
Abstract
Although arachidonic acid (AA) has diverse vascular effects, the mechanisms that mediate these effects are incompletely defined. The goal of our study was to use genetic approaches to examine the role of hydrogen peroxide (H2O2), glutathione peroxidase (Gpx1, which degrades H2O2), and CuZn-superoxide dismutase (SOD1, which produces H2O2 from superoxide) in mediating and in determining vascular responses to AA. In basilar arteries
Introduction
Arachidonic acid (AA) plays a major role in vascular biology and elicits a variety of effects, including changes in vascular tone and permeability. Such effects may be mediated by multiple mechanisms. Arachidonic acid can potentially alter cell function without being metabolized by acting on specific targets, such as ion channels (Meves, 2008). Moreover, AA can also be metabolized through several major pathways, including cyclooxygenase, cytochrome P450 monooxygenase, and lipoxygenase pathways (Meves, 2008). In addition to producing eicosanoids, the metabolism of AA also results in the generation of reactive oxygen species (ROS) (Kontos, 1990), which have both acute and chronic vascular effects (Faraci, 2006a; Chrissobolis and Faraci, 2008).
In blood vessels from several organs, including the heart and the brain, AA increases ROS levels (Didion et al, 2001a; Oltman et al, 2003; Zafari et al, 1999). Earlier studies using pharmacological approaches have provided evidence both for (Oltman et al, 2003; Sobey et al, 1998) and against (Bryan et al, 2006; Pomposiello et al, 1999) the role for ROS as mediators of changes in vascular tone in response to AA. In this study, we examined the role of ROS, and specifically hydrogen peroxide (H2O2), as a mediator of the vascular effects of AA using pharmacological and new genetic approaches. Steady-state levels of H2O2 are determined in part by the activity of glutathione peroxidases (Gpx), which metabolizes H2O2 to water (Arthur, 2000; Briggelius-Flohe et al, 2003; Faraci, 2006a). The functional importance of Gpx in most vascular beds, including the cerebral circulation is unknown.
The goal of these experiments was to examine the role of H2O2 in mediating the effects of AA on vascular tone. We sought to define the functional importance of the cytosolic form of Gpx (Gpx1) as a determinant of this vascular response. We tested the hypothesis that vasodilation to AA is mediated by H2O2 and that genetic overexpression of Gpx1 would inhibit this response. As part of these experiments, we also tested whether overexpression of Gpx1 affected dilation of cerebral arteries to the classic endothelium-dependent agonist, acetylcholine.
Superoxide dismutases (SODs) are a major cellular source of H2O2 as they convert superoxide to H2O2 (Faraci and Didion, 2004). To further define mechanisms involved, we tested the hypothesis that vascular responses to AA depend on the expression of the cytosolic form of SOD (CuZn-SOD or SOD1). In vascular tissue, SOD1 accounts for the majority of total SOD activity (Faraci and Didion, 2004; Didion et al, 2001b). Overall, our findings provide the first evidence that Gpx1 has functional effects in the cerebral circulation and that the vascular effects of AA, but not acetylcholine, are mediated by H2O2 produced by SOD1.
Materials and methods
Experimental Animals
Glutathione peroxidase transgenic (Gpx1 Tg) mice (Chrissobolis et al, 2008) were obtained from breeding C57B1/6 and Gpx1 Tg mice. The Gpx1 Tg mice and their nontransgenic littermates (non-Tg) were studied. SOD1-deficient mice were obtained from breeding heterozygous SOD1-deficient mice (Didion
Studies of Cerebral Arteries In Vitro
After an overdose of anesthesia (pentobarbital, 150mg/kg intraperitoneally), the brain was rapidly removed and placed in an ice-cold Krebs buffer. As described earlier (Faraci et al, 2006a, Yamada et al, 2001), the basilar artery was isolated, cannulated onto glass micropipettes filled with Krebs buffer in an organ chamber, and secured using a nylon monofilament. Arteries were pressurized to 60 mm Hg. Using a microscope and a video camera, vessel images were projected on a video monitor. An electronic dimension analyzer was used to measure lumen diameter.
Once prepared, basilar arteries were allowed to equilibrate for at least 30mins at a distending pressure of 60 mm Hg before protocols were initiated. We examined changes in diameter of the basilar artery in response to KCl (50mmol/L). For studies testing vasodilator responses, basilar arteries were constricted by ∼30% (∼60% of the response to 50mmol/L of KCl) with U-46619, a thromboxane A2 mimetic. Acetylcholine was used as an endothelium-dependent agonist and is known to produce a nitric oxide (NO)-mediated dilation of the basilar artery (Faraci and Heistad, 1998; Faraci et al, 2006b). After development of a stable baseline diameter, cumulative dose-response curves were obtained. At the end of the protocols, papaverine (an endothelium-independent vasodilator, 100 μmol/L) was used to produce maximal vasodilation.
Studies of Cerebral Arterioles In Vivo
Mice were anesthetized with pentobarbital sodium (75 to 90mg/kg intraperitoneally), supplemented regularly at ∼20mg/kg per h. Animals were ventilated mechanically with supplemental oxygen, and the arterial blood pressure and blood gases were monitored as described earlier (Sobey and Faraci, 1997). A cranial window was made over the left parietal cortex; the window constantly suffused with artificial cerebrospinal fluid, and a pial arteriole (branches of the middle cerebral artery) was exposed. The arteriolar diameter was measured using a microscope equipped with a camera coupled to a video monitor and to an image-shearing device. The diameter of one arteriole per animal was measured under control conditions and during topical application of drugs. The mean arterial pressure was similar in all groups of mice and averaged 69 ± 1 mm Hg (mean ± s.e.). Arterial blood gases were monitored and were also similar (PCO2 = 41 ± 1 mm Hg, PO2 = 128 ± 10 mm Hg, and pH = 7.28 ± 0.01). The gases and pH of the artificial cerebrospinal fluid were PCO2 = 40 ± 1 mm Hg, PO2 = 73 ± 2 mm Hg, and pH = 7.32 ± 0.02. In these studies, responses of arterioles to topical application of AA (1 and 10 μmol/L), acetylcholine (1 and 10 μmol/L), and papaverine (10 and 100 μmol/L) were examined in the absence or presence of catalase (300 U/mL, a scavenger of H2O2) or diethyldithiocarbamate (10 mmol/L, an inhibitor of SOD1 and SOD3) (Faraci and Didion, 2004). These latter experiments were carried out to examine whether responses to AA and the mechanisms involved in the mouse were similar to earlier studies in other species (Didion et al, 2001a; Sobey et al, 1998).
Statistical Analysis
For the basilar artery, constriction in response to KCl was determined by calculating the percentage reduction in vessel diameter from the baseline control level. For vasodilator responses, results are expressed as percentage dilation (% of induced tone), with 100% representing the difference between the resting value under basal conditions and the constricted value with U-46619. For cerebral arterioles
Results
Dilation of Cerebral Arterioles to Arachidonic Acid is Inhibited by Catalase
The baseline diameter of cerebral arterioles in all C57Bl6/J mice
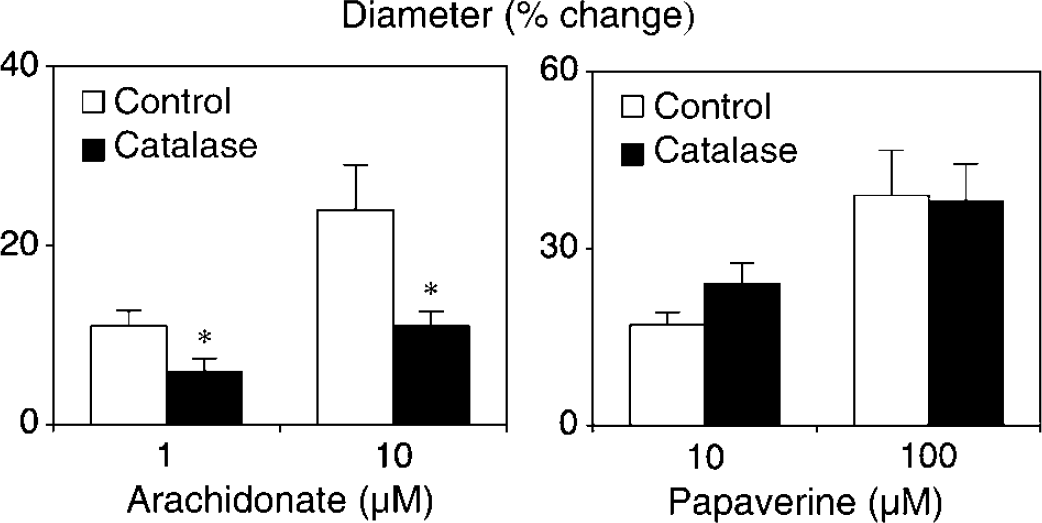
Changes in the diameter of cerebral arterioles in response to arachidonic acid (left panel) and papaverine (right panel) in C57BI/6 mice in the absence (control) and presence of catalase (300 U/mL). Values are means ± s.e.; *
Vasodilation to Arachidonic Acid is Inhibited by Indomethacin
The baseline diameter of the basilar artery in C57Bl6/J mice was 128 ± 10 μm. Dilation of the basilar artery in response to AA was completely inhibited by indomethacin (10 μmol/L,
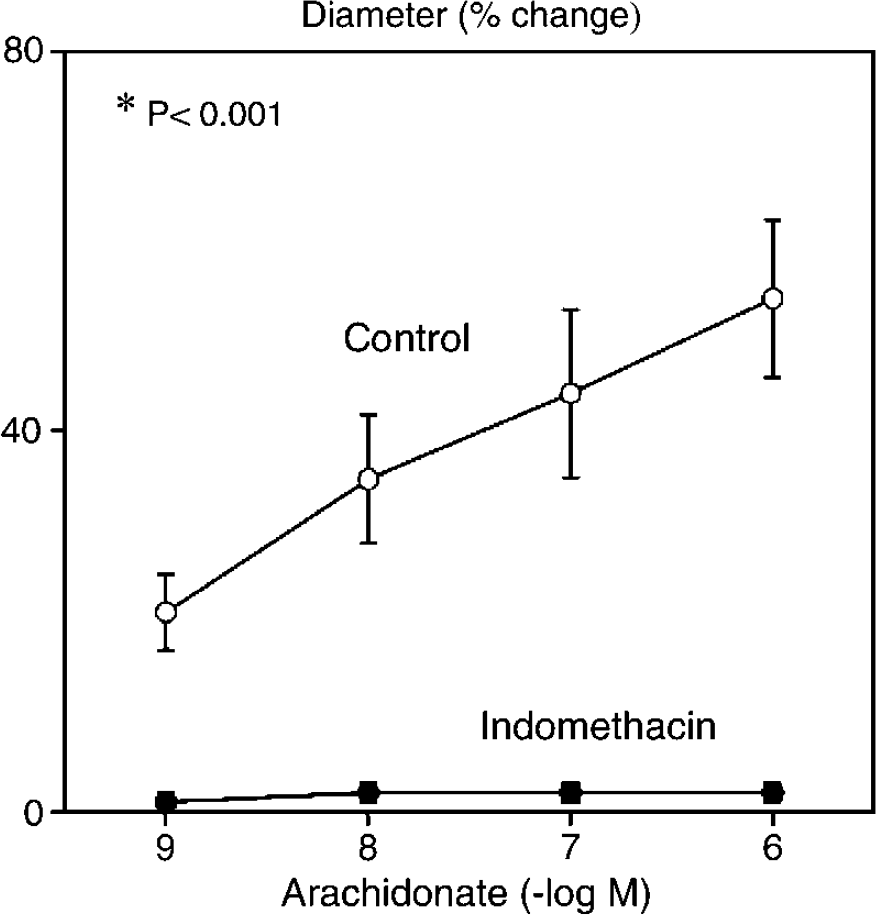
Changes in the diameter of the basilar artery in response to arachidonic acid in the absence and presence of indomethacin (10 μmol/L,
Overexpression of Glutathione Peroxidase Markedly Reduces Dilation of the Basilar Artery and Cerebral Arterioles to Arachidonic Acid
We have shown earlier that vascular expression of Gpx1 is increased approximately three-fold in Gpx1 Tg mice compared with that in non-Tg controls (Chrissobolis et al, 2008). Baseline diameters of the basilar artery in non-Tg and Gpx1 Tg mice were 132 ± 1 and 130 ± 2 μm (
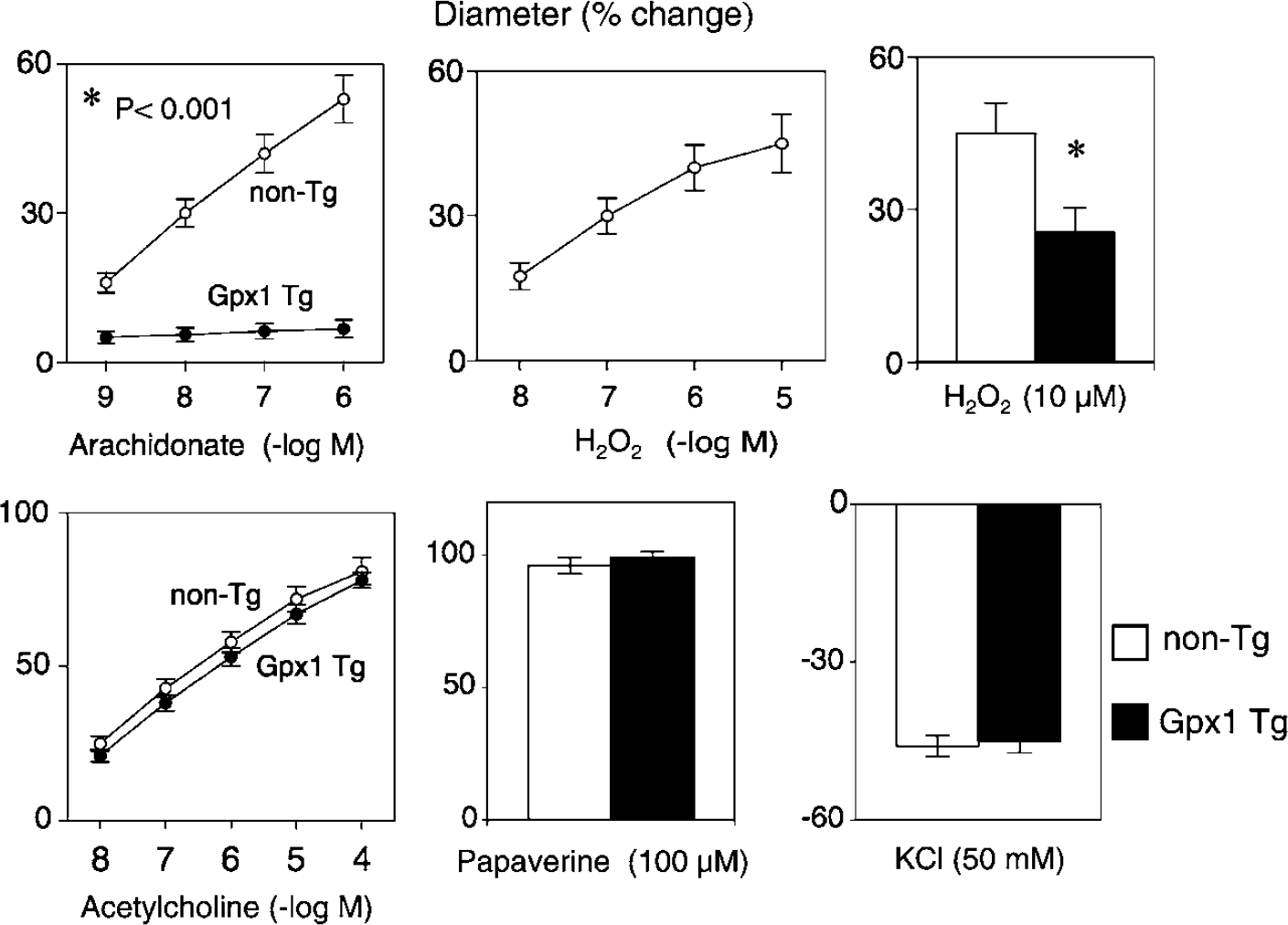
Changes in the diameter of the basilar artery in response to arachidonic acid (upper left panel) and acetylcholine (lower left panel) in non-Tg (nontransgenic) and glutathione peroxidase Tg (Gpx1 Tg) mice. Effects of hydrogen peroxide on vessel diameter in non-Tg mice (upper middle panel) and in non-Tg and Gpx1 Tg mice (upper right panel). Vascular responses to papaverine and KCI are also shown (lower middle panel and lower right panel, respectively). In non-Tg and Gpx1 Tg mice, respectively, the vessel diameters were 131 ± 4 and 130 ± 5 μm under baseline conditions and 92 ± 4 and 91 ± 4 μm after preconstriction with U46619. Values are means ± s.e.; *
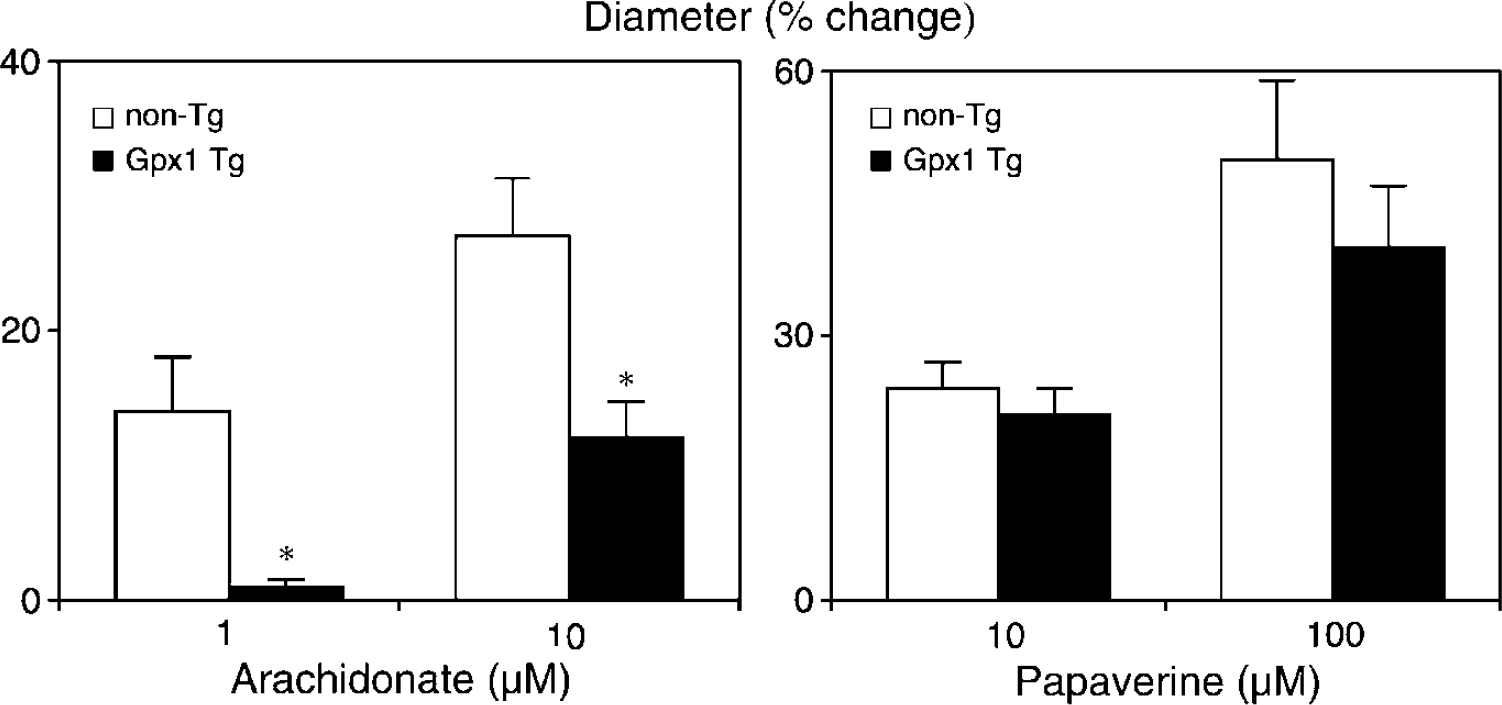
Changes in the diameter of cerebral arterioles in response to arachidonic acid (left panel) and papaverine (right panel) in nontransgenic controls (non-Tg) and glutathione peroxidase (Gpx1) Tg mice. Values are means ± s.e.; *
Vasodilation to Arachidonic Acid is Greatly Attenuated in SOD1-Deficient Mice
Baseline diameters of the basilar artery in wild-type and SOD1-deficient mice were 135 ± 2 and 133 ± 2 μm (
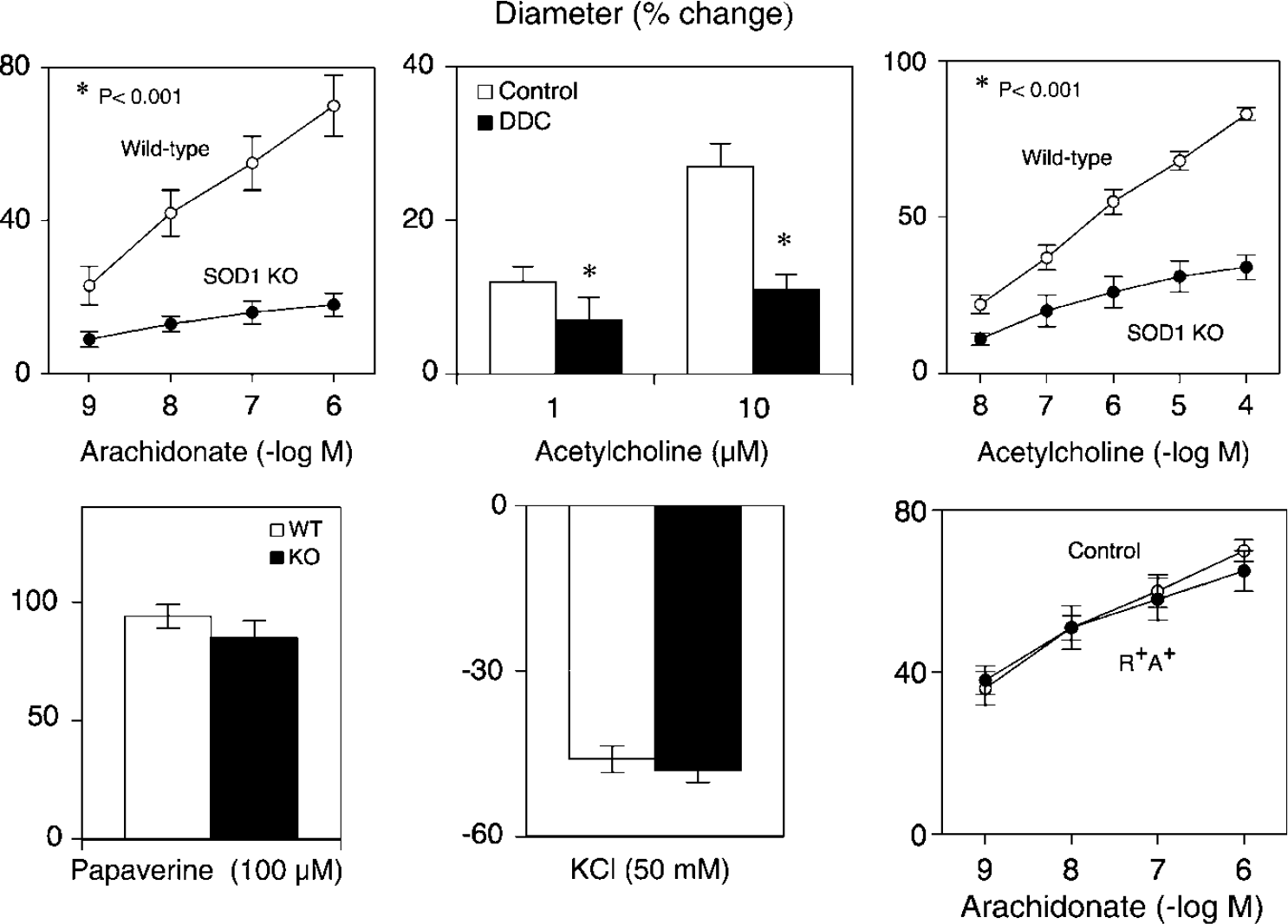
Changes in the diameter of the basilar artery in response to arachidonic acid (upper left) and acetylcholine (upper right) in wild-type and in SOD1-deficient mice. The effect of DDC on changes in the diameter of cerebral arterioles in response to acetylcholine in C57BI/6 mice are also shown (upper middle panel). Vascular responses to papaverine and KCl are also shown (lower left and lower middle, respectively). KO = knockout. Effects of arachidonic acid on basilar artery diameter in a control R+ A+ mice are shown in the lower right panel. In wild-type and SOD1-deficient mice respectively, vessel diameters were 136 ± 6 and 136 ± 6 μm under baseline conditions and 97 ± 5 and 102 ± 5 μm after preconstriction with U46619. In control and R+ A+ mice respectively, vessel diameters were 141 ± 4 and 140 ± 3 μm under baseline conditions and 99 ± 3 and 95 ± l μm after preconstriction with U46619. Values are means ± s.e.; *
We and others have shown that genetic deficiency in SOD1 reduces formation of H2O2, increases superoxide, and impairs NO-mediated vascular responses (Didion et al, 2001b; Baumbach et al, 2006; Yada et al, 2008). Consistent with earlier studies in cerebral arterioles (Didion et al, 2001a), diethyldithiocarbamate inhibited dilation of cerebral arterioles in response to acetylcholine (Figure 5). Dilation of the basilar artery in response to acetylcholine was reduced in SOD1-deficient mice (Figure 5). This change was selective as constriction of the basilar artery to KCl (Figure 5) and U46619 (data not shown), and dilation to papaverine was similar in both groups of mice (Figure 5).
As deficiency in SOD1 produces oxidative stress, we considered the possibility that reductions in the response to AA in SOD1-deficient mice were simply a consequence of oxidative stress. To examine this possibility, we studied basilar arteries from a genetic model of angiotensin II-dependent hypertension (R+A+ mice) (Didion et al, 2002). R+A+ mice have oxidative stress, increased vascular superoxide, and superoxide-mediated vascular dysfunction (Didion et al, 2002; Faraci et al, 2006a). Vasodilation in response to AA was not altered in R+A+ mice compared with that in littermate controls (Figure 5), suggesting that the presence of oxidative stress
Discussion
There are several novel findings in this study. Arachidonic acid dilated cerebral blood vessels both
Cerebrovascular Effects of Arachidonic Acid and the Role of Reactive Oxygen Species
Arachidonic acid is generally a vasodilator, including in the cerebral circulation. Although AA has the potential to produce vascular effects through direct actions on ion channels and following metabolism through multiple pathways, most earlier studies in multiple species, including primates indicate that AA produces changes in the tone of cerebral arteries and arterioles through a cyclooxygenase-dependent mechanism (Busija and Heistad, 1983; Ellis et al, 1990; Hayashi et al, 1985; Niwa et al, 2001; Sobey et al, 1998). In this study, we found that indomethacin completely inhibited the vascular effects of AA, indicating that the response was also dependent on the activity of cyclooxygenase in the mouse basilar artery. As the vascular effects of AA are reduced by cyclooxygenase inhibitors, some investigators assume that prostacyclin or other eicosanoids mediates these responses (Hayashi et al, 1985; Ospina et al, 2003).
The metabolism of AA is known to be a major source of ROS (Kontos, 1990). Superoxide is produced during cyclooxygenase activity (i.e., the conversion of AA to prostaglandin H2) (Kontos, 1990). For example, AA produces cyclooxygenase-dependent superoxide formation in cerebral and coronary blood vessels (Didion et al, 2001a; Oltman et al, 2003). Once produced, superoxide may have direct effects (Faraci, 2006b; Faraci and Didion, 2004) or may be converted to H2O2 primarily by SODs (Faraci and Didion, 2004; Faraci, 2006a). Although some earlier studies suggest that vasodilation in response to AA is inhibited by scavengers of ROS, there is controversy regarding the role of ROS and which ROS mediate the response (Bryan et al, 2006; Kontos et al, 1984; Niwa et al, 2001; Pomposiello et al, 1999; Sobey et al, 1998). Our finding that dilation of cerebral arterioles in response to AA is inhibited by catalase supports the concept that ROS are key components and that H2O2 is a mediator of this response (Kontos et al, 1984; Sobey et al, 1998).
In the cerebral circulation and other vascular beds, H2O2 is generally a dilator in both large arteries and microvessels (see Faraci, 2006a, b for reviews). A similar effect was observed in this study, as H2O2 was a potent dilator of the basilar artery. Although there is little evidence to suggest that ROS affect cerebral vascular tone under resting conditions, some pharmacological studies suggested that dilation of cerebral arterioles to both AA and bradykinin is mediated by an endogenous formation of ROS. We provided the first evidence that H2O2 dilates cerebral arterioles by activation of potassium channels (Sobey et al, 1997, 1998) and thus, may function as an endothelium-derived hyperpolarizing factor. In addition to these stimuli, H2O2 may contribute to flow-mediated vasodilation (Paravicini et al, 2006) and increases in cerebral blood flow in diseases with an inflammatory component, including meningitis (Hoffmann et al, 2007).
Is SOD1 a Major Source of Hydrogen Peroxide in the Cerebral Circulation?
As noted above, H2O2 is formed primarily from superoxide as a result of activity of the various SODs (Faraci and Didion, 2004). In blood vessels, there are three isoforms of SOD, namely SOD1, manganese SOD localized in mitochondria (SOD2), and an extracellular form of CuZn-SOD (SOD3) (Faraci and Didion, 2004). Vasodilation of cerebral arterioles in response to AA was markedly reduced by diethyldithiocarbamate (Didion et al, 2001a), an inhibitor of SOD1 and SOD3 (Faraci and Didion, 2004), suggesting the source of H2O2 that mediates the response is a copper-containing SOD. However, this approach is limited and provides no insight into the relative importance of the specific isoforms of SOD. In addition, diethyldithiocarbamate is a chelator of copper and thus, may have nonselective effects (Faraci and Didion, 2004). In these experiments, we found that vasodilation to AA was markedly reduced in mice deficient in SOD1, suggesting this response is mediated very predominately by H2O2 produced by SOD1.
We and others have shown that pharmacological inhibition of SOD1 and SOD3 or genetic deficiency in SOD1 increase vascular superoxide (Baumbach et al, 2006; Didion et al, 2001a, b ), reduce vascular H2O2, (Yada et al, 2008), and impair NO-mediated vascular responses (Baumbach et al, 2006; Didion et al, 2001a, b ). Consistent with this concept, we found that dilation of cerebral arterioles to acetylcholine was inhibited by diethyldithiocarbamate and that responses of the basilar artery to acetylcholine were impaired in SOD1-deficient mice. In both these vascular segments, multiple lines of evidence suggest that vasodilation to acetylcholine is normally mediated by endothelium-derived NO (Faraci and Heistad, 1998; Sobey and Faraci, 1997).
Glutathione Peroxidase is a Major Determinant of Vascular Responses to Arachidonic Acid
Glutathione peroxidases are part of a family of selenium-dependent enzymes that use glutathione to metabolize H2O2 and lipid peroxides to water and respective alcohols (Arthur, 2000; Briggelius-Flohe et al, 2003). Similar to the SODs, there are multiple isoforms of Gpx, including the cytosolic Gpx1 (Arthur, 2000; Briggelius-Flohe et al, 2003). Gpx is expressed in vascular cells and blood vessels, including cerebral arteries (Chrissobolis et al, 2008; Kobayashi et al, 2002; Lapenna et al, 1998; Napoli et al, 1999). The expression of Gpx is relatively high in endothelium (Kobayashi et al, 2002), and may change within the vessel wall depending on genetic factors and in disease (Lapenna et al, 1998; Ulker et al, 2003). Although catalase also degrades H2O2, human vascular cells reportedly express little or no catalase (Shingu et al, 1985).
Although exogenous catalase had inhibitory effects on vascular responses to AA, that approach may underestimate the role of intracellularly produced H2O2 and provides no insight into the subcellular action of H2O2 or into the functional importance of GPx1. The existence of multiple isoforms of SOD and GPx implies that specific functions exist in different subcellular locations. Increasing evidence suggests that the effects of ROS are compartmentalized with different signaling consequences at different locations. Our findings with Gpx1 Tg mice provide genetic evidence that H2O2 is the mediator of the majority of the vascular response to AA, particularly with lower concentrations of arachidonate. We earlier provided evidence that Gpx1 has functional effects in the carotid artery and was important in protecting against angiotensin II-induced vascular dysfunction (Chrissobolis et al, 2008). The present study provides the first evidence that Gpx1 has functional effects in the cerebral circulation (in intracranial blood vessels). It is perhaps not surprising that the response to AA is dependent on the activities of both SOD1 and Gpx1 as both these enzymes are cytosolic as opposed to other forms of SOD and Gpx.
Role of Hydrogen Peroxide in Mediating Endothelium-Dependent Relaxation
Many studies suggest that NO produced by eNOS (endothelial isoform of NO synthase) affects basal tone and mediates responses to varied endothelium-dependent stimuli (Faraci and Heistad, 1998; Cipolla and Bullinger, 2008; Cipolla et al, 2004), In contrast, a recent study suggests that under normal conditions, eNOS is uncoupled in cerebral arteries and thus, produces superoxide because of a deficiency in substrate (Drouin et al, 2007). In this scenario, the superoxide is produced by eNOS and is then converted to H2O2 by SOD(s). With this postulated mechanism, H2O2 is the actual mediator of endothelium-dependent relaxation (Drouin et al, 2007). Our finding that overexpression of Gpx1 greatly reduces vascular responses to AA and exogenous H2O2, but not acetylcholine, do not support the concept that H2O2 is the mediator of responses to this neurotransmitter and muscarinic agonist. These findings are consistent with earlier pharmacologically based studies that concluded that responses to acetylcholine in the cerebral circulation are not mediated by ROS (see Katusic et al, 1989; Kontos et al, 1988 for examples). Uncoupled eNOS may well contribute to oxidative stress in vascular disease, including the brain, but there has been little evidence to suggest that this form of dysfunction occurs under normal conditions
In summary, this study provides the first genetic evidence that the vascular effects of AA are mediated by H2O2 produced by SOD1. Our study suggests that Gpx1 is a major determinant of the vascular effects of AA both