
Abstract
Overpressure blast-wave induced brain injury (OBI) leads to progressive pathophysiologic changes resulting in a reduction in brain blood flow, blood brain barrier breakdown, edema, and cerebral ischemia. The aim of this study was to evaluate cerebral vascular function after single and repeated OBI. Male Sprague-Dawley rats were divided into three groups: Control (Naive), single OBI (30 psi peak pressure, 1 to 2 msec duration), and repeated (days 1, 4, and 7) OBI (r-OBI). Rats were killed 24 hours after injury and the basilar artery was isolated, cannulated, and pressurized (90 cm H2O). Vascular responses to potassium chloride (KCl) (30 to 100 mmol/L), endothelin-1 (10−12 to 10−7 mol/L), acetylcholine (ACh) (10−10 to 10−4 mol/L) and diethylamine-NONO-ate (DEA-NONO-ate) (10−10 to 10−4 mol/L) were evaluated. The OBI resulted in an increase in the contractile responses to endothelin and a decrease in the relaxant responses to ACh in both single and r-OBI groups. However, impaired DEA-NONO-ate-induced vasodilation and increased wall thickness to lumen ratio were observed only in the r-OBI group. The endothelin-1 type A (ETA) receptor and endothelial nitric oxide synthase (eNOS) immunoreactivity were significantly enhanced by OBI. These findings indicate that both single and r-OBI impairs cerebral vascular endothelium-dependent dilation, potentially a consequence of endothelial dysfunction and/or vascular remodelling in basilar arteries after OBI.
INTRODUCTION
Blast overpressure injury is a common problem among military personnel, and these individuals experience chronic health problem that persists into civilian life. Besides soldiers, civilians can also experience blast injuries from terrorist attacks with explosives. The primary injury is due to the physical impact, which may manifest in a form of polytrauma involving multiple organs. This phenomenon can cause blast overpressure-induced brain injury (OBI) and long-term neurologic problems. The overall clinical presentation results from the combination of all blast effects, i.e., primary, secondary, tertiary, and quaternary mechanisms.1,2 One consequence of OBI is an increase in the production of reactive oxygen species (ROS) that compromise cell function, leading to cell death.3,4 Reactive oxygen species such as superoxide radicals and nitric oxide (NO) can form a powerful oxidant, peroxynitrite. 5 These oxidants may directly damage vessels and can trigger inflammatory pathways that cause a deformation in the arterial structure, 6 mainly the endothelium,7,8 leading to impaired blood flow. Thus, trauma alters the cerebral vascular function and impairs oxygenation of the brain due to ischemia.9,10 Additionally, human studies showed that brain edema and increased intracranial pressure contribute to the decreased blood flow and hypoperfusion of the brain tissue. 11 Ischemic tissue is prone to oxidative damage that triggers other deleterious effects such as failure of Na/K ATPase pumps, increased blood brain barrier permeability, and modification of AQP4 proteins. 12 As a result, brain edema occurs, intracranial pressure increases, and the circulatory failure worsens. Sympathetic hyperactivity, along with the oxidative stress, is another contributory factor to cerebral vascular dysfunction after OBI.13–15 Decreased cerebral blood flow, autonomic dysfunction, edema, increased blood brain permeability, oxidative stress, and inflammatory responses are multiple pathways leading to neuronal injury, neurodegeneration, and cognitive decline.3,4
The basilar artery is the most important artery in the posterior cerebral circulation. It supplies blood to the medulla, cerebellum, pons, midbrain, thalamus, occipital cortex, as well as several areas of the brain that are critical for mediating episodic memory, including portions of the hippocampus and the parahippocampal gyrus. Reduced posterior brain blood flow contributes to vertebrobasilar insufficiency, 16 resulting in a set of symptoms including vertigo, headaches, sleep disturbances, pupillary and oculomotor abnormalities, dysarthria, and dysphagia to quadriparesis. Furthermore, it leads to memory impairment and neurologic disorders. 17
Vasospasm of the basilar artery is frequently observed after subarachnoidal hemorrhages, but little is known regarding the OBI-induced functional changes of the basilar artery. Specifically, to our knowledge, this is the first study to evaluate the vasoreactivity of the basilar artery to pharmacological agents after OBI.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats, 7 weeks old, weighing 250 to 300 g were used. All animals were housed at the University of Florida (UF) Animal Care Services Center and experiments were performed according the NIH, UF Institutional Animal Care and Use Committee (IACUC; protocol no: 201207693) guidelines. This manuscript was written up in accordance with the Animal Research Reporting in Vivo Experiments (ARRIVE) guidelines. Animals were maintained on a 12:12-hour light-dark cycle and provided food (AIN93 diet) and water ad libitum throughout the experimental protocol. Animals were divided into three groups (6 animals/group) as follows: Control (Naive); single OBI: 30psi peak pressure, 1 to 2 msec duration; repeated OBI (r-OBI): 30psi peak pressure, 1 to 2 msec duration, on days 1, 4, and 7 during a week period.
Induction of overpressure blast-wave injury. Anesthetized (isoflurane 3%) rats were mounted in the prone position on the animal holder. The head was laid on a flexible mesh surface to diminish any surface reflection from the blast waves and decrease the formation of secondary waves that would potentially exacerbate the injury. The neck, head, torso, and abdomen of the animal were fixed to the animal holder in an effort to avoid any movement, thus minimizing tertiary blast effects. The design features a 4-inch inner diameter shock tube that is 7 ft in length with the fixture positioning of the rat nose tip at 53 cm downstream from the driven opening to the driver section, and the end of the rat body about 10 cm from the exit opening of the driven section.18,19 Control of burst and incident pressure was achieved by adjusting the thickness of Mylar membranes of 0.03 in thicknesses (pressure changes are achieved by using differing thicknesses). A blast wave (30psi peak pressure, 1 to 2 msec duration) was then directed at the animal lying below within the driven section. The peak pressure was recorded by a computer system.
The single injury group (OBI) was exposed only once, whereas the repeated injury group (r-OBI) was exposed a total of three times, once every 3 days over a week period (i.e., on days 1, 4, and 7). Both groups were killed 24 hours after the last injury (either single or repeated); i.e., for the r-OBI group, it is the 8th day after the first injury but it is also 24 hours after the last (3rd) injury.
Microvessel Preparations
Rats were anesthetized (isoflurane 3%/O2 balance) and euthanized by the removal of the heart. The brain was rinsed and placed in cold (4°) physiologic saline solution containing 145.0 mmol/L NaCl, 4.7 mmol/L KCl, 2.0 mmol/L CaCl2, 1.17 mmol/L MgSO4, 1.2 mmol/L NaH2PO4, 5.0 mmol/L glucose, 2.0 mmol/L pyruvate, 0.02 mmol/L EDTA, 3.0 mmol/L MOPS buffer, and 1 g/100mL bovine serum albumin, pH 7.4. The basilar arteries were isolated with the aid of a dissection microscope (Olympus SVH10, Leeds Instruments, Minneapolis, MN, USA) as previously described.8,20 The arteries were transferred to a Lucite chamber containing physiologic saline solution equilibrated with room air. The ends of the artery were cannulated with micropipettes and secured with nylon sutures. The chamber containing the cannulated artery was then placed on an inverted microscope (Olympus IX70, Leeds Instruments) equipped with a video camera and micrometer (Panasonic BP310; Texas A&M Cardiovascular Research Institute) to measure intraluminal diameter. The basilar arteries were then pressurized at 90cm H2O (≅66 mm Hg) with two hydrostatic columns. 21 Arteries unable to hold pressure due to leaks or branches were discarded. Arteries without leaks were warmed to 37° and allowed to equilibrate for 40 minutes before beginning the assessment of vasoconstrictor or vasodilator responses. 8
Potassium Chloride-Induced Vasoconstriction
Non-receptor dependent contractile responses were evaluated with three different doses of KCl (30, 50, and 100 mmol/L, isotonic substitution for NaCl) administered at 2-minute intervals. The change in the internal diameter of the vessel was recorded and a dose response curve was generated.
Endothelin-1 Induced Vasoconstriction
Endothelin-1 (ET-1) (Phoenix Pharmaceuticals, catalog no: 023-01, Burlingame, CA, USA) was used as a receptor-dependent contractile agent. It was added to the bath cumulatively in increasing concentrations (10−12 to 10−7 mol/L) at 2-minute intervals. The change in the internal diameter of the vessel was recorded and a concentration-response curve was generated.
Acetylcholine Vasodilation
Endothelium-dependent dilation to acetylcholine (ACh) (Sigma, A6625, St. Louis, MO, USA) was evaluated by the addition of ACh to the bath in incremental doses (10−10 to 10−4 mol/L) at 2-minute intervals.
Diethylamine-NONO-ate Vasodilation
Endothelium-independent dilation was evaluated by the addition of a NO donor, diethylamine NONO-ate (DEA-NONO-ate) (Sigma, D184) to the bath in incremental doses (10−10 to 10−4 M) at 2-minute intervals.
Calculations
Vasoconstrictor responses to KCl and ET-1 are expressed as percent constriction as calculated by the formula:
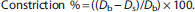
For vasodilation experiments, a minimum of 15% spontaneous tone was necessary before assessment of concentration-response curves.
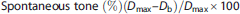
Relaxation responses to ACh and DEA-NONO-ate are expressed as percent relaxation as calculated by the following formula: Relaxation (%) = ((Ds - Db)/(Dmax - Db)) × 100.
where Ds is the arteriolar diameter measured after addition of the vasodilatory agent, Db is the diameter recorded immediately before initiation of the concentration-diameter curves, and Dmax is the maximal diameter for the arteriole.
Hematoxylin & Eosin Staining of Basilar Artery
Isolated arteries were fixed in formalin solution, immersed in fast green, embedded in optimal cutting temperature compound, and then immediately stored at − 80°, until the cutting of 5 μm sections occurred. Sections were then stained by Hematoxylin & Eosin method.
Endothelial, wall, and lumen thicknesses were determined under the microscope (Axiovert 40 CFL, Zeiss, Göttingen, Germany) connected to a computerized system using the Image J/Lab program.
Endothelin-1 Type A receptor and Endothelial Nitric Oxide Synthase Immunohistochemistry
Five μm sections were rinsed three times for 5 minutes with phosphate-buffered saline at room temperature, then incubated in 1% sodium dodecyl sulfate in phosphate-buffered saline for 5 minutes. After rinsing, sections were incubated in blocking solution (phosphate-buffered saline containing 5% goat serum, and 0.3% Triton) for 1 hour at room temperature followed by overnight incubation with anti-ETA (1:400, Sigma, E9780) or anti-eNOS (1:100, Sigma, E9905) primary antibodies at 4°. After rinsing, sections were incubated in fluorescein isothiocyanate-conjugated secondary antibody (1:400, Abcam, ab6717, Cambridge, MA, USA) for fluorescence development. The cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Images were taken using a Nikon TiE-PFS-A1R (Tokyo, Japan) confocal microscope equipped with a 488-nm laser diode with a 510- to 560-nm band pass filter, and a 561 laser with a 575- to 625-nm band pass filter. The fluorescence intensity was analyzed using Image J software (NIH, Bethesda, MD, USA).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). Each group consisted of six animals. All data were expressed as means ± s.e.m. Sigmoidal dose-response curves and groups/doses were compared using two-way analysis of variance (ANOVA) followed by Bonferroni post hoc tests. The histologic evaluation was performed by one-way ANOVA followed by Tukey's multiple comparison. Values of P<0.05 were regarded as significant.
RESULTS
Contractility Responses
Contractility responses to three concentrations of KCl (30, 50, and 100 mmol/L) were evaluated, and did not differ between groups. EC50 of control, OBI, and r-OBI were 49.85, 50.04, and 49.97 mmol/L, respectively (Figure 1a).
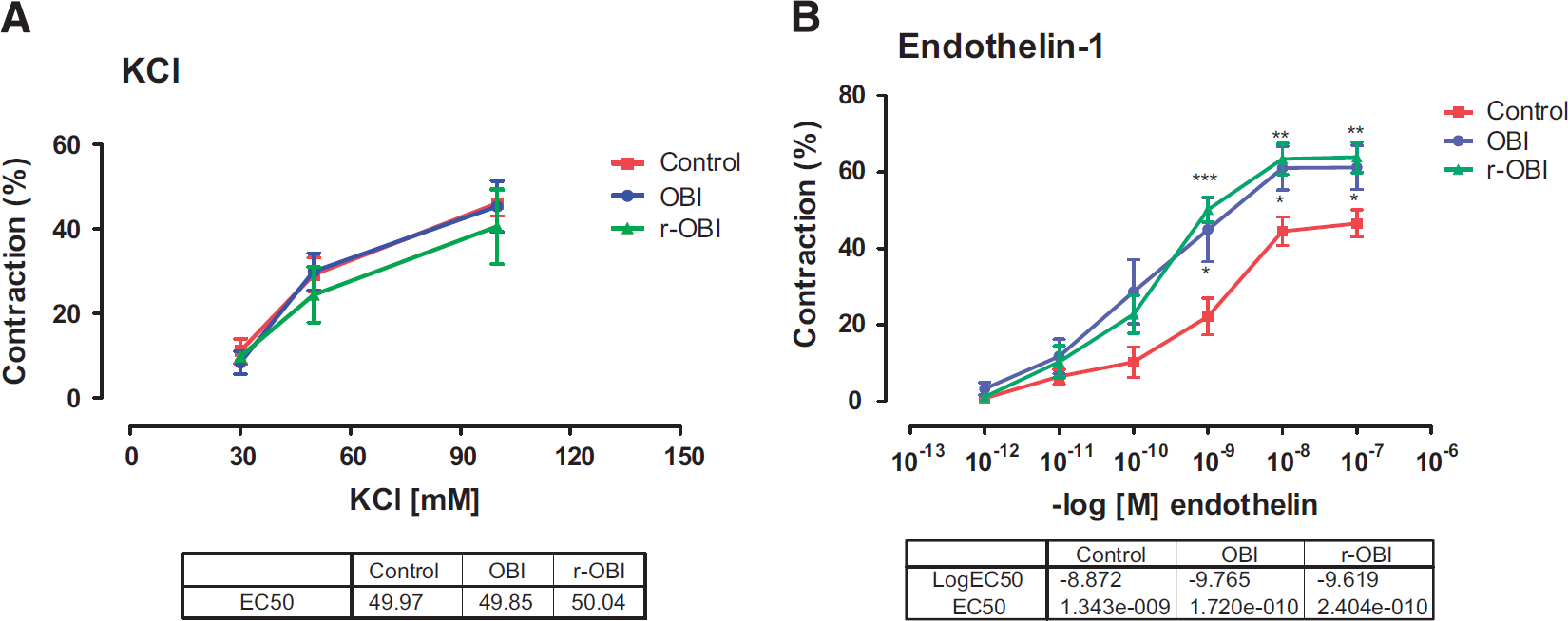
Contractile responses of the basilar arteries to (
In contrast, basilar arteries from both OBI groups showed increased sensitivity to ET-1 when compared with basilar arteries from the control group. The dose-response curve in OBI and r-OBI was shifted to the left, with EC50 1.72×10−10 and 2.40×10−10, respectively, versus control (EC501.34×10−9). Emax was also significantly (P<0.01 to 0.05) higher in arteries from both OBI groups when compared with arteries from the control group.
Relaxation Responses
Basilar arteries from both OBI groups showed significantly (P<0.01 to 0.05) impaired endothelium-dependent vasodilation to cumulative ACh (10−10 to 10−4 mol/L). The EC50 values in control, OBI, and r-OBI were 3.7×10−7, 6.9×10−6, and 1.90×10−6, respectively (Figure 2a).
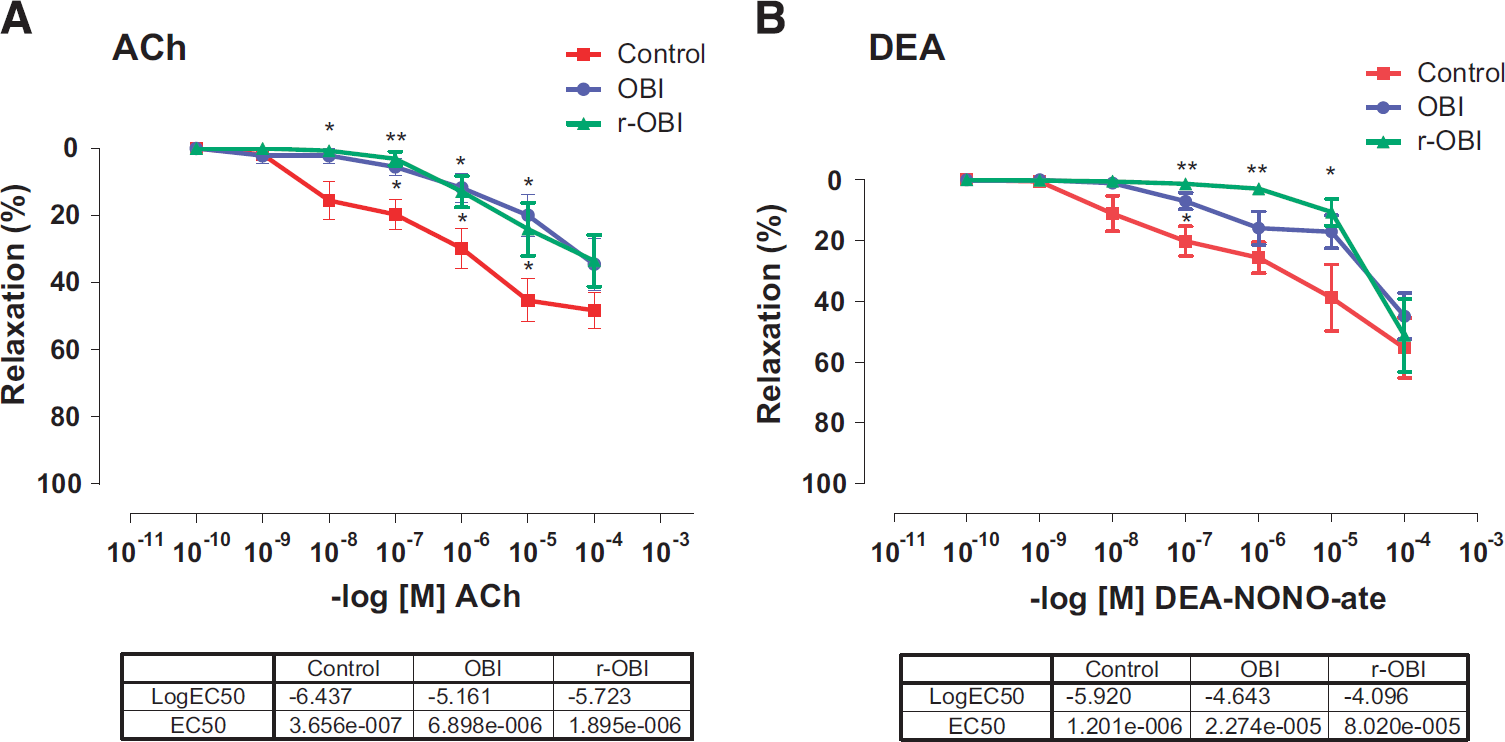
(
Relaxation to the NO donor, DEA-NONO-ate (10−10 to 10−4 mol/L) was significantly (P<0.01 to 0.05) decreased in basilar arteries from the r-OBI group as compared with those from the control group, with an EC50 of 8×10−5 in the r-OBI group versus 1.2 × 10−6 in the control group (Figure 2b).
General Characteristics of Vessels and Histomorphology The maximum diameters (Table 1), spontaneous diameters, and endothelium thickness of basilar arteries from the OBI and r-OBI groups were similar to those in arteries from the control group (Figure 3). There was a significant increase (P<0.05) in wall thickness of basilar arteries in the r-OBI groups as compared with those of the control group. Although the wall thickness of arteries in the OBI group increased, the change did not reach statistical significance. The wall/lumen ratio also increased significantly (P < 0.05) in r-OBI group versus the control group (Table 1). The wall/endothelium ratios in OBI and r-OBI groups were not significantly different when compared with the ratio in the control group.
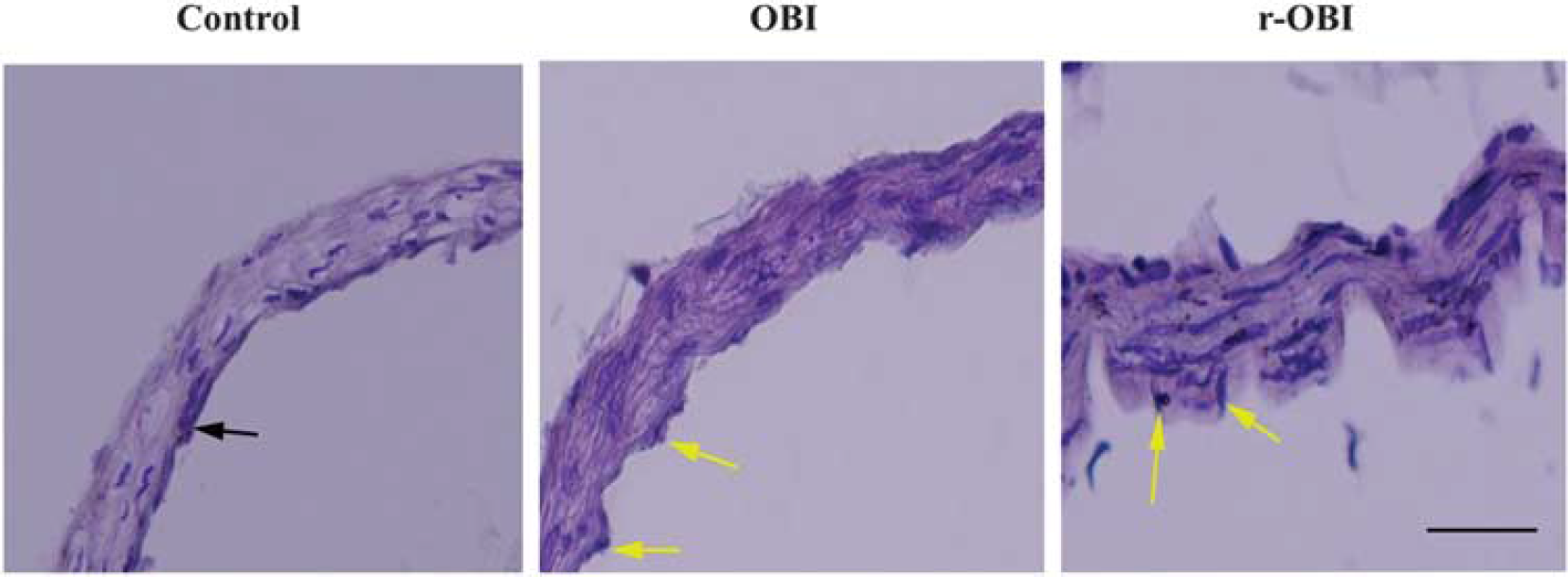
Hematoxylin & Eosin staining of basilar arteries. The wall thickness and the wall/lumen ratio were increased in both overpressure blast injury (OBI) groups. Internal elastic lamina was severely corrugated (arrows). Bar: 100 μm. r-OBI, repeated OBI.
General characteristics of the basilar arteries
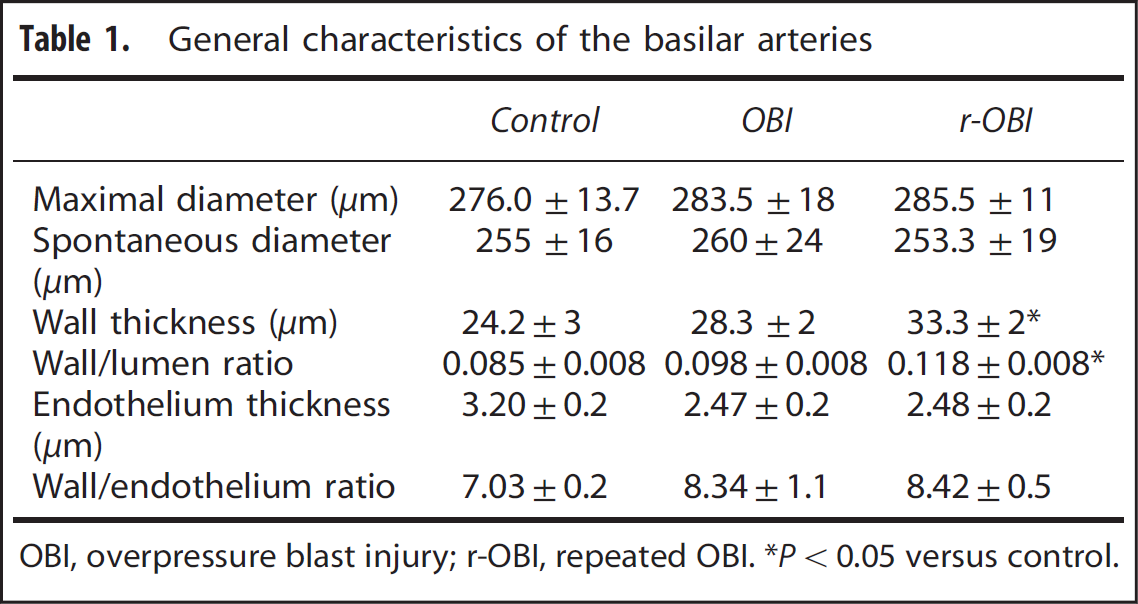
OBI, overpressure blast injury; r-OBI, repeated OBI. *P< 0.05 versus control.
Endothelin-1 Type A and Endothelial Nitric Oxide Synthase Immunohistochemistry
The cell nuclei counterstained with DAPI (blue fluorescence, first column) were merged with endothelin-1 type A (ETA) (green fluorescence, middle column in Figure 4) or endothelial nitric oxide synthase (eNOS) (green fluorescence, middle column in Figure 5). The ETA immunofluorescence in basilar artery cross-sections, an indicator of ETA receptor expression, was significantly increased in both OBI groups as compared with the control group (Figure 4), consistent with the increased contractile responses to ET-1 in arteries from the OBI groups. In contrast, although there was a decreased relaxation response to ACh, indicating impairment of endothelium-dependent NO-mediated dilation, eNOS immunoreactivity (green fluorescence) was significantly increased in crosssections of arteries from both OBI groups. This increase in fluorescence was more apparent in the single OBI group, possibly resulting from the activation of compensatory mechanisms (Figure 5).
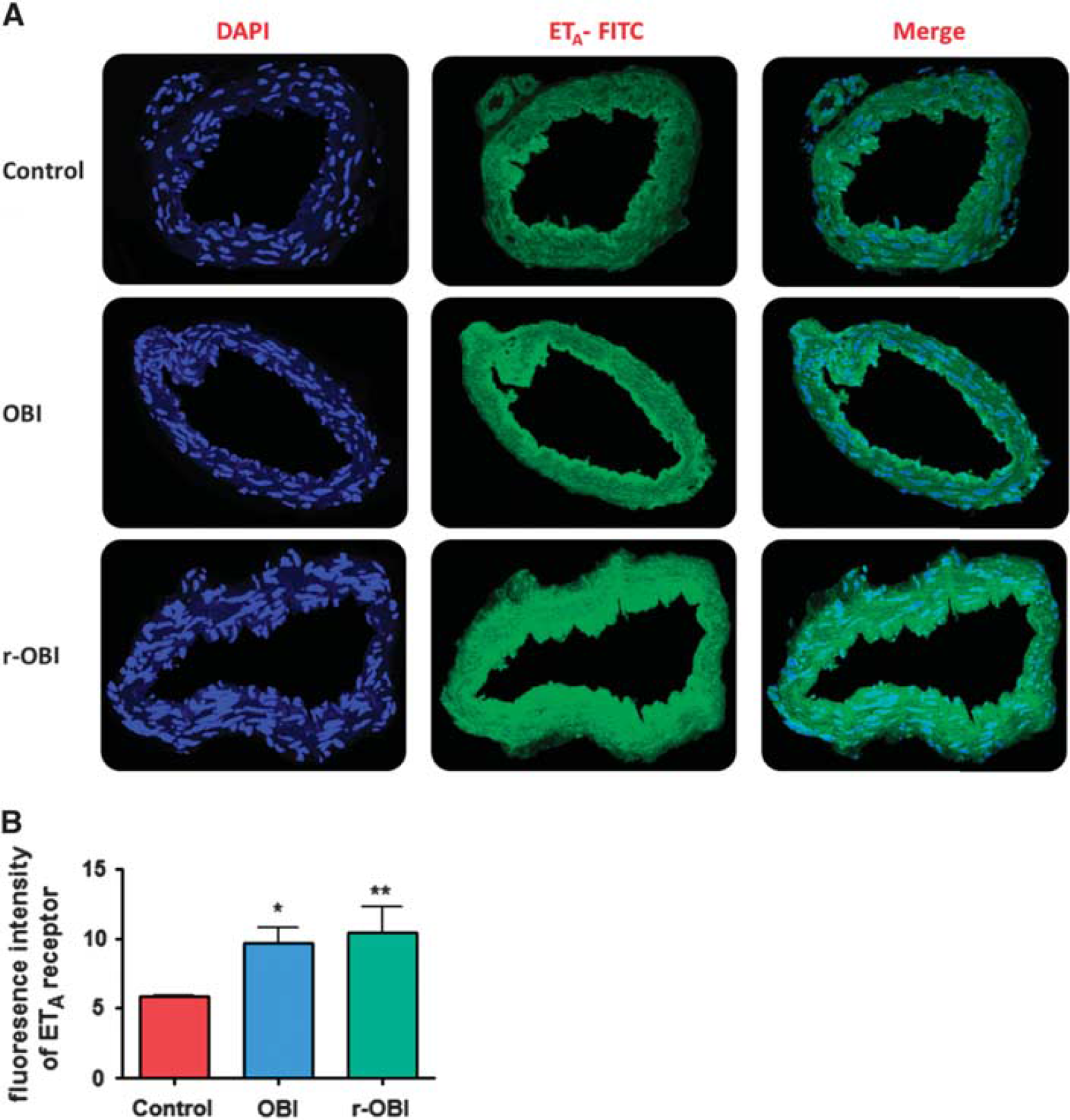
(
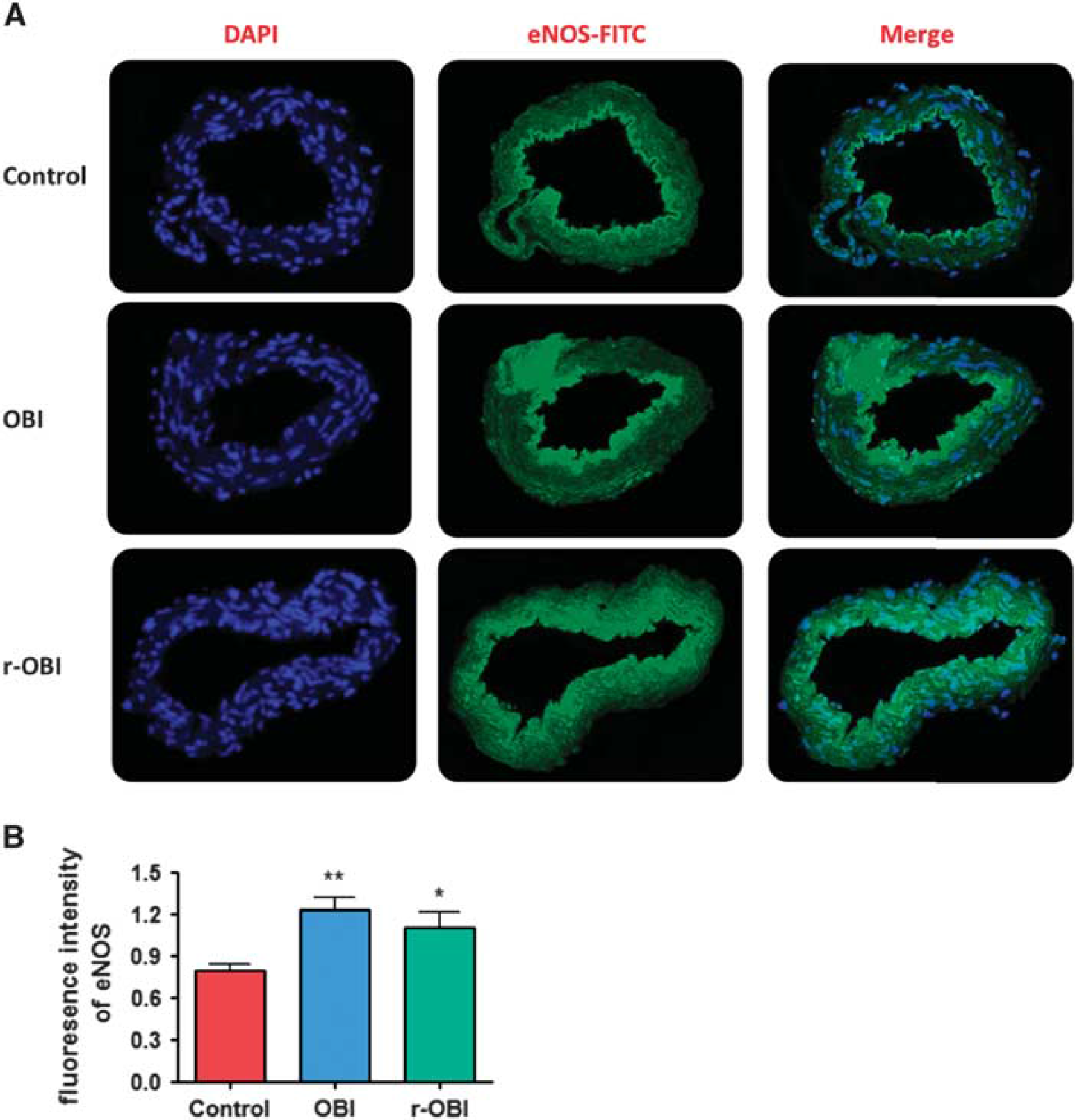
(
DISCUSSION
Experiments using whole-body or head protection in animals subjected to blast showed that head protection failed to prevent inflammation in the brain or reduce neurologic deficits, whereas whole-body protection was successful in alleviating blast-induced functional and morphologic impairments in the brain. From these results, it was concluded that in addition to the localized brain response, systemic responses to the blast are also involved in the pathobiology of blast neurotrauma. 18
In patients with subarachnoid hemorrhage, vasospasm of the basilar artery is the major incapacitating or morbid complication. Although the underlying pathogenic mechanisms of cerebral vasospasm are not completely understood, experimental and clinical studies have shown that the acute symptoms are attributed to increased intracranial pressure, decreased cerebral perfusion pressure and blood flow and the resulting ischemic brain injury.22–24 Since the basilar artery is the most important vessel in the posterior cerebral circulation, decreased blow flow to this region is associated with cognitive deficits, psychosocial impairments, sleep disturbances, and a decreased quality of life after brain injury.17,25 Although we did not observe evidence of hemorrhage or measurable vasospasm (resting and maximal diameters were not altered by OBI) in the basilar artery, impaired endothelial function and hypersensitivity to ET-1 could contribute to impaired perfusion of the brain regions served by the basilar artery during periods of greater functional activity. Future in vivo measurements of basilar artery flow will be needed to establish the impact of OBI-induced basilar artery dysfunction during periods of increased neural activity in these brain regions.
Decreased cerebral blood flow may lead to ischemia and is accompanied by increased generation of ROS leading to neuronal damage, by promoting lipid peroxidation, protein breakdown, DNA damage, and astrocytic cell death. 26 Ischemia also leads to inhibition of NA/K ATPase pumps and increases edema,12,27 which in turn worsen the circulatory failure. The subsequent inflammatory response further contributes to endothelial dysfunction and related impairment of vascular function. 7 Furthermore, this model of OBI we used in our experiments has been shown to cause a neuropathology leading to neuroglial perturbation, taurine hyperphosphorylation, TDP-43 proteinopathy, gliosis, and microgliosis in brain.19,28,29
In our experimental setup to evaluate cerebrovascular function, we used two contracting and two relaxing agents for receptor-dependent and -independent contractility plus endothelium-dependent and -independent relaxation. KCl-mediated contraction was used as a reference contraction for each individual artery because it is a nonreceptor-mediated contractile agonist. Previous studies with KCl have shown that the contractions are not influenced by the presence or absence of the endothelium. 30 In our experiments, the contractile response to KCl was similar among groups. In contrast, contractile response to ET-1, which is receptor dependent, was increased in the OBI groups, indicating an increased sensitivity to ET-1. Endothelin-1 type A receptors are the predominant subtype found in the vascular smooth muscle of the blood vessels. Activation of ETA receptors by ET-1 induces vasoconstriction, whereas the ETB1 receptors mediate vasodilation via NO release. 31 When we observed that the contractility due to ET-1 was increased in basilar arteries from the OBI groups, we hypothesized either ETA was upregulated or ETB1 was downregulated. The immunohistochemistry assays showed an increased fluorescence to ETA receptors consistent with ETA upregulation and greater ET-1-induced vasoconstriction in isolated basilar arteries.
For the evaluation of vasodilation, we used two pharmacological agents, ACh and DEA-NONO-ate, to assess endothelium-dependent NO-mediated relaxation and endothelium-independent relaxation to exogenous NO, respectively. ACh-induced endothelium-dependent relaxation is largely attributed to NO, whereas endothelium-independent relaxation is likely to be mediated by ion channels, and regulation of intracellular calcium within the vascular smooth muscle. 32 In addition, the endothelium has a significant role in preventing platelet aggregation and clot formation. 30 In the present study, vasodilation to ACh was significantly decreased in both OBI groups, indicating endothelial dysfunction. Endothelium-independent relaxation to the NO donor, DEA-NONO-ate, was only impaired in the r-OBI group, indicating that dysfunction of the vascular smooth muscle in basilar arteries only occurred with repeated injury.
Although relaxation responses to ACh decreased in basilar arteries from both OBI groups, eNOS immunofluorescence increased in the arteries from the OBI groups. The increase in eNOS immunofluorescence may be the result of an acute reduction in NO availability and the subsequent production of ROS leading to a compensatory increase in eNOS expression. For example, H2O2 has been shown to be critical for exercise training-induced increases in eNOS expression. 33 Although the low concentrations of NO produced by eNOS are necessary to maintain good endothelial function, high levels of NO can worsen the pathologic processes, by promoting inflammation or an immune response, and thus can be detrimental to the cells. 6 Alternatively, the increase in eNOS expression may reflect an uncoupling of eNOS expression with NO production,34,35 as has been shown to occur with old age. 36 Regardless of the mechanism, the higher eNOS expression in the OBI group appears to be due to an acute response to injury that is not sustained with r-OBI.
In the present study, we observed that the wall/endothelium ratio had a tendency toward an increase while the increase in wall thickness in the r-OBI group was significant. This may be an indicator of vascular remodelling—due to the repeated trauma exposure—that was not apparent in the single OBI group. Also, endothelial thickness was decreased in basilar arteries from the OBI groups, but failed to reach significance. Because the vessel consists of three layers, if the wall thickness is increased while the endothelial thickness remained unchanged, it shows that the media-intima thickness is increased. This likely occurs through proliferation of smooth muscle cells causing vascular remodelling and stiffness. Vascular stiffness is well known to lead to a decreased relaxation of the vessels, which is consistent with the observed decrease relaxation to both endothelium-dependent and -independent agents. Since the endothelium is critical for the release of vasoactive factors that modulate vascular smooth muscle tone, endothelial dysfunction is characterized by decreased production and/or bioavailability of NO. Overpressure blast injury resulted in impaired endothelium-dependent dilation, despite greater eNOS expression, suggesting decreased NO bioavailability. According to the free radical hypothesis of vascular dysfunction, excessive ROS generation leads to an increased formation of peroxynitrite (formed by the binding of superoxide with NO), which activates prostaglandin metabolism and vascular remodelling. 6 Our data indicate that the arterial wall thickness and the wall/lumen ratio increased significantly in basilar arteries from the r-OBI group, demonstrating remodelling of the vascular wall. Nitric oxide is known to inhibit smooth muscle proliferation in vitro 37 and the loss of a functionally intact endothelial layer would prevent this inhibitory effect. If r-OBI causes persistent endothelial dysfunction, then this could contribute to smooth muscle proliferation and wall thickening in the basilar arteries from the r-OBI group. Thus, in addition to potential changes in NO signalling mechanisms, our data suggest that remodelling of the arterial media also contributes to OBI-induced vascular dysfunction of the basilar arteries.
In conclusion, our results indicate that although no apparent change was observed in terms of diameter (resting and maximum) after OBI, the physiologic response of the vessels to endogenous agents like ET-1 or endothelium-dependent vasodilator stimuli was altered, potentially as a result of the molecular changes in receptors or enzymes. Upregulation of ETA receptors may further contribute to basilar artery vasospasm after OBI. Moreover, impairment of a vasodilation response as a consequence of endothelial dysfunction and/or vascular remodelling in basilar arteries after OBI may lead to decreased cerebral blood flow, which is a precipitating factor for neurologic disorders.
Footnotes
NT, HZT, JM-D, and KKWW designed the experiments. HZT, JM-D, ZY, JO, YS, KS, and PG performed the experiments and data analysis. NT, HT, MDD, PJS, and KKWW contributed to the composition of the manuscript.
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors express their special thanks to Prof. Russell M.Bauer for his leadership for the support of the project.
Notes
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit ![]()