairly homogeneously and induce injury continuously along the length of fibers in our in vitro model of anoxia. Equally important is our finding that key functional proteins such as voltage-gated Na channels are also targets of proteolytic cleavage, which will conspire to render an axon nonfunctional, eventually leading to its degeneration.