
Abstract
Very little is known regarding the mechanisms of action of angiotensin II (Ang II) or the consequences of Ang II-dependent hypertension in the cerebral circulation. We tested the hypothesis that Ang II produces constriction of cerebral arteries that is mediated by activation of AT1A receptors and Rho-kinase. Basilar arteries (baseline diameter ~130 µm) from mice were isolated, cannulated and pressurized to measure the vessel diameter. Angiotensin II was a potent constrictor in arteries from male, but not female, mice. Vasoconstriction in response to Ang II was prevented by an inhibitor of Rho-kinase (Y-27632) in control mice, and was reduced by ~85% in mice deficient in expression of AT1A receptors. We also examined the chronic effects of Ang II using a model of Ang II-dependent hypertension, mice which overexpress human renin (R+) and angiotensinogen (A+). Responses to the endothelium-dependent agonist acetylcholine were markedly impaired in R+ A+ mice (P < 0.01) compared with controls, but were restored to normal by a superoxide scavenger (PEG-SOD). A-23187 (another endothelium-dependent agonist) produced vasodilation in control mice, but no response or vasoconstriction in R+ A+ mice. In contrast, dilation of the basilar artery in response to a NO donor (NONOate) was similar in R+ A+ mice and controls. Thus, Ang II produces potent constriction of cerebral arteries via activation of AT1A receptors and Rho-kinase. There are marked gender differences in cerebral vascular responses to Ang II. Endothelial function is greatly impaired in a genetic model of Ang II-dependent hypertension via a mechanism that involves superoxide.
Introduction
Angiotensin II (Ang II) plays a major role in vascular disease. For example, in peripheral blood vessels (or vascular cells in culture), Ang II alters gene expression, activates components of the inflammatory response, and produces oxidative stress as well as changes in vascular structure (Dzau, 2001; Griendling et al, 2000; Nickenig and Harrison, 2002). In the cerebral circulation, Ang II produces impairment of functional hyperemia and inward remodeling of cerebral blood vessels (Kazama et al, 2004; Baumbach et al, 2003).
Although the effects of Ang II on vasomotor tone have been studied, relatively little is known regarding the mechanisms that mediate constrictor effects of Ang II in the cerebral circulation. The present study had several goals. First, we determined if Ang II produces constriction of cerebral arteries in the mouse. Because vascular responses can exhibit sexrelated differences (Lamping and Faraci, 2001), we also determined if the effects of Ang II vary with gender. Second, we examined the role of Rho-kinase in mediating vasoconstriction in response to Ang II. Activation of Rho-kinase is thought to be a key mechanism of calcium sensitization that mediates responses to several vasoconstrictors (Miao et al, 2002; Nishikawa et al, 2003; Wickman et al, 2003; Luykenaar et al, 2004).
The effects of Ang II occur via activation of two receptor subtypes, AT1 and AT2 (Hein, 1998; Crowley et al, 2004). The vasoconstrictor effects of Ang II are generally considered to result from activation of AT1 receptors. In rodents, two AT1 receptor subtypes (AT1A and AT1B) exist, but cannot be distinguished using pharmacological inhibitors (Hein, 1998; Crowley et al, 2004). Thus, our third goal was to use genetically altered mice deficient in expression of AT1A receptors to examine the importance of the AT1A receptor in mediating the vascular effects of Ang II.
In the peripheral circulation, Ang II produces oxidative stress and impairment of endothelium-dependent relaxation (Dzau, 2001; Griendling et al, 2000; Nickenig and Harrison, 2002). We are not aware of studies that have examined the effects of chronic Ang II-dependent hypertension on vascular function in brain. Thus, our last goal was to examine endothelial function in a genetic model of chronic hypertension, transgenic mice that overexpress human renin and human angiotensinogen (R+ A+) (Baumbach et al, 2003; Merrill et al, 1996; Didion et al, 2000, 2002).
Methods
Animal Preparation
The protocol used in these experiments was reviewed and approved by the University of Iowa Animal Care and Use Committee. Mice were provided with regular chow and water ad libitum. Male or female C57BL/6J mice were used for a portion of the study. A colony of AT1A receptor mice was maintained on a mixed genetic background (~87% 129P3/J and ~13% C57BL/6J) (Ryan et al, 2004) and AT1A+/+ littermates were used as controls. We included AT1A+/+ mice in our experiments because studies of heterozygousdeficient mice may lead to studies of genetic polymorphisms in humans (Takahashi and Smithies, 2004).
Double-transgenic (R+ A+) mice were generated by crossbreeding single transgenic human renin (R+) mice with single transgenic human angiotensinogen (A+) mice (Baumbach et al, 2003; Merrill et al, 1996; Didion et al, 2000, 2002). Only double-transgenic animals have elevated circulating Ang II and increased blood pressure (Merrill et al, 1996). Because blood pressure and vascular responses to acetylcholine are not different between R−A−, R−A+, and R+ A− mice (Didion et al, 2000), responses from these three groups are pooled and simply referred to as the control group. Genotyping of AT1A and R+ A+ mice was performed using PCR of DNA from tail biopsies.
Cerebral Arteries In Vitro
After induction of deep anesthesia (pentobarbital, 200 mg/kg intraperitoneally), the brain was rapidly removed and placed in ice-cold Krebs buffer. As described previously (Yamada et al, 2001; Lamping et al, 2004), the basilar artery was isolated using a dissecting microscope, mounted onto glass micropipettes filled with Krebs buffer in an organ chamber, and secured with nylon monofilament suture. Arteries were pressurized to 60 mm Hg. Using a microscope and a video camera, vessel images were projected on a video monitor. An electronic dimension analyzer was then used to measure the lumen diameter.
Basilar arteries were allowed to equilibrate and reach a stable diameter for at least 30 min at a distending pressure of 60 mm Hg before protocols were initiated. We examined changes in the diameter of the basilar artery in response to abluminal KCl (50 mmol/L) and cumulative doses of Ang II. In one group, we examined the effects of two applications of KCl and Ang II (with a 30-min recovery period between applications). The purpose of these experiments was to determine if responses to these stimuli were reproducible. In a second group, we examined the effects of KCl and Ang II on vascular tone. Then, after the recovery period, responses to Ang II and KCl were repeated in the presence of an inhibitor of Rho-kinase (Y-27632, 3 µmol/L).
For studies in which responses to acetylcholine or other vasodilators were examined, basilar arteries were preconstricted by approximately 30% (~60% of the response to 50 mmol/L KCl) with the thromboxane mimetic U-46619. Acetylcholine and A-23187 are endothelium-dependent agonists (Faraci, 1992; Faraci and Heistad, 1998), and thus were used to examine endothelial function. In some studies, the nitric oxide donor NONOate was also used.
In R+ A+ mice, we also determined whether superoxide contributes to the alterations in vascular responses. For this experiment, responses to acetylcholine were examined after a 30-min treatment with PEG-SOD (50 U/mL). We have shown previously that PEG-SOD does not alter the responses of cerebral vessels under control conditions, but is efficacious in reducing superoxide levels in blood vessels (Didion et al, 2002, 2005).
Statistical Analysis
Constriction in response to KCl and Ang II was determined by calculating the percent reduction in vessel diameter from the baseline control level. For vasodilator responses, results are expressed as percent dilation (% of induced tone), with 100% representing the difference between the resting value under basal conditions and the constricted value with U-46619. As appropriate, comparisons were made using paired or unpaired t-tests or ANOVA with repeated measures, followed by Student–Newman–Keuls test to detect individual differences. P < 0.05 was defined as significant.
Results
Vasoconstrictor Responses to Angiotensin II are Greater in Male than in Female Mice
Baseline diameters of the basilar arteries in male and female C57BL/6J mice were 138 ± 2 and 137 ± 6 µm, respectively (n = 19 and 7). Angiotensin II was a potent constrictor of basilar arteries from male mice, with reductions in diameter starting at a concentration of 0.01 nmol/L and maximum vasoconstriction occurring at approximately 1 nmol/L (Figure 1). In contrast, basilar arteries from female mice were relatively unresponsive to Ang II (Figure 1). This difference in responsiveness was selective for Ang II, as vasoconstriction to KCl (Figure 1) and U-46619 (data not shown) as well as vasodilation in response to acetylcholine were similar in male and female mice (Figure 1).
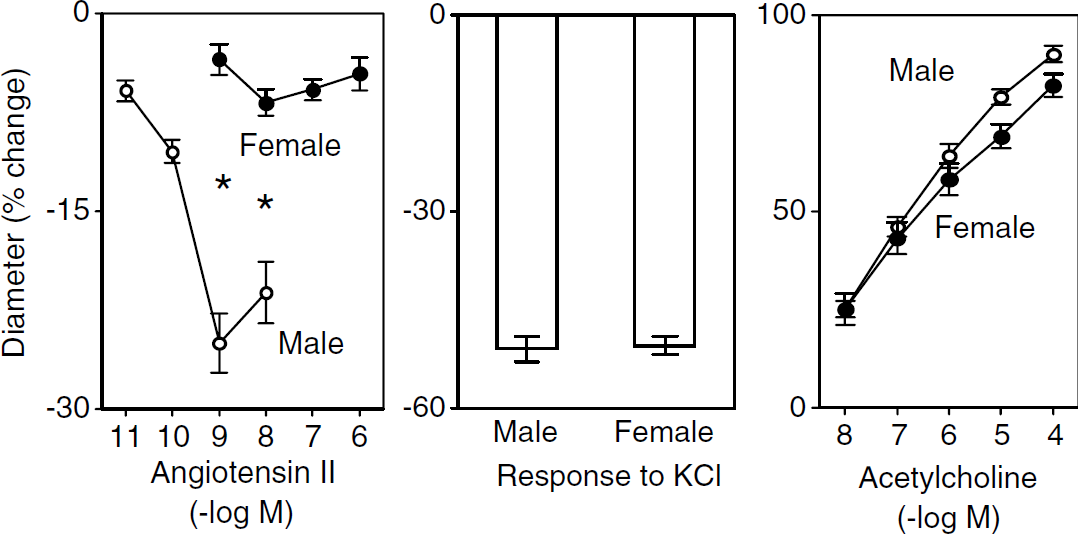
Constriction of the basilar artery in response to Ang II (left panel) and KCl (middle panel) and vasodilation in response to acetylcholine (right panel) in male and female mice. Values are mean ± s.e. Responses to 1 and 10 nmol/L angiotensin II were significantly less in female mice (* P < 0.01 versus males).
Vasoconstriction in Response to Angiotensin II is Mediated by Rho-Kinase
Because responses to Ang II were very modest in arteries from female mice, experiments examining the mechanisms that mediate vasoconstriction were performed using male mice. Under control conditions, responses to Ang II and KCl were reproducible. For example, 1 nmol/L Ang II produced 21% ± 4% and 18% ± 3% constriction of the basilar artery during the first and second applications, respectively (n = 5). Application of Y-27632 completely prevented vasoconstriction in response to Ang II (n = 14) (Figure 2). This inhibitory effect of Y-27632 was selective, as constriction of the basilar artery in response to KCl was similar in the absence and presence of Y-27632 (n = 5) (Figure 2).
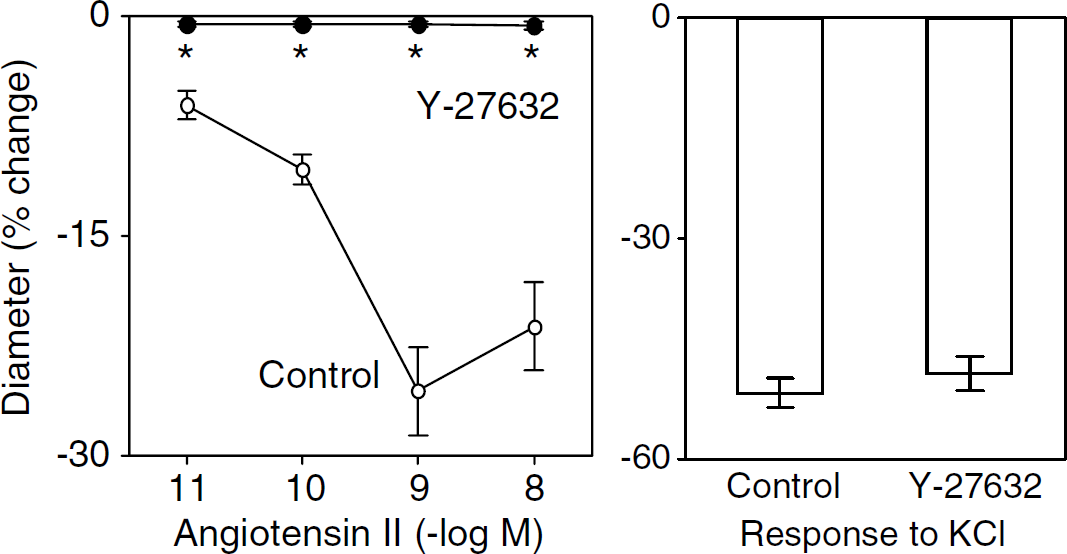
Constriction of the basilar artery in response to Ang II (left panel) and KCl (right panel) in the absence (Control) and presence of Y-27632. Values are mean ± s.e. *P < 0.05 versus Control.
Vasoconstrictor Responses to Angiotensin II are Mediated by AT1A Receptors
Baseline diameters were similar in basilar arteries from male mice with the three different AT1A genotypes (128 ± 5, 127 ± 6, and 130 ± 7 µm in AT1A+/+ (n = 8), AT1A+/+ (n = 7), and AT1A+/+ mice (n = 7), respectively). In littermate controls (AT1A+/+), Ang II produced constriction of the basilar artery (Figure 3). This response was similar in AT1A+/+ and AT1A+/+ (data not shown) mice, but was almost completely absent in AT1A+/+ mice (Figure 3). This marked difference in vascular responses between mice with different AT1A genotypes was selective for Ang II, as vasoconstriction in response to KCl was similar in AT1A+/+, AT1A+/+ (data not shown), and AT1A+/+ mice (Figure 3). Similarly, dilation of the basilar artery in response to acetylcholine was similar in AT1A+/+, AT1A+/+ (data not shown), and AT1A+/+ mice (Figure 3).
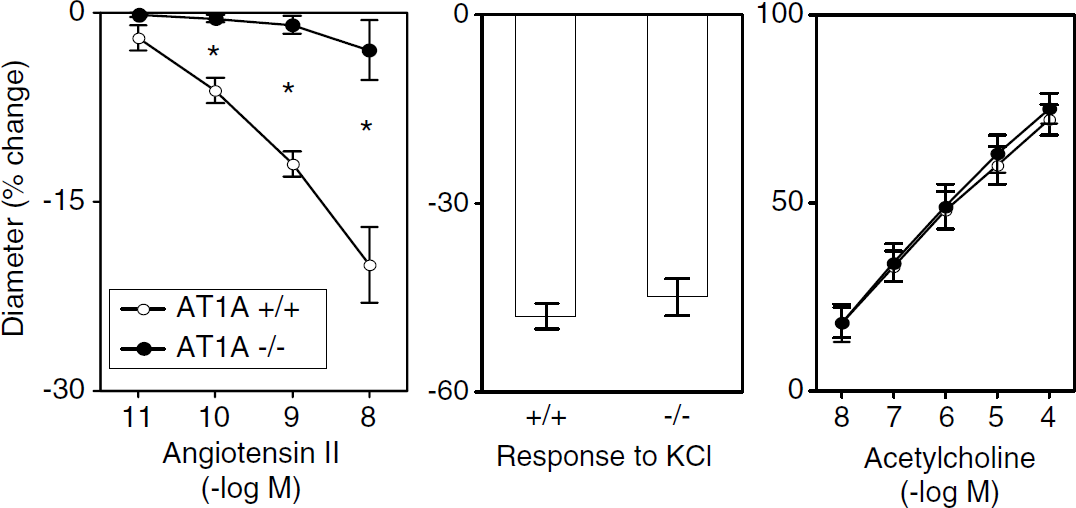
Constriction of the basilar artery in response to Ang II (left panel) and KCl (middle panel) and vasodilation in response to acetylcholine (right panel) in AT1A+/+ and AT1A+/+ mice. Values are mean ± s.e. *P < 0.05 versus AT1A+/+ mice.
Endothelial Function is Markedly Impaired in R+ A+ Mice
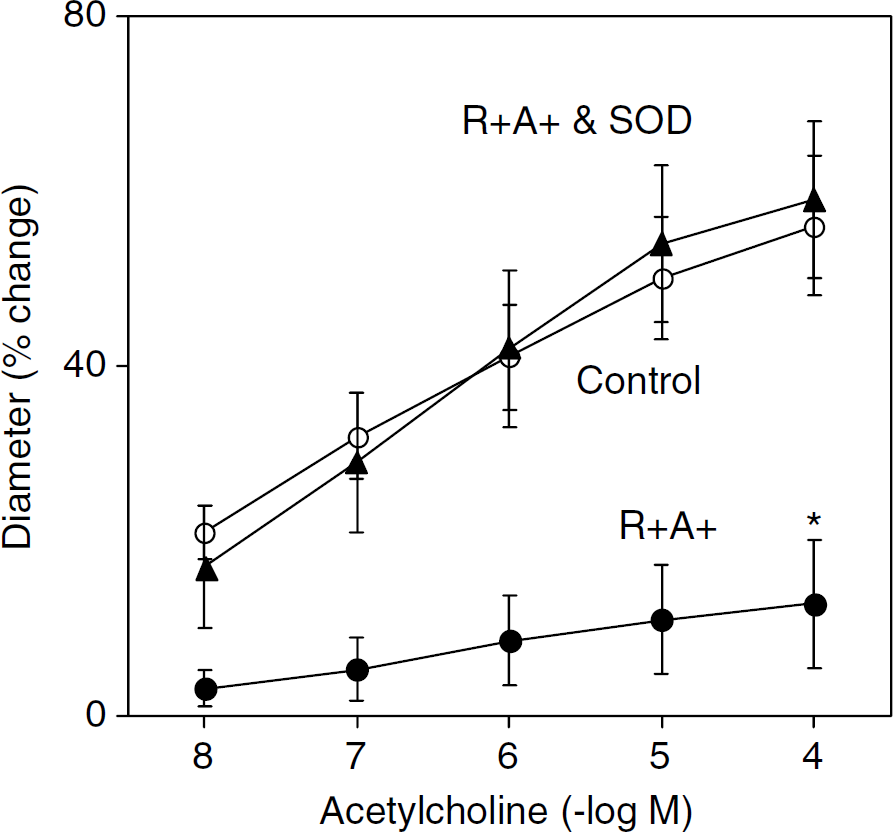
Baseline diameters were similar in basilar arteries from control and R+ A+ mice (138 ± 3 and 141 ± 2 µm in control (n = 13) and R+ A+ mice (n = 10), respectively). Dilation of the basilar artery in response to acetylcholine was greatly impaired in R+ A+ mice (n = 10) compared with controls (n = 13) (Figure 4). Impaired responses to acetylcholine in R+ A+ mice were restored to normal by PEG-SOD (n = 4) (Figure 4). Similar to acetylcholine, A-23187 produced dilation in cerebral arteries from control mice (n = 8), but no response or constriction in R+ A+ mice (n = 5) (Figure 5). In contrast to endothelium-dependent agonists, vasodilator responses to NONOate were similar in control and R+ A+ mice (Figure 5).
Dilation of the basilar artery in response to acetylcholine in control and R+ A+ mice and R+ A+ mice in the presence of PEG-SOD. Values are mean ± s.e. *P < 0.05 versus Control for all concentrations of acetylcholine.
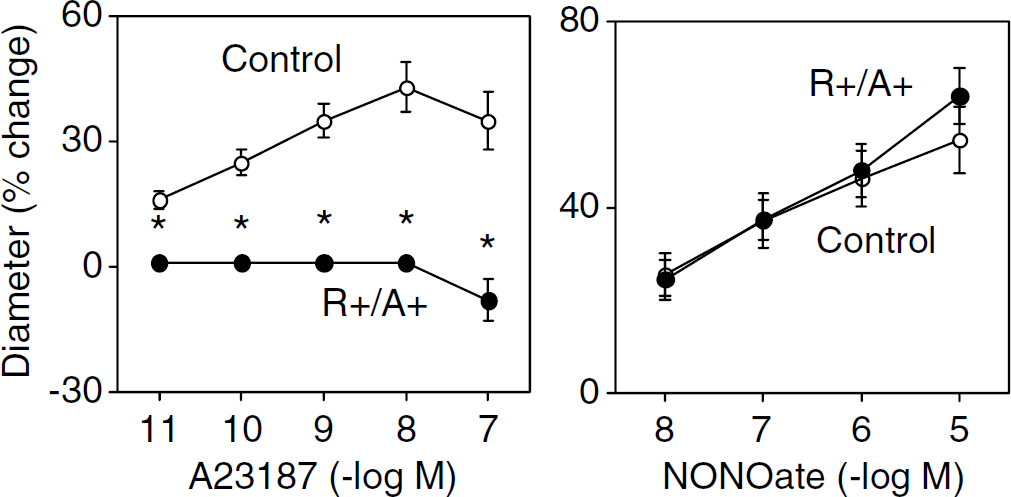
Dilation of the basilar artery in response to A-23187 (left panel) and NONOate (right panel) in Control and R+ A+ mice. Responses to all concentrations of A-23187 were significantly less in R+ A+ mice (*P < 0.05 versus Controls).
Discussion
There are several major findings in this study. First, Ang II was a potent constrictor of the basilar artery from male mice, whereas this response was markedly, but selectively, reduced in female mice. Second, vasoconstrictor responses to Ang II were completely inhibited by Y-27632, suggesting that the response was mediated by activation of Rho-kinase. Third, responses of the basilar artery to Ang II were similar in AT1A+/+ and AT1A+/+ mice, but were greatly reduced in AT1A+/+ mice. This finding provides direct evidence that AT1A receptors mediate the majority of the constrictor response to Ang II in cerebral arteries. These experiments also indicated that responses to acetylcholine are not affected by deletion of AT1A receptors. Fourth, endothelial function is greatly impaired in a genetic model of chronic Ang II-dependent hypertension (R+ A+ mice) via a mechanism that involves increased oxidative stress.
Effects of Angiotensin II in the Cerebral Circulation
Several previous studies have examined the acute effects of Ang II on tone in cerebral blood vessels. In almost all studies of cerebral arteries, including those in humans, Ang II produced constriction (Edvinsson et al, 1979; Paul et al, 1982; Shimizu et al, 1986; Juul et al, 1987; Whalley et al, 1985; Naveri et al, 1994; Stenman and Edvinsson, 2004; Krueger and Cook, 1985; Toda et al, 1990; Toda and Hayashi, 1979). In the present study, we found that Ang II was a potent constrictor of the basilar artery in male mice. In a previous study, Ang II (10−8 and 10−5 mol/L) was reported to produce constriction of pial arterioles in mice (Rosenblum et al, 1994).
One of the major findings of this study was that vasoconstrictor responses to Ang II were much smaller in arteries from female mice compared with male mice. This difference was selective as responses to KCl, acetylcholine, and U-46619 were similar in the two groups. In previous studies using rats, constrictor responses to serotonin (Chrissobolis and Sobey, 2004), U-46619 (Sanz et al, 2003), and arginine vasopressin (Garcia-Villalon et al, 2003) were unaffected by gender in the basilar artery. The mechanisms that account for the marked gender difference in responsiveness of the basilar artery to Ang II are not clear at present. One possibility could be differences in the influence of endothelial NO synthase (eNOS) on vasoconstrictor responses (Lamping and Faraci, 2001). This possibility seems unlikely, however, as expression of eNOS is similar in cerebral arteries (Chu et al, 2002) and responses to acetylcholine (which are NO-mediated) are similar in basilar arteries from male and female mice (present study).
Constrictor Responses of Cerebral Arteries to Angiotensin II: Role of Rho-Kinase
Activation of RhoA and its target Rho-kinase is thought to be a key mechanism to modulate the sensitivity of contractile proteins to calcium and produce contraction of vascular muscle (Somlyo and Somlyo, 2003; Sward et al, 2003). Relatively few studies have examined the functional importance of Rho-kinase in cerebral circulation, but recent evidence suggests that this signaling pathway mediates vasoconstrictor responses to serotonin, endothelin-1, UTP, and oxyhemoglobin (Miao et al, 2002; Nishikawa et al, 2003; Wickman et al, 2003; Luykenaar et al, 2004).
In the present study, we found that constriction of the basilar artery in response to Ang II was eliminated by Y-27632, suggesting that activation of this signaling pathway is the main mechanism by which Ang II increases cerebral vascular tone. Importantly, the effect of Y-27632 was selective, as the inhibitor had no effect on vasoconstrictor responses to KCl. Our findings are consistent with previous studies in extracranial blood vessels which suggested that activation of Rho-kinase was a major mediator of Ang II-induced vasoconstriction (Sward et al, 2003; Fagan et al, 2004; Matrougui et al, 2001).
Role of AT1A Receptors in Cerebral Circulation
Previous studies have described the expression of receptors for Ang II (ATa or AT1A and AT1B) in cerebral arteries of several species, including humans (Zhang et al, 2002; Viswanathan et al, 2000; Stenman and Edvinsson, 2004; Ohkuma et al, 2003). Pharmacological studies using losartan have implicated a role of AT1 receptors as mediators of vasoconstriction in brain (Stenman and Edvinsson, 2004; Naveri et al, 1994). There are limitations with the use of losartan (and other AT1 receptor antagonists) as it does not distinguish AT1A and AT1B receptors (Hein, 1998), and can exhibit nonspecific effects (Sadoshima, 2002).
Mice completely deficient in expression of the AT1A receptors exhibit little pressor response and no impairment of endothelial function in response to Ang II (Ryan et al, 2004). In contrast, some reports suggest a potential role for AT1B receptors in the regulation of vascular tone. For example, AT1B receptors have been implicated as the predominant mediator of Ang II-induced contraction in the abdominal aorta and femoral artery (Zhou et al, 2003). In renal afferent arterioles, more than half the constrictor response to Ang II is preserved in AT1A+/+ mice, and thus presumably mediated by AT1B receptors (Harrison-Bernard et al, 2002).
In cerebral arteries, we found that vasoconstriction in response to Ang II was markedly reduced in AT1A+/+ mice, providing direct evidence that this receptor subtype mediates the majority of the effect of Ang II. Our results in cerebral arteries are similar to findings obtained recently in efferent arterioles of the kidney, where Ang II-induced constriction was absent in AT1A+/+ mice (Harrison-Bernard et al, 2002).
As a part of these experiments, we examined the responses of cerebral arteries in heterozygote AT1A-deficient mice. We observed that responses of the basilar artery to Ang II were similar in AT1A+/+ and AT1A+/+ mice, indicating that the presence of only one copy of the AT1A gene is sufficient to elicit a normal cerebral vasoconstrictor response to Ang II. We also found that dilation of the basilar artery to acetylcholine was not altered in AT1A+/+ or AT1A+/+ mice, suggesting that the absence of this receptor does not alter endothelial function.
Endothelial Dysfunction in Cerebral Arteries of R+ A+ Mice
Although it has been known that responses of cerebral arteries to endothelium-dependent agonists are impaired in spontaneously hypertensive rats (Faraci and Heistad, 1998; Chrissobolis et al, 2002; Sobey et al, 1999), the mechanism that accounts for this impairment has not been defined (Faraci and Heistad, 1998). Gerzanich et al (2003) provided evidence for reduced local levels of NO in cerebral arteries in response to acetylcholine in rats induced hypertension by infusion of Ang II for 4 weeks. The potential role of oxidative stress was not addressed in that study.
In the present study, we observed marked endothelial dysfunction in the cerebral arteries of R+ A+ mice to both receptor and nonreceptor-mediated agonists. This dysfunction appears to be greater in magnitude than that observed in peripheral blood vessels in the same model (Didion et al, 2000, 2002), but was selective as responses to NONOate were normal. The mechanism which accounts for this dysfunction involves superoxide, as treatment with PEG-SOD completely restored responses to acetylcholine in R+ A+ mice. To our knowledge, this is the first demonstration of superoxide-mediated endothelial dysfunction in the cerebral circulation in a model of chronic Ang II-dependent hypertension. Related to these findings, recent work indicated that Ang II can also impair neurovascular coupling via a superoxide-dependent mechanism (Kazama et al, 2004).
In summary, hypertension is a major risk factor for carotid artery disease and stroke, and has been linked with vascular cognitive impairment and Alzheimer's disease (Gorelick, 2004). Angiotensin II contributes importantly to some forms of hypertension. The present study provides the first evidence that Ang II is a potent constrictor of cerebral arteries in mice and that this effect is mediated very predominantly by AT1A receptors and activation of Rho-kinase. There are marked, but selective, gender differences in cerebral vascular responses to Ang II. Endothelial function is greatly impaired in a genetic model of chronic Ang II-dependent hypertension via a mechanism that involves superoxide.
Footnotes
Acknowledgements
Dr Didion was supported by DK-25295 and a National Scientist Development Grant from the American Heart Association (0230327N). Dr Ryan was supported by a grant from the American Heart Association, Heartland Affiliate (BGIA, 0460046Z).