
Abstract
Heat shock proteins (HSPs) are molecular chaperones with essential roles in modulating the proteolytic machinery and accelerating cell repair. Heat shock protein overexpression has been observed in vivo and in vitro under stresses including heat, nutrient deprivation and ischemia. Experiments in in vivo models of stroke indicate that transgenically overexpressed or virally delivered HSPs can enhance cell survival, but cannot always reduce lesion size. This study aims to assess the effects of virally delivered HSPs in a rat middle cerebral artery occlusion model of reversible focal cerebral ischemia using noninvasive magnetic resonance imaging. Attenuated herpes simplex virus carrying HSP27, HSP70, or a LacZ control was microinjected into the striatum 3 days before ischemia. Multislice T2-weighted images at 24 h after ischemia indicated that lesion volume was reduced by 44% in HSP27-treated animals compared with controls (P 0.019). No significant differences were found between HSP70-treated and control animals (P 0.88). Immunohistochemistry and Western blots revealed that HSP27 and HSP70 expression levels were equally high in injected hemispheres, but only the former had an effect on lesion size. This is the first evidence of the efficacy of gene therapy with any viral vector expressing HSP27 in an experimental model of stroke.
Introduction
Heat shock proteins (HSPs) were discovered in 1962 as part of an endogenous protective mechanism by which cells respond to environmental stress (Ritossa, 1962). Their name was initially derived from the observation that their expression is enhanced on exposure to high temperatures that lead to protein denaturation. Subsequent experiments in vitro and in vivo have established that these proteins are upregulated during a variety of stresses (Lindquist and Craig, 1988; Higashi et al, 1994; Wagstaff et al, 1996; Ehrnsperger et al, 1997; Feder and Gretchen, 1999). The HSP family consists of proteins with different subcellular anatomical and temporal distributions and functions, and is divided into subfamilies named according to their molecular weight (Latchman, 1998). Some of the most studied HSPs include ubiquitin, HSP27, HSP56, HSP70, and HSP90. Although these proteins have variable roles, it is clear that they are essential for the maintenance of cell viability under stressful conditions.
Under normal conditions, many HSPs are constitutively expressed and responsible for the correct folding and localization of nascent polypeptides. They can also be involved in antigen presentation, steroid receptor function, intracellular trafficking of proteins to different organelles, multiprotein complex assembly, nuclear receptor binding, and apoptosis among other housekeeping functions (Jakob et al, 1993; Ehrnsperger et al, 2000; Richter-Landsberg and Goldbaum, 2003; Latchman, 2004; Katschinski, 2004). The inducible forms, however, are expressed upon stress and can enhance cell survival by binding to denatured proteins to prevent their misfolding or aggregation with other proteins. Their main role is refolding aberrant proteins or targeting them for disposal into aggresomes (Kiang and Tsokos, 1998; Sharp et al, 1999; Latchman, 2004).
HSP overexpression has been observed in vivo and in vitro under a range of conditions, including hypoxia, nutrient deprivation, ischemia, and thermal stress. Briefly, upregulation of HSP27, HSP70, and HSP90 mRNA and protein after several insults has been detected in numerous neuronal cultures and brain tissue extracted from animals exposed to stress (Kawagoe et al, 1993a, b; Higashi et al, 1994; Wagstaff et al, 1996; Feder and Gretchen, 1999). In vitro evidence of HSP-transfected neurons supports the protective role of HSP27 and HSP70, although only the former seems to have significant antiapoptotic effects in neuronal cells (Uney et al, 1993; Mailhos et al, 1994; Amin et al, 1996; Wyatt et al, 1996; Fink et al, 1997; Carper et al, 1997; Wagstaff et al, 1999, Rajdev et al, 2000; Lee et al, 2001a; Kelly et al, 2001a; Benn et al, 2002; Zourlidou et al, 2004). Experiments in in vivo models of epilepsy and stroke indicate that transgenically overexpressed or virally delivered HSPs are not always able to reduce lesion size, but are able to enhance cell survival (Plumier et al, 1997; Yenari et al, 1998; Rajdev et al, 2000; Lee et al, 2001a, b; Kelly et al, 2001a, b, 2002; Hoehn et al, 2001; Akbar et al, 2003; Kalwy et al, 2003; Tsuchiya et al, 2003). The great majority of these studies have involved HSP70, while the protective effect of HSP27 in vivo has only been investigated in a model of kainate-induced cell death in the hippocampus (Wagstaff et al, 1999).
Since HSP27 and HSP70 are the main inducible HSPs in the central nervous system (Yenari, 2002; Latchman, 2004), we investigated the effect of gene therapy with these HSPs in an in vivo model of transient cerebral ischemia using noninvasive magnetic resonance imaging (MRI) techniques in conjunction with immunohistochemistry and Western blotting. Here we report that preischemic injections with HSP27 delivered by a herpes simplex virus (HSV-HSP27) had protective effects that were not apparent with HSV-HSP70. This is the first direct evidence of the efficacy of gene therapy with any viral vector expressing HSP27 in reducing lesion size in an experimental model of stroke.
Materials and methods
Experimental Design
All animal care and procedures were performed in accordance with the UK Animals (Scientific Procedures) 1986 Act.
Our experiment involved 4 different studies, A, B, C, and D, all using adult male Sprague-Dawley rats (220 to 250 g, Charles Rivers).
For study A, 18 rats were divided into 3 subgroups microinjected with HSV-HSP27 (n = 6), HSV-HSP70 (n = 6), or HSV-LacZ as a control (n = 6). All rats underwent occlusion of the middle cerebral artery (MCA) 3 days after microinjection and MRI scanning 24h later.
For study B, 15 rats were microinjected with control vector HSV-LacZ. Viral spatial distribution and expression levels in the brain were assessed by X-gal staining in 5 subgroups (n = 3) at different time points after microinjection (3 days, 1 week, 2 weeks, 3 weeks, and 4 weeks).
Rats in studies C (n = 12) and D (n = 12) were divided into subgroups microinjected with HSV-HSP27 (n = 4), HSV-HSP70 (n = 4), or HSV-LacZ as a control (n = 4). All rats underwent middle cerebral artery occlusion (MCAO) 3 days after microinjection. T2-weighted multislice scans were run 24 h later to confirm the presence of a lesion. At 48 h after MRI, brains were removed for immunohistochemistry (study C) or Western blotting (study D).
Virus Growth
Herpes Simplex virus strains 4 ± /27-/pRl9-HSP27, 4 ± / 27-/pRl9-HSP70, and 4 ± /27-/pRl9-LacZ have been described previously (Coffin et al, 1996; Wagstaff et al, 1999; Wolfe et al, 1999; Latchman, 2001; Lilley et al, 2001). Briefly, the HSV-1 backbone 1764/4 ± /27-/RL1 was attenuated by a disabling mutation in Vmw65, deletion of ICP34.5 and ICP4. An expression cassette containing cDNA inserts from either Chinese hamster HSP27, inducible human HSP70, or β-galactosidase under the control of a cytomegalovirus immediate-early promoter was inserted into the backbone via the pl9/20.5/UL43 vector by homologous recombination. Replication of such incompetent viruses was obtained in the complementing B130/2 BHK cell line (stably overexpressing ICP27 gene of HSV-l). Concentrated virus was titrated by counting plaque formation on B130/2 BHK cells. Titers of 1–2 × 1010 plaque-forming units (pfu) were routinely obtained.
Intracerebral Microinjections
Rats were anesthetized with 2% isoflurane in 100% oxygen. All rats in study B were microinjected with 2.5 μL of 1 × 106pfu/μL HSV-LacZ. Subgroups of rats in studies A, C, and D were stereotaxically injected with 2.5μL of 1 × 106pfu/μL HSV-LacZ, HSV-HSP70, or HSV-HSP27. The virus suspensions were delivered into the right striatum (bregma coordinates: AP = + 1.0 mm; L = −3.5 mm; V = −5.0 mm) at a rate of 0.25μL/min. Animals were allowed to recover for 3 days before being subjected to focal reversible MCAO.
MCAO
All rats in studies A, C, and D were anesthetized with 3% isoflurane in 100% O2 and maintained at 2% isoflurane in a 70:30 O2:N2O mix delivered by a nose cone. The origin of the right MCA was occluded with an intravascular suture as described elsewhere (Longa et al, 1989). Briefly, the common, internal, and external carotid arteries were exposed by a cervical midline incision. A 3/0 (290 μm) monofilament suture (Monocryl, Ethicon, Edinburgh, UK) with an araldite (Sil-Mid, West Midlands, UK)-coated rounded tip was introduced into the lumen of the common carotid artery and advanced approximately 17 mm to occlude the MCA. After 30 mins, reperfusion occurred by retracting the embolus. Rectal temperature was maintained at 37°C ± 1°C via a heating blanket controlled by a thermocouple. The whole procedure took 1.5 h and the animals were left to recover for 24 h before scanning with free access to food and water.
Magnetic Resonance Imaging
Animals in studies A, C, and D were anesthetized with 2% halothane in a 70:30 O2:N2O mix delivered via a nose cone and placed on a probe with bite and ear bars securing the head to minimize motion artifacts. Physiological monitoring included electrocardiography (ECG) recordings and rectal temperature recordings. Temperature was maintained at 37°C ± 1°C using an air-warming system.
All scans were performed 24 h after MCAO using a 2.35-T horizontal bore magnet (Oxford Instruments, Oxford, UK) interfaced to a SMIS console (Guildford, UK). Images were acquired using a volume transmitter coil and a separate decoupled surface receiver coil. All sequences were run on a single 2-mm-thick coronal slice located in the MCA territory (0.5 mm from bregma). The matrix size was 128 × 64 and the field of view was 40 × 20 mm. The imaging protocol included two echo planar imaging (EPI) sequences, T1 (TR = 2000 ms, TE = 18 ms, TI array = 284, 434, 634, 834, 1234, 1734, 2734, 3734 ms, 16 averages), and continuous arterial spin labeling (CASL) (TR = 1000 ms, TE = 18 ms, 44 averages). The inversion slice was positioned approximately 13 mm from the back of the cerebellum, and labeling and reference scans were obtained with a 500-ms delay time (Alsop and Detre, 1996). A multislice T2-weighted spin echo (SE) sequence (TR = 1500 ms, TE = 120 ms, 8 averages, 9 slices) with a 1-mm-slice thickness was also run to determine the lesion volume. Total scan time was 35 mins.
Image Processing and Data Analysis
All images were reconstructed with IDL Software Version 5.2 (Research Systems Inc., Boulder, CO, USA) to obtain quantitative maps. Labeled and reference CASL images were subtracted and combined with T1 measurements to calculate cerebral blood flow (CBF) maps. Lesions and regions of interest were delineated on all processed images and analyzed with SMIS Image Display Version 3.7 and Scion Image software. Lesion size was then expressed as a percentage of total pixel number in the brain slice. Data analysis was performed by an observer masked to the groups, and resulting lesion areas per slice were used to determine lesion volume across the whole brain. An analysis of the group differences in lesion spread from the front to back of the brain was performed using a random coefficients model, as required to deal with the expected spatial correlation in the data. A comparison based on Akaike's Information Criterion indicated that a parallel response model provided an adequate description of the lesion data over slices 3 to 9 inclusive, covering the MCA territory. These calculations were performed using SAS PROC MIXED (SAS Institute, 1999). Student's t-tests assuming unequal variances and Mann-Whitney tests were used to compare all other measurements (SPSS 12.0.1). Data are presented as mean ± standard error and considered significant at P<0.05.
Histology
X-Gal Staining: Study B investigated LacZ expression patterns after HSV delivery using X-gal staining to detect enzymatic activity. Animals were deeply anesthetized with 3% to 5% halothane in O2 and transcardially exsanguinated with 0.9% saline, followed by 4% paraformaldehyde (PFA) at 4°C. Brains were embedded in 3% agarose and sliced into 250-μ-thick- slices. Sections were stained with X-gal solution (5 mmol/L potassium ferricya-nide, 5 mmol/L potassium ferrocyanide, 1 mmol/L MgCl2, 0.2% Igepal in phosphate-buffered saline (PBS) and 40mg/ml X-gal, 5'bromo-4chloro-3indolyl-B-
Immunohistochemistry: HSP-treated animals in study C were stained with HSP27 and HSP70 antibodies to verify the expression levels of microinjected proteins in the brain. After scanning, animals were deeply anesthetized with halothane for decapitation. Brains were removed and immediately snap frozen in isopentane (2-methyl-butane, BDH, Leicester, UK) cooled in liquid nitrogen. Cryostat sections (l0-μ-thick) were stained with rabbit polyclonal anti-HSP27 (1:200, product #SPA-801, Stressgen Biotechnologies, York, UK) and mouse monoclonal anti-HSP70 (1:500, product #SPA-810, Stressgen Biotechnologies, York, UK) primary antibodies overnight at 4°C. StreptABC Complex/horseradish peroxidase (HRP) Duet Mouse/Rabbit kit (DAKO, Glostrup, Denmark) was used as secondary antibody. Signal was amplified with 3,3-diaminobenzidine tetrahydrochloride (DAB, Sigma, Dorset, UK). Sections were counterstained with hematoxylin. All slides were viewed under an Axioskop 2 Plus microscope (Carl Zeiss Ltd, Hertfordshire, UK) and pictures captured with an AxioCam HRc camera and viewed with Axiovision 3.0.6 SP2 software.
Western Blots: Brains of all rats in study D were quickly isolated and snap-frozen. Basal ganglia and cortical regions were dissected and homogenized in 0.1% Tween extraction buffer. Bradford assay was used to determine the protein concentration, and 30 μg of total lysate was subjected to sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis. Membranes were probed with polyclonal anti-HSP27 (1:1000, Stressgen, York, UK) and monoclonal anti-HSP70 (1:1000, Stressgen, York, UK) primary antibodies, and secondary anti-rabbit (1:2000, DAKO, Glostrup, Denmark) and anti-mouse HRP (1:2000, DAKO, Glostrup, Denmark) antibodies, respectively. Blots were then stripped and reprobed with GAPDH antibody (1:3000, Chemicon International, Hampshire, UK), followed by anti-mouse secondary to confirm equal protein loading. The signal was visualized with an enhanced chemoluminiscence kit (ECL, Amersham Biotechnologies, Buckinghamshire, UK), followed by exposure of the membranes to an X-ray film. Band densities were measured in a GS-800 calibrated densitometer using Quantity One Version 4.2.1 software (BIO-RAD, Hertfordshire, UK).
Results
HSP Effect on Lesion Size
Figure 1 shows multislice T2-weighted SE images obtained 24 h after MCA occlusion. The three groups of six animals had been microinjected with HSV-HSP27 HSV-HSP70, or, as controls, HSV-LacZ, 3 days before occlusion (study A). Averaged total lesion volumes measured from the images were: 223.5 ± 5.2 mm3 for HSV-LacZ controls, 218.3 ± 4.6 mm3 for HSV-HSP70-injected rats, and 100.1 ± 3.7 mm3 for HSV-HSP27-injected rats. A marked lesion volume reduction of 44.9% (confidence interval (CI) (26.4%, 76.0%)) was seen in HSP27-injected animals compared with LacZ-injected rats (P= 0.019). HSP70-treated animals showed no significant difference in lesion size from controls (P=0.88).
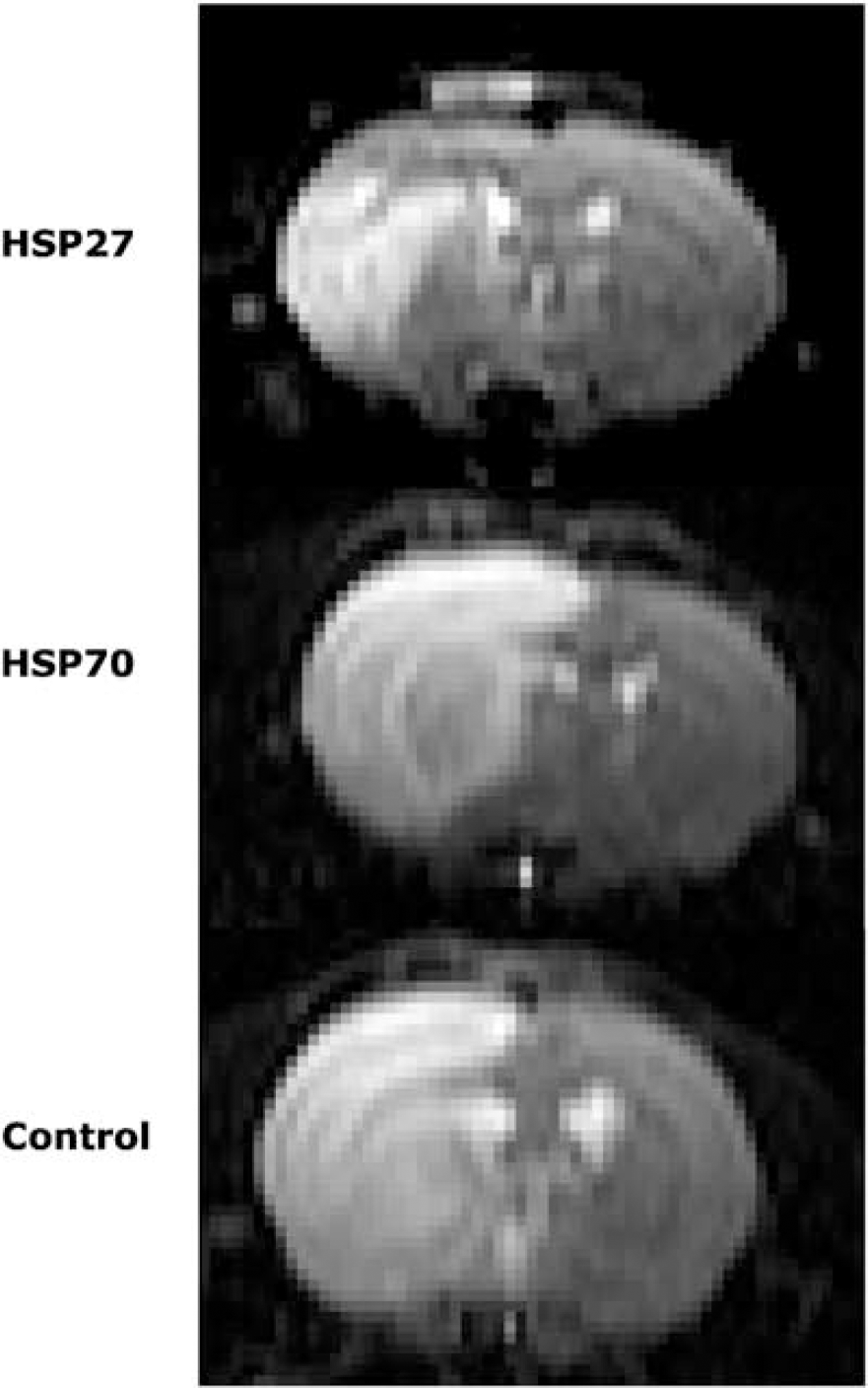
Representative center slice T2-weighted images of treated and control animals obtained 24 h after occlusion of the MCA. Intracerebral microinjections into the rat striatum with HSV-HSP27, HSV-HSP70 and HSV-LacZ were performed 3 days before ischemia. Lesion area appears as bright signal on these T2-weighted images.
Figure 2 shows the lesion volume per slice from front to back of the brain in HSV-HSP27-, HSV-HSP70- and HSV-LacZ-injected rats. Results yielded by the parallel response statistical model indicated that there was a constant volume difference between the HSP27-treated group and the other two groups in slices 3 to 9 of the multislice dataset (P< 0.003, difference = 14.35 mm3, CI (5.08, 23.62)). No lesion volume differences were found between HSP70-treated and control animals.
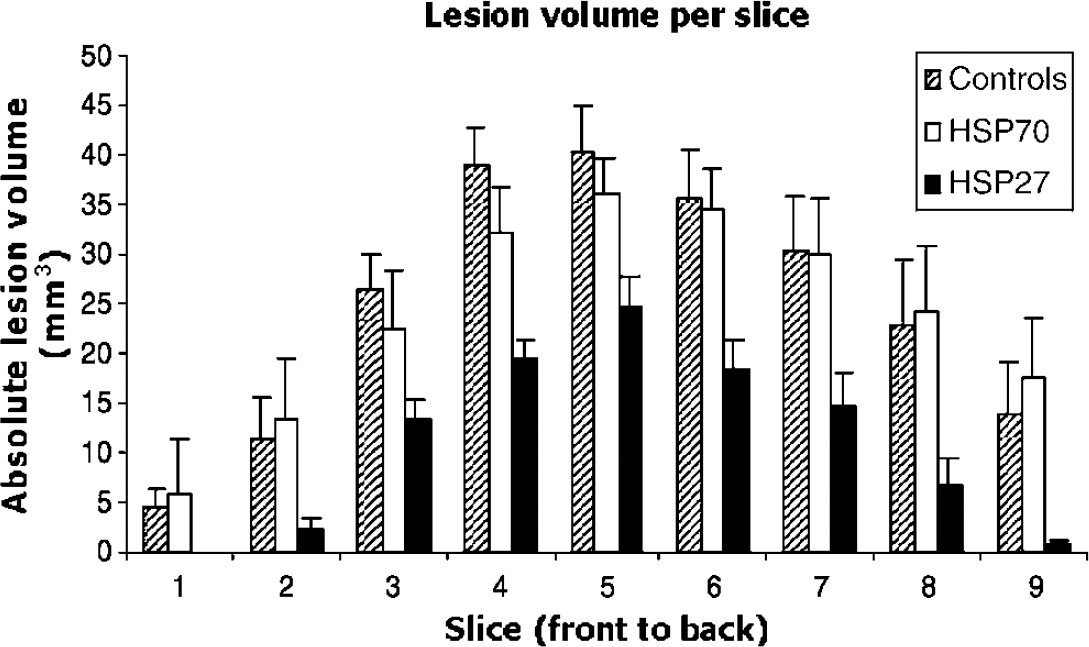
Absolute lesion volume per slice from the front to the back of the brain in HSP27, HSP70, and control groups. Total lesion size was calculated from multislice T2-weighted scans at 24 h after ischemia and expressed as volume (mm3) ± s.e.m.
Cerebral blood flow was measured 24 h after occlusion-reperfusion. Values in the ischemic hemisphere were expressed as a percentage of flow in the contralateral hemisphere: 82.3% ± 5.3% for HSV-HSP27, 77.1% ± 1.4% for HSV-HSP70, and 78.2% ± 5.0% for control animals. Complete recovery of CBF was not achieved, but all values were above the ischemic threshold (Kohno et al, 1995). Statistical analysis showed that there were no significant differences between animals in each group (Controls versus HSV-HSP27 P>0.2; Controls versus HSV-HSP70 P>0.7) at 24 h after reperfusion.
Herpes Simplex Virus Expression
Study B was designed to assess HSV-LacZ virus spatial distribution and timecourse of expression after intracerebral microinjection in a separate group of 15 animals. Previous studies by our group have characterized the vector's ability to infect different neuronal populations in vivo and its expression levels (Wagstaff et al, 1998, 1999; Howard et al, 1998; Palmer et al, 2000; Lilley et al, 2001; Kalwy et al, 2003). Determination of the regional distribution of the virus was necessary to compare it with the area affected by MCAO. A β-galactosidase staining assay with X-gal was used to detect LacZ expression at 5 different time points, ranging from 3 days to 1 month after injection. Blue signal was clearly detected in the basal ganglia of all microinjected rats (Figure 3). No staining was observed in the cortex, with the exception of traces of expression along the needle tract. Protein expression was strongest at 3 days and was sustained just below maximum levels for up to 1 month (data not shown). Mild β-galactosidase expression was detected along the needle tract at early time points, but this disappeared within a maximum of 10 days. This same assay was used to visualize LacZ expression in three of the six rats used as controls in our MRI study, and the results confirmed that protein expression overlapped with the ischemic lesion area in the basal ganglia, but not in the cortex.
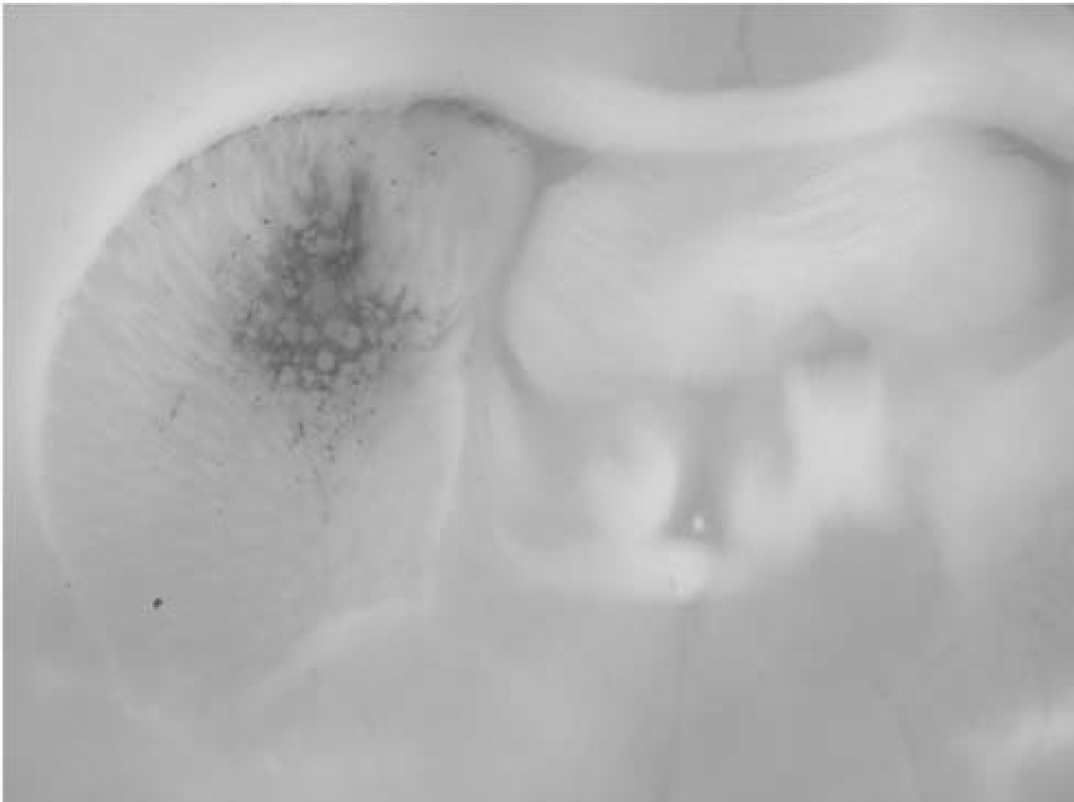
LacZ expression in the basal ganglia peaks 3 days after HSV microinjection. X-gal assay showing HSV-LacZ spatial distribution and expression levels at 72 h after intracerebral microinjection in the rat basal ganglia.
Histological Detection of HSP27 and HSP70
Studies C and D assessed the spatial distribution and levels of HSP expression by immunohistochemistry and Western blotting, respectively. These studies were performed in separate groups of animals due to differences in brain extraction and processing for the two histological techniques.
Western blots were performed for all animals in study D to test whether total HSP levels were elevated due to both ischemic insult and viral overexpression. Four different regions were dissected from each microinjected and ischemic brain: ipsilateral basal ganglia (BGI), ipsilateral cortex (CTXI), contralateral basal ganglia (BGC), and contralateral cortex (CTXC). Figure 4A shows protein extracts from an HSP27-injected animal, where HSP27 expression levels in BGI and CTXI were higher than both its contralateral counterparts and the equivalent brain regions in a LacZ-injected control. Similar results were seen in HSP70-treated animals. Membranes were rinsed and reprobed with GAPDH antibody to control for protein loading, and no differences were detected between samples (Figure 4B).
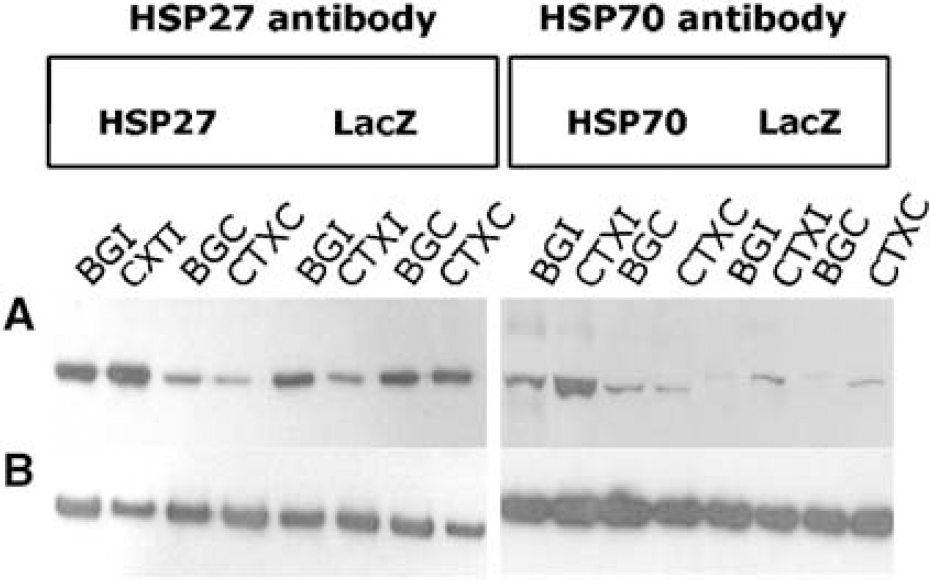
Western blots showing HSP27 and HSP70 protein expression levels in all microinjected and MCA occluded rats. Four brain regions were dissected from each rat brain: BGI, CTXI, BGC, and CTXC. (
Quantification of the bands revealed that both HSV-HSP27- and HSP70-microinjected rats had an approximate fourfold increase in protein levels of the appropriate HSP in ipsilateral brain regions compared with their contralateral counterparts. Similarly, a significant fourfold increase was seen when comparing the BGI and cortex of HSP-treated and HSV-LacZ-injected animals. Ratios of protein increase in the basal ganglia and cortex within and between animals are summarized in Table 1. These results indicated that the injected vectors were successfully expressing HSPs and raising the total levels of expression of the appropriate HSP after ischemic insult. Immunohistochemistry results were consistent with these findings, indicating that total HSP levels were higher in HSP-injected hemispheres than in LacZ-injected controls. Figure 5 shows expression of HSPs in the representative brain sections of ipsilateral cortex and basal ganglia of HSP27-, HSP70- and LacZ-injected animals.
Heat shock protein (HSP) levels in BG and CTX, expressed as ratiosipsilateral/contralateral
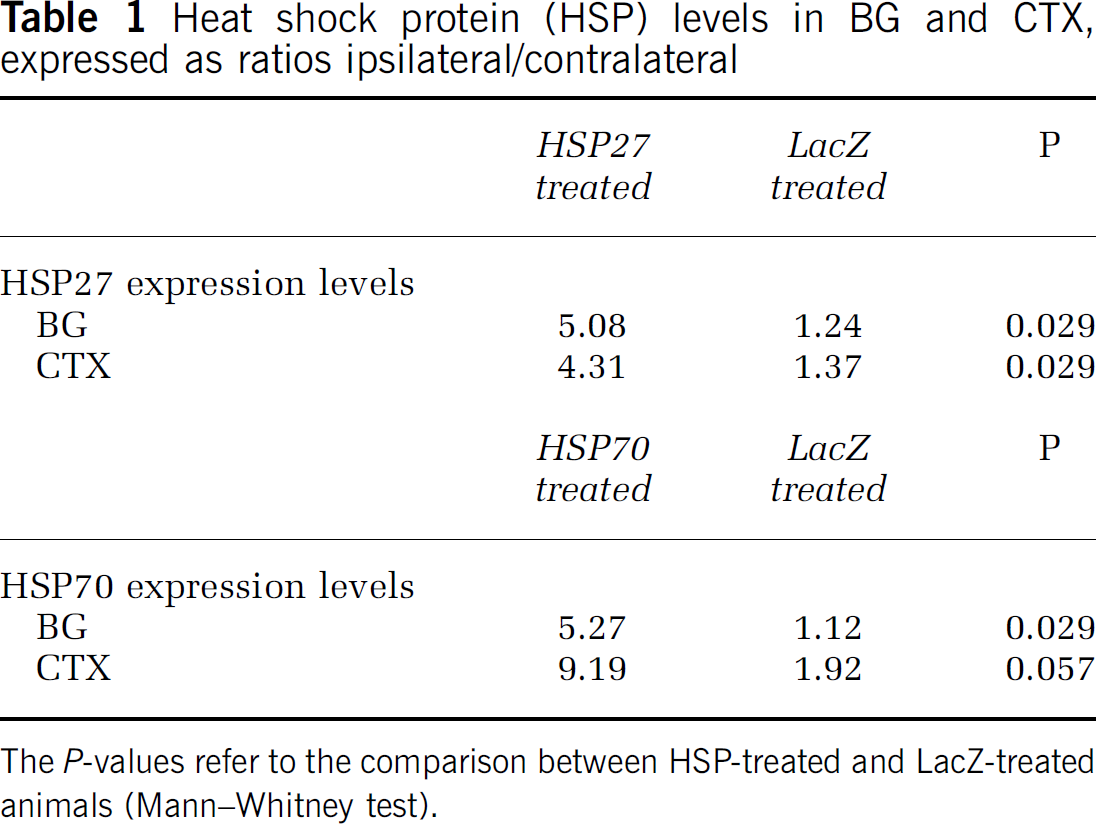
The P-values refer to the comparison between HSP-treated and LacZ-treated animals (Mann-Whitney test).
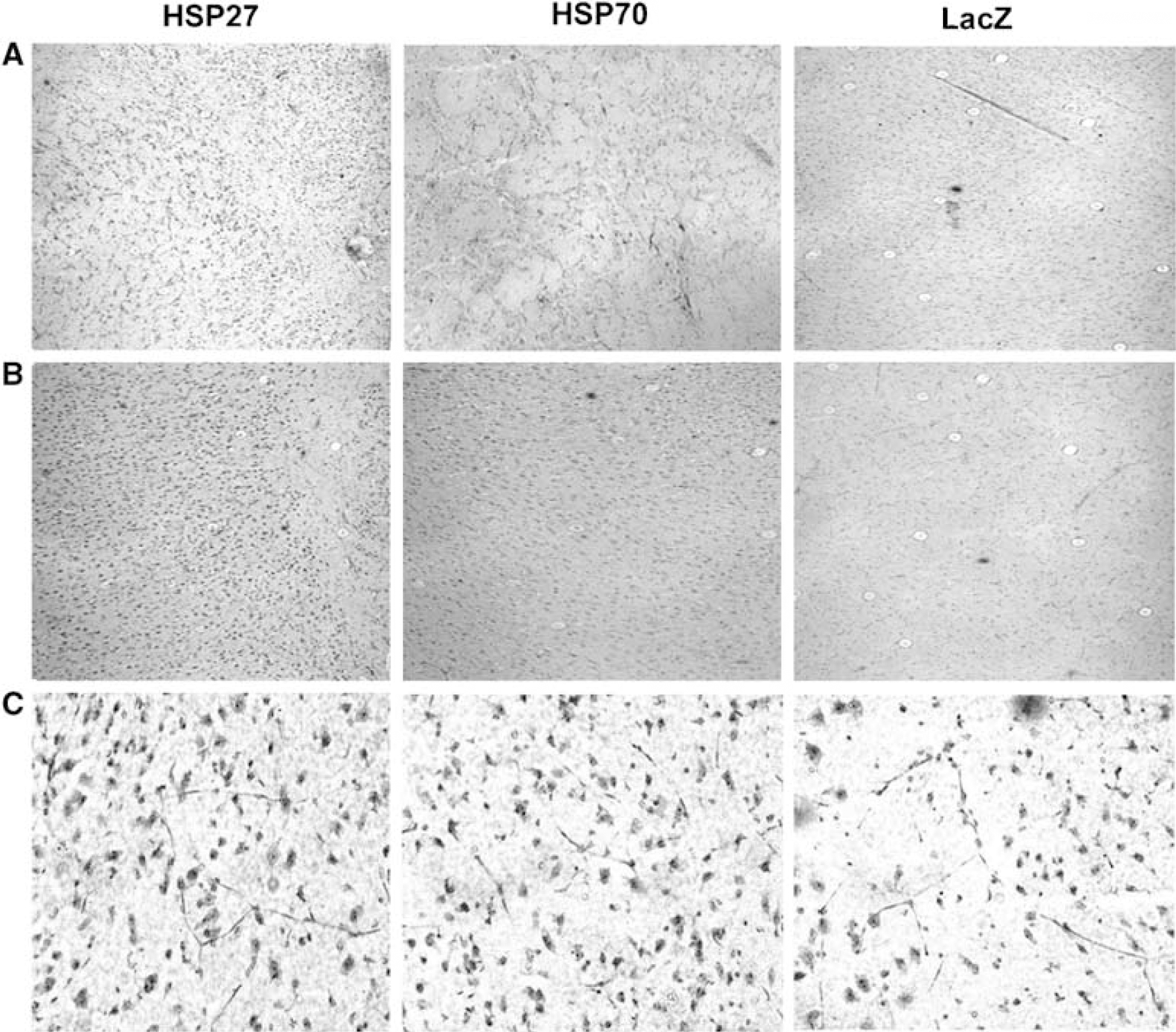
HSP27 and HSP70 protein expression in the rat brain. Immunohistochemistry on 10-μ cryosections taken 3 days after ischemia induction shows that both HSPs are overexpressed in the BGI and CTXI of microinjected and MCA-occluded rats. HSV-HSP27- and HSV-HSP70-injected rat brain sections were stained for HSP27 or HSP70, respectively. HSV-LacZ-injected controls shown were stained with HSP27 antibody for comparison. (
Discussion
The aim of the study was to investigate neuroprotection in vivo after delivery of an HSV-based vector expressing HSP27 or HSP70 in a rat model of cerebral ischemia. Magnetic resonance imaging enabled us to measure the effect of HSP27 and HSP70 treatment on lesion size at 24 h after stroke. For this in vivo study, we have chosen transient occlusion of the middle cerebral artery, a well-established and reproducible rat model of stroke. This creates a relatively large area of vasogenic edema in the cortex and basal ganglia that can be detected by multislice T2-weighted scans (Figure 1). These images showed a marked reduction in lesion size in HSV~HSP27-treated animals compared with HSV-LacZ controls. In contrast, HSV-HSP70 treatment had no effect on lesion size. All CBF values obtained 24 h after occlusion-reperfusion were above the ischemic threshold and similar in the three groups. Immunohistochemistry and Western blotting were used to examine the expression profile of the HSP27 and HSP70 vectors in tissue sections. The absence of a specific neuronal marker in our sections did not allow us to establish whether the virus was infecting neurons, glial cells, or both. Thus, histology revealed widespread staining for both proteins in ipsi- and contralateral brain hemispheres of injected and MCA occluded rats. Western blots, however, showed a significant increase in HSP27 and HSP70 expression in the ipsilateral cortex and basal ganglia of treated animals compared with LacZ-injected and MCA-occluded controls.
Interestingly, T2-weighted images showed that protection was not only localized to the basal ganglia where the virus was injected, but also extended to the cortex. Based on our HSV-LacZ profiles of spatial distribution and expression timecourse, we would have only expected HSPs to be present within the basal ganglia. The reason for this increase in the cortex is unclear and needs to be further investigated. Several studies have shown that HSPs can either be locally produced in axonal compartments away from the neuronal cell body (Willis et al, 2005) or transferred from glia to neuronal axons (Tytell et al, 1986; Sheller and Bittner, 1992; Guzhova et al, 2001). Moreover, in vitro studies in neurons suggest that uptake of exogenous HSPs added to the culture medium can protect these cells from stress (Tidwell et al, 2004; Guzhova et al, 2001). Our observation could therefore reflect the secretion of proteins from virally transfected cells and their recruitment to the injured cortex.
We report HSP27 caused a significant 44% reduction in lesion size. This protein is naturally expressed in glial cells and not in neurons, which previously suggested that it did not directly contribute to neuroprotection (Wagstaff et al, 1996). However, glial cells appear to be crucial for structural and metabolic support of neurons, maintaining synapse homeostasis and regulating the rate of neuronal repair (Jessen, 2004). Thus, their ability to secrete HSPs on stress might be a crucial mechanism to protect neurons, although this has not yet been investigated for HSP27. Regardless of the function of the endogenous protein, we observe that viral microinjections successfully induce expression of exogenous HSP27 across the brain, thus raising total HSP levels and possibly enhancing the neuroprotective effect. A ‘preconditioning’ or ‘tolerance’ effect of HSPs in the brain has been reported in in vitro and in vivo studies, showing that exposure to an initial mild stress raises HSP expression levels and can render cells more resistant by enhancing protection against a second more severe insult (Chen and Simon, 1997; Kirino, 2002; Dirnagl et al, 2003). The protection we observe is therefore likely to be due to a preischemic induced tolerance after microinjection, combined with the effect of the endogenous HSPs.
HSP27 has been shown to protect cells in vitro by interfering with the stress-triggered apoptotic cascades (Mehlen et al, 1996; Paul et al, 2002; Benn et al, 2002; Akbar et al, 2003; Latchman, 2004; Zourlidou et al, 2004). It has been suggested that it can interfere with both caspase-dependent and -independent apoptotic pathways. Several experiments show that this protein is able to block caspase-3 activation after ischemia (Pandey et al, 2000; Whitlock et al, 2005). Interestingly, replacing HSP27 in rat neurons with a caspase-blocking agent does not prevent cytotoxicity, suggesting that, in addition to caspase-3 blockage, HSP27 must exert chaperoning functions that prevent necrosis and caspase-independent events (Whitlock et al, 2005). Other groups have shown that HSP27 overexpression can sequester cytochome c after release from the mitochondria and prevent its binding to the apoptosome (Bruey et al, 2000; Garrido et al, 1997, 1999). Overexpression of HSP27 can also decrease the amount of apoptosis-inducing factor (AIF), which is released by mitochondria in the caspase-independent pathway of cell death (Rashmi et al, 2003).
Other possible mechanisms of protection by this HSP could be related to its chaperone functions. Some experiments have linked cytoskeleton stability to mitochondria and apoptosis. They suggests that HSP27 acts further upstream and prevents cytochrome c release via Bid relocalization, which is in turn associated with F-actin filaments modulated by this chaperone (Paul et al, 2002). It has also been shown that phosphorylated and unphosphorylated HSP27 binds actin and tubulin filaments and can promote axonal growth and regeneration after injury (Costigan et al, 1998; Williams et al, 2005). In contrast to other HSPs including HSP70, HSP27 has the ability of binding partially or completely denatured proteins in an ATP-independent fashion, which could be crucial in preserving cellular stability during the metabolic depletion caused by ischemia (Rogalla et al, 1999; Garrido, 2002). Earlier experiments have shown translocation of HSP27 into the nucleus on stress signals where it can stabilize mRNAs, and limit denaturation of proteins involved in transcription (Carper et al, 1997). Since protein synthesis is impaired by cerebral ischemia, protection may work by enabling accelerated recovery of protein synthesis rate after stress.
We also report that preischemic injections with HSP70 had no effect on lesion size in this model. Most of the literature concentrates on HSP70 as the major constitutive and inducible HSP in the brain (Yenari et al, 1999; Kelly and Yenari, 2002). It is widely accepted that HSP70 protects cells from apoptosis, but it is important to note that many in vitro experiments use toxic substances to induce apoptosis without causing protein denaturation, misfolding, and/or aggregation. This may therefore mask the chaperone functions of HSP70 and emphasize how it can interfere with the apoptotic cascade. HSP70's ability to accelerate cell survival by repairing the protein machinery is more related to necrotic forms of death. Several studies have shown that binding of this chaperone to denatured proteins prevents their aberrant aggregation and restores their functional conformation, thus increasing stabilization and reactivation of protein machinery after stress (Kabakov et al, 2002; Beaucamp et al, 1998).
As far as its role in apoptosis is concerned, there is conflicting evidence that HSP70 regulates upstream events by inhibiting the formation of the apoptosome, or downstream, by binding either the apoptosome complex or the effector caspases. In fact, HSP70 can bind to different components of this complex, such as Apaf-1 (Saleh et al, 2000), cytochrome c (Tsuchiya et al, 2003; Mosser et al, 2000; Creagh et al, 2000; Steel et al, 2004), and dATP. If the apoptosome complex forms, it then binds to procaspase-9 and activates a downstream cascade of effector caspases such as caspase-3. Beere et al (2000) have shown that HSP70 prevents the binding of procaspase-9 to the apoptosome complex on cytochrome c release. Some studies show that HSP70 is unable to prevent cytochrome c release from mitochondria, but it can inhibit activation of caspase 3 or act downstream of the caspase cascade by downregulating cytosolic phospholipase activity (Jaattela et al, 1998; Li et al, 2000). There is also some evidence that this chaperone can bind to the AIF and stop its release from mitochondria (Ravagnan et al, 2001), whereas other groups suggest that inhibition of apoptosis occurs via the JNK/SAPK pathway (Gabai et al, 1997; Meriin et al, 1999).
Interestingly, there is also evidence that increased levels of HSP70 can be detrimental to neurons by binding to survival elements that could otherwise block TNF-induced apoptosis (Ran et al, 2004). This may explain why HSP70 treatment fails to reduce lesion size in our model since viral injections may induce activation of cytokines such as TNF. It is important to note that HSP27 can protect cells from TNF-induced apoptosis, which may account for the differences in protection observed in our study (Mehlen et al, 1997). In vitro evidence of protection with HSP70, however, remains extremely dependent on the type and severity of insult and the susceptibility to different environmental insults of the cell population affected (Amin et al, 1996; Jaattela et al, 1998; Lee et al, 2001a; Ran et al, 2004; Latchman, 2004).
In vivo experiments have also yielded confounding results about the extent of protection of this HSP. Viral delivery of HSP70 in ischemia and epilepsy models in rats has shown no reduction in lesion size, whereas transgenic mice overexpressing HSP70 seem to be protected against ischemia. Viral models, however, provide support for a neuroprotective role of HSP70 at a cellular level because survival or morphology of certain neurons, like CA1 neurons in the hippocampus, is improved even if overall lesion size is not reduced (Yenari et al, 1998; Kelly et al, 2002). It could be argued that, when using viral microinjections as a delivery method, the virus itself may render infected cells more or less responsive to HSP treatment. Previous studies, however, suggest that microinjection of our vector does not cause any obvious damage to hippocampal cells in vivo and other neuronal cell types in vitro (Wagstaff et al, 1999; Kalwy et al, 2003; Zourlidou et al, 2004).
Literature on transgenic animals overexpressing HSP70 should help circumvent the problem of unwanted effects of virally delivered proteins. Some studies show reduction in lesion size both in stroke and epilepsy mouse models (Rajdev et al, 2000; Tsuchiya et al, 2003; van der Weerd et al, 2005), while others show improved neuronal survival with no effect on the lesion size (Plumier et al, 1997; Lee et al, 2001a; Kelly et al, 2001a, b). The variability of protection against ischemia reported in these HSP70 transgenic mice studies has been partially attributed to differential levels of expression. In our case, the levels of endogenous and virally delivered HSP70 expression could have simply not reached a required threshold for lesion size to be reduced. However, histological analysis of our HSP70-injected and MCA-occluded animals showed widespread staining across the brain, suggesting high levels of expression after ischemia. Moreover, in vitro experiments have shown that our viral vector yields the same levels of expression of both HSP27 and HSP70 (Wagstaff et al, 1998), although only the former protein has a potent protective effect in this experimental model.
To the best of our knowledge, our study is the first to show that viral delivery of HSP27 has a significant effect in reducing lesion volume after focal cerebral ischemia in rats. The fact that HSP70 treatment was unable to decrease lesion size may reflect different protective activities of the two chaperones. In this particular model, HSP27 might be more efficient due to its links with cytoskeletal stability and its ability to bind proteins in an ATP-independent fashion in conjunction with its antiapoptotic functions. The high expression levels of both HSPs detected in the cortex suggested that these proteins could either be transported to or secreted in lesion areas. There could also be signaling events leading to cooperation between endogenous and exogenous HSPs that enhanced the brain's natural response to stress. The difficulty in elucidating a precise cascade of events might simply be due to the variability of experimental systems, cell lines and species employed, or the complexity and specificity of the cell repair machinery in response to stress.
In conclusion, these findings provide evidence supporting the role of HSP27 as a major protective protein in the brain, and highlight the need for further investigation of the mechanisms underlying its neuroprotective effect.
Footnotes
Acknowledgements
The authors thank Dr Samantha Alsbury, Liz Ensor, Dr Alexandra Zourlidou, Dr Miratul Muqit and Dr Martin D King for their helpful assistance and discussions.