
Abstract
In the human setting, it has been shown that acute increase in the concentration of ketone bodies by infusion of β-hydroxybutyrate increased the cerebral blood flow (CBF) without affecting the overall cerebral metabolic activity. The mechanism by which this effect of ketone bodies was mediated is not known. Alterations in several parameters may possibly explain the increase in CBF and the resetting of the relation between CBF and cerebral metabolism. To study this phenomenon further, we measured global CBF and global cerebral metabolism with the Kety–Schmidt technique in the wakeful rat before and during infusion of ketone bodies. During acute hyperketonemia (average concentration of β-hydroxybutyrate: 6 mmol/L), global CBF increased 65% from 108 to 178 mL/***100 g min and the cerebral metabolic rates for both oxygen and glucose remained constant. This resetting of the relation between CBF and cerebral metabolism could not be explained by alterations in blood pH or arterial CO2 tension. By measuring cerebral intracellular pH by 31P nuclear magnetic resonance spectroscopy, it could further be concluded that the brain pH was unchanged during acute hyperketonemia. These observations indicate that the mechanism responsible for the increase in CBF is rather a direct effect on the cerebral endothelium than via some metabolic interactions.
Keywords
Introduction
Under normal conditions cerebral metabolic rate, functional activity, and cerebral blood flow (CBF) are tightly coupled. When the functional activity is increased, an increase in the metabolic rate of glucose can be detected (Sokoloff, 1981; Fox et al, 1988). This increase in metabolic rate is accompanied by an increase in blood flow to establish adequate supply of nutrients to maintain energy-demanding synaptic activity (Magistretti et al, 1999). These coupling mechanisms are not readily known in detail, and a great variety of changes are observed during functional activity. Cerebral blood flow changes seem to be related to changes in the rate of the glycolysis and depletion of high-energy phosphate stores (Breier et al, 1993; Dirnagl et al, 1994), and several putative mechanisms have been suggested to account for the coupling, including direct neuronal control (Yang et al, 2000), changes in local pH (Faraci and Heistad, 1998), and astrocyte-mediated control of arterioles (Zonta et al, 2003). Several drugs affect the cerebral circulation; some cause vasoconstriction, others cause vasodilatation, and studies on the effect of drugs on cerebral circulation have supplied important information on the basal mechanisms of regulation of CBF. Therefore, it can be important to discover new effects on the CBF and metabolism by drugs or even by endogenously produced substances, for example, ketone bodies during diabetic ketoacidosis.
In the human setting, Hasselbalch et al (1996) have investigated the effect of intravenous infusion of ketone bodies on global CBF and global carbohydrate metabolism. In that study, Hasselbalch et al observed a 39% increase in global CBF during infusion without changes in cerebral metabolic activity. The CMRO2 was unchanged, but ketone bodies were substituting glucose as fuel for the oxidative metabolism, in line with previous findings in long- and short-term starvation (Owen et al, 1967; Hasselbalch et al, 1994). The changes in CBF could not be explained by alterations in arterial CO2 tension nor blood pH, and it was concluded that the mechanism of this ketone-body-induced resetting of the coupling between CBF and metabolism was unknown. This unexplored mechanism may act as a good tool for further investigations on the regulation of CBF.
The study by Hasselbalch et al (1996) did not fully investigate all the possible explanations of the ketone-body-induced increase in CBF, for example, the study only measured the changes in blood pH and not brain pH, which could possibly influence the CBF. A main purpose of the present study was therefore to establish an animal model that allows measurement of CBF and metabolism before and during infusion of ketone bodies. A second major purpose of our study was to evaluate whether the known regulators of CBF, for example, arterial O2 and CO2 tension and pH in blood and brain, were implicated in the CBF regulation during hyperketonemia.
Materials and methods
Animals and Surgical Procedures
Cerebral blood flow, cerebral metabolic rate and arteriovenous difference studies: Experiments were performed on male Sprague–Dawley rats (Møllegarden, Lille Skensved, Denmark) weighing 400 to 450 g. The animals were fed with standard laboratory chow and water ad libitum. The dark/light cycle was set for equal periods of light (1800 to 0600 h) and darkness (0600 to 1800 h). All experiments were performed in accordance with the National Institutes of Health Guide for and Use of Laboratory Animals and were approved by The Danish Animal Experiments Expectorate.
Surgical procedures were performed under anesthesia with halothane, 4% for induction and 1.0% to 1.5% for maintenance in 70% nitrous oxide (N2O) and 30% O2. Polyethylene catheters (ID (inner diameter) 0.58 mm, OD (outer diameter) 0.96 mm) (Holm & Halby, Denmark) filled with heparinized saline (30 IU/mL) were inserted in the right and left femoral arteries (catheter length, 20 cm) and vein (catheter 30 cm). The surgical incisions were filled with lidocaine/noradrenalin (10 mg/mL, injection substance) and closed with sutures. The closed incisions were infiltrated with lidocaine/prilocaine (EMLA creme, Astra, Södertalje, Sweden). The head of the rat was fixed in a stereotactic frame and a midline scalp incision was made to expose the lambdoid suture. A 1 mm burr hole was drilled 1 mm posterior to the lambdoid suture in the midline. A screw was placed in the borehole, leaving the dura intact. The dura was punctuated through the screw and a polyethylene catheter identical to the catheters inserted in the femoral vessels was inserted into the confluens sinuum (length, 20 cm). The catheter was secured to the screw with cyanoacrylate (Scoth-Weld, Germany). The above-described procedures were all performed without touching the surface of the brain. A plaster cast was applied around the lower torso for restraint, and at least 2.5 h of recovery was allowed before initiation of experimental procedures. In the recovery period, the rat was kept in a shelter specially designed to minimize external stimulation. The shelter was a closed triangular box with apertures permitting passage of blood sample catheters. In the shelter, the rat was monitored by closed circuit video surveillance, and a small ventilator assured air change. Rectal temperature was monitored with a rectal probe and mean arterial blood pressure (MABP) was continuously measured with a pressure transducer connected to one of the arterial catheters.
31P nuclear magnetic resonance (31P NMR) spectroscopy studies: Animals as described above were used for this series of experiments. However, in this part of the study, animals were housed in alternating light and dark cycle (light (0600 to 1800 h) and dark (1800 to 0600 h)). In this part of the study, animals were anesthetized by a subcutaneous injection of an anesthesia mix (fentanyl, fulanisone, midazolam, and suxamethonium). Arterial and venous femoral vessels were catheterized with polyethylene catheters (ID 0.58 mm, OD 0.96 mm) (Holm & Halby, Denmark) filled with heparinized saline (30 IU/mL); long catheters were used, allowing to place the animal in the center of the catheter. The animals were intubated and the animals were fixed in a stereotactic device and placed in the magnet. The animals were mechanically ventilated using an animal ventilator with a tidal volume at approximately 3 mL, at a rate of 57 breaths/min. As the effect of the anesthesia mix faded out, the inspired gases were shifted from air to 1% halothane in N2O/oxygen 60/40 (vohvol).
Experimental Protocols
The effect of infusion of β-hydroxybutyrate on global CBF and global cerebral metabolic rates (CMRs) was determined during three different conditions: (1) in nonanesthetized animals, (2) in animals anesthetized with halothane, and (3) in animals anesthetized with pentobarbital.
The effect of infusion of sodium bicarbonate on global CBF and global CMRs were determined in nonanesthetized animals.
Consecutive measurements of cerebral arteriovenous differences for oxygen, glucose and lactate were performed in a series of nonanesthetized animals before and during infusion of β-hydroxybutyrate to obtain a more exact determination of the oxygen/glucose index (OGI) before and during infusion.
In another line of experiments, average intracellular brain pH was determined by 31P NMR before and during infusion of β-hydroxybutyrate and before and during infusion of sodium bicarbonate.
Determination of Global Cerebral Blood Flow
Global CBF was measured with a modification of the Kety–Schmidt technique (Kety and Schmidt, 1948) in the desaturation mode using 133Xenon as the flow tracer. A detailed description is given in a previous paper (Linde et al, 1999); in brief, the methodological approach can be described as follows: The brain was saturated by intravenous infusion of 133Xenon dissolved in saline at a constant rate of approximately 2.5 MBq/min. After 30 mins of constant 133Xenon infusion, three paired blood samples were obtained from the arterial and cerebral venous catheter. Infusion with 133Xenon was terminated, and during the first 9 mins of tracer desaturation arterial and cerebral venous blood was sampled at a constant rate of 0.50 mL/min, using a mechanical double-action withdrawal pump (Harvard Apparatus, model 55-2226, Harvard apparatus, South Natich, MA, USA). After completion of continuous blood sampling, paired blood samples were obtained from the two catheter systems. The concentration of 133Xenon in the blood samples was determined in a gamma counter (Packard Auto-Gamma Cobra II, Meriden, CT, USA), and CBF was calculated using a slightly modified version of the Kety–Schmidt equation (Kety and Schmidt, 1948; Lassen and Klee, 1965):
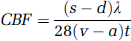
in which s is the tracer concentration in cerebral venous blood at the end of 133Xenon infusion, d is the tracer concentration in cerebral venous blood at the end of the period of the measurement, λ is the tissue/blood partition coefficient for whole rat brain of 1.015 (Gjedde et al, 1975; Mallett and Veall, 1965), v the tracer concentration in cerebral venous blood obtained during the period of measurement (integrated sample), a the tracer concentration in arterial blood obtained during the period of measurement (integrated sample), and t the duration of the measurement. The tracer concentration in cerebral venous blood at the end of 133Xenon infusion was calculated as the average of the tracer concentration in the three samples obtained before the initiation of the period of measurement. To perform a second measurement of CBF, the saturation of the brain with 133Xenon infusion was re-initiated and the procedures described above were repeated. After the second CBF measurement, the rat was killed by intravenous injection of 100 rng pentobarbital.
Consecutive Measurements or Cerebral Arteriovenous Differences
Determination of cerebral arteriovenous differences for oxygen [(a – v)O2], glucose [(a–v)glc] and lactate [(a–v)lac] were performed on blood obtained simultaneously from the arterial catheter and from the cerebral venous catheter. Before blood sampling, a dead-space volume of 0.150 mL blood was drawn. Immediately after this, 0.300 mL blood was drawn into ice-cold gas-tight glass Microliter Syringes (Hamilton Bonaduz AG, Bonaduz, Switzerland) prepared with heparin and NaF The rate of sampling was the same from both the arterial and the venous catheter system: 0.5 mL/min. The blood samples were stored on ice under strict anaerobic conditions and after sampling the blood loss was replaced by intravenous infusion of blood obtained from a donor rat.
31P Nuclear Magnetic Resonance Spectroscopy
The measurement of pH by 31P NMR spectroscopy is a standard technique (Petroff et al, 1985) that relies on the pH dependence of the chemical shift of the Pi signal. The chemical shift of the Pi frequency varies with intracellular pH, and is the basis for measurement of pH by 31P NMR. Phosphocreatin (Per) is insensitive to pH changes in the physiological range, and provides therefore a reference for measurement of the Pi chemical shift. 31P nuclear magnetic resonance experiments were performed on a SISCO 4.7 T unit (Varian, Palo Alto, USA), equipped with a 10 G/cm insert gradient coil. The rats were positioned in a double-tuned 31P/1H head coil with an integrated stereotactic device. The animals' brains were centered in the magnet and the coil tuned to 1H. The global shim was optimized, and a multi-slice transaxial spin-echo sequence was used to obtain scout images, to position the voxel of interest (VOI) for localized 31P NMR. A 560 μL voxel (7.5 × 7.5 × 10.0 mm3) was selected, and the shim in this volume of the brain was optimized using a STEAM localized spectroscopy sequence (Frahm et al, 1987). After tuning the coil to 31P, the power was optimized using an array of hard pulses, and by focusing on the intensity of the Pcr signal. The XBR sequence (Topp et al, 1995) was used to obtain 31P spectra. A reference spectrum was acquired (number of acquisitions = 64) to calibrate the magnetic resonance (MR) frequency. The carrier frequency was set between the Pcr and Pi frequency, thus minimizing the chemical shift disturbances at both frequencies. Measurements used to calculate pH were obtained with the following parameters: TR = 5 secs, SW = 5000 Hz, and number of acquisitions = 512. Fourier transformations of the FIDs were performed after exponential weighting (3 to 5 Hz). The frequencies of Pcr and Pi were determined using the scanner software.
8-Hydroxybutyrate Infusion
Sodium (DL)-β-hydroxybutyrate (Sigma Chemical Company Ltd, Poole, UK) was dissolved in sterile water, filtered, and tested for pyrogens. The concentration of the solution was 166.5 mg β-hydroxybutyrate/kg body weight mL, and the pH was secured to be approximately 7.1. β-Hydroxybutyrate was infused in a femoral vein at a rate of 0.225 mL/min, resulting in an infusion of 37.5 mg β-hydroxybutyrate/kg body weight min. In a pilot study in two rats, the concentration of β-hydroxybutyrate in the arterial blood and blood pH was measured every 15 min during β-hydroxybutyrate infusion to determine the time for reaching a sufficient concentration of β-hydroxybutyrate in the blood and the effect on blood pH. Based on data from this line of investigation, measurements were initiated after 45 mins of infusion.
Bicarbonate Infusion
A commercial preparation of sodium bicarbonate was used (Natriumbikarbonate ‘DAK’, Nycomed DAK, Denmark). The bicarbonate was infused at a concentration of 56 mg/kg body weight mL in a femoral vein at a rate of 0.225 mL/min, resulting in an infusion of 12.5 mg/kg body weight-min. The infusion regime was tested by changing the bicarbonate concentration to reproduce a similar increase in blood pH introduced by the β-hydroxybutyrate infusion.
Biochemical Substrate Analysis
All biochemical analysis procedures were performed on arterial and venous blood from the two samples collected during the desaturation period. The blood samples were stored on ice under strict anaerobic conditions until analyzed. Within 20 mins, the arterial and venous blood were analyzed in duplicate for oxygen saturation and hemoglobin concentration with an OSM3 Hemoximeter (Radiometer, Copenhagen, Denmark). Blood for determination of glucose and lactate concentrations in whole blood was transferred to vials containing dried fluoride-EDTA. These vials were stored on ice and analyzed within 20 mins in triplicate on a YSI 2700 Select Biochemistry analyzer (Yellow Springs Instruments Co., Inc., Yellow Springs, OH, USA). β-Hydroxybutyrate concentrations were determined enzymatically (Williamson et al, 1962). Arterial blood was anaerobically collected in glass capillaries, and PaCO2, PaO2, and pH were analyzed on an ABL30 acid–base analyzer (Radiometer, Copenhagen, Denmark).
Calculations
Cerebral (a — v)Q2 was calculated using the hemoglobin concentration and oxygen saturation of hemoglobin in arterial and cerebral venous blood. Cerebral (a –v)glc, cerebral (a – v)lac, and cerebral (a –v)β- hydroxybutyrate were calculated from glucose and lactate β-hydroxybutyrate concentrations in arterial and venous whole blood. Global CMRs were calculated for each substrate according to the Fick principle, for example, CMRO2 = CBP × (a – v)O2. The proportion of glucose uptake being oxidized, OGI, was calculated as OGI = 100 × (a – v)O2/6 × (a –v)glc (%) (Cohen et al, 1964). The proportion of glucose uptake appearing as lactate in cerebral venous blood was calculated as the lactate/glucose index (LGI): LGI = −100 × (a – v)lac/2 × (a – v)glc (%) (Cohen et al, 1964).
Statistics
Paired and unpaired two-tailed Student's t-test was used in the statistical evaluation of the data.
Results
Cerebral Blood Flow and Metabolism in the Awake Animal
Results obtained during control conditions in the awake animal using the Kety–Schmidt technique were all within normal range (Table 1). Global CBF was 108 mL/100 g min (s.d.: 15 mL/100 g min, respectively, and the OGI was 109% (s.d.: 23%)). In the series of consecutive measurements of cerebral arteriovenous differences for glucose, oxygen and lactate in the awake animal were very similar to the result obtained by the Kety–Schrnidt technique, described above.
Physiological variables, global CBF, global CMR and arteriovenous differences for oxygen, glucose and lactate before (control) and during infusion of β-hydroxybutyrate (37.5 mg β-HB/kg body weight min) (acute hyperketonemia) in awake animals
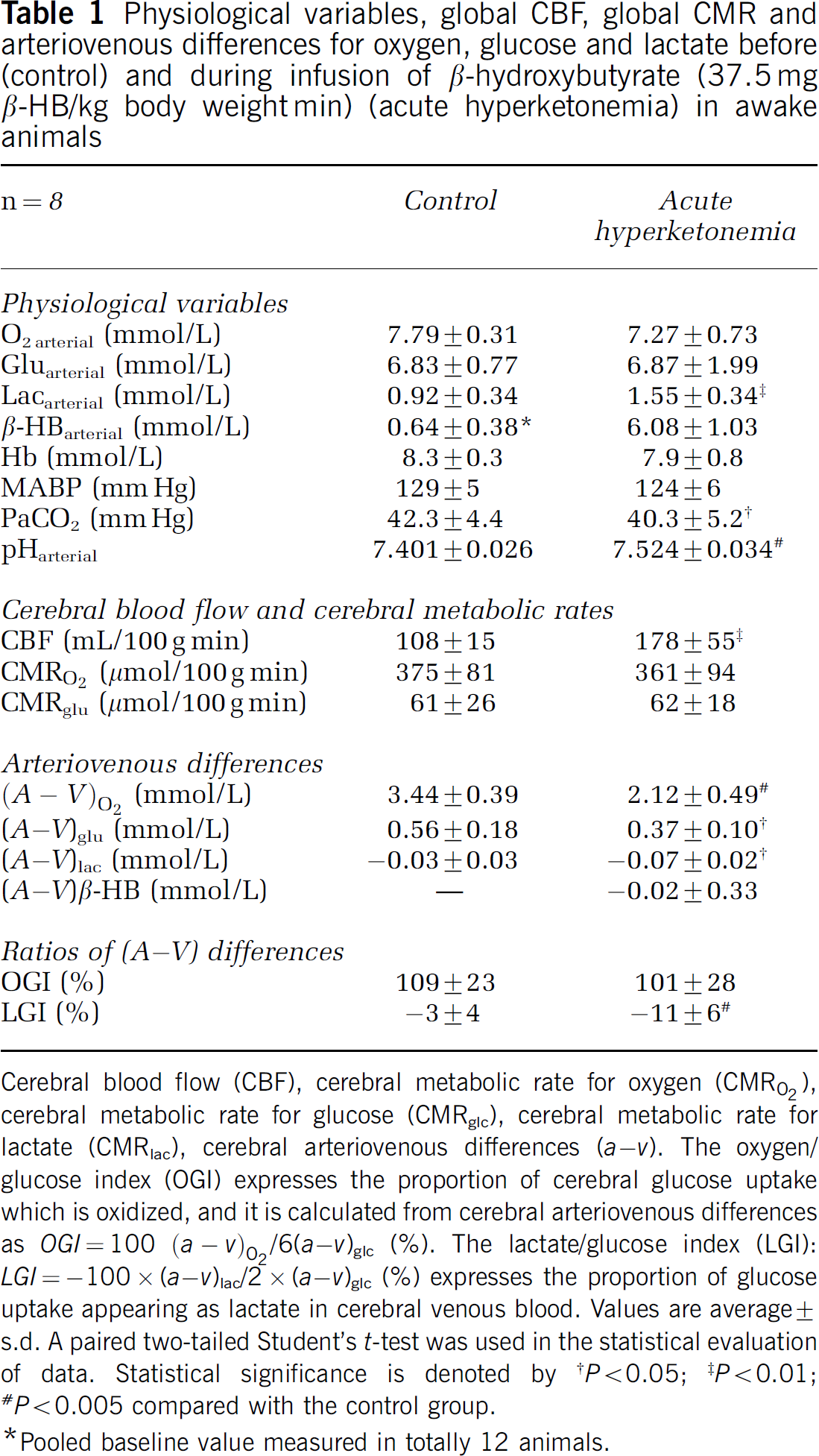
Cerebral blood flow (CBF), cerebral metabolic rate for oxygen (CMRO2), cerebral metabolic rate for glucose (CMRglc), cerebral metabolic rate for lactate (CMRlac), cerebral arteriovenous differences (a – v). The oxygen/glucose index (OGI) expresses the proportion of cerebral glucose uptake which is oxidized, and it is calculated from cerebral arteriovenous differences as OGI =100 (a – v)O2/6(a – v)glc (%). The lactate/glucose index (LGI): LGI = −100 × (a –v)lac/2 × (a – v)glc (%) expresses the proportion of glucose uptake appearing as lactate in cerebral venous blood. Values are average ± s.d. A paired two-tailed Student's t-test was used in the statistical evaluation of data. Statistical significance is denoted by
P < 0.05
P < 0.01
P < 0.005 compared with the control group.
Pooled baseline value measured in totally 12 animals.
β-Hydroxybutyrate Concentration and pH during Infusion
Results from the preliminary experiments are shown in Figure 1. The infusion of 37.5 mg β-hydroxybutyrate/kg body weight min raised the concentration of β-hydroxybutyrate to approximately 6 mmol/L during the first 45 mins of infusion. In the MR studies where the setup for technical reasons (MR computability) was somewhat different, the concentration reached somewhat higher values. After the 45 mins of infusion, the concentration only very slightly showed a tendency to increase further. All experiments were performed in the interval between 45 and 90 mins of infusion. The figure also shows that the pH in the blood increased to approximately 7.55 during the infusion. The same infusion regime was used in the rest of the study series.
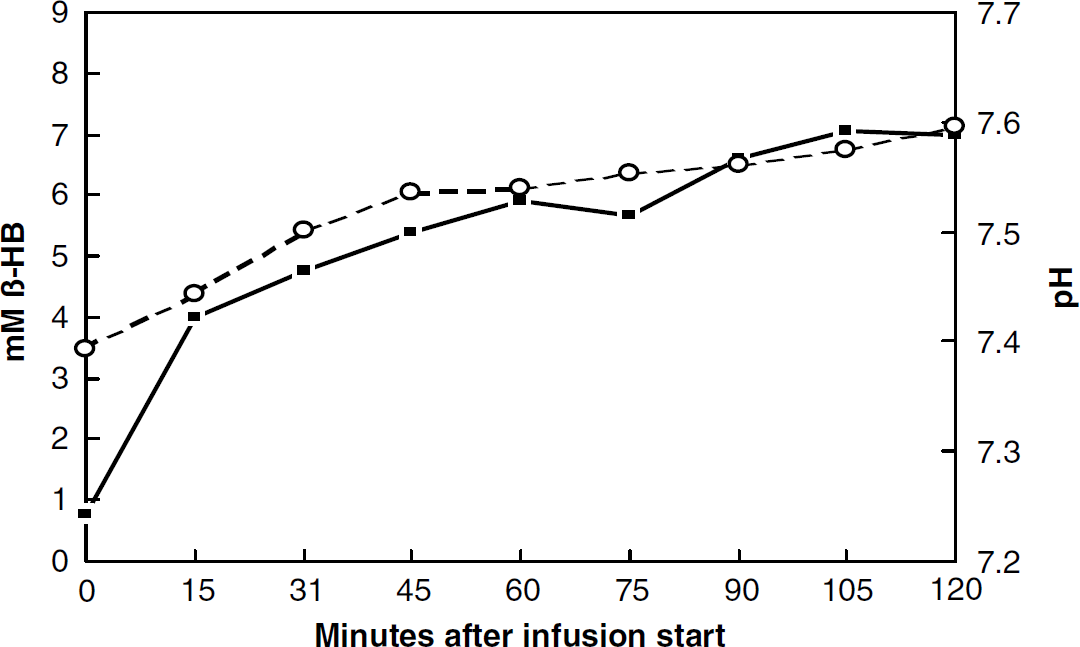
Relationship between blood pH and blood β-hydroxybutyrate concentration during infusion of β-hydroxybutyrate (37.5 mg β-hydroxybutyrate/kg body weightmin) as a function of time in a typical animal. The infusion started at time zero. The straight line represents the β-hydroxybutyrate concentration and the dotted line represents the pH.
Cerebral Blood Flow and Metabolism in the Awake Animal during Hyperketonemia
The arterial blood concentration of β-hydroxybutyrate increased in these animals to 6.08 mmol/L (s.d.: 1.03 mmol/L) and arterial lactate increased to 1.55 mmol/L (s.d.: 0.34 mmol/L) (P = 0.001), while the arterial glucose and arterial oxygen concentrations remained constant. Global CBF increased by 65% to 178 mL/100 g min (s.d.: 55 mL/100 g min) during acute hyperketonemia (P = 0.008). The PaCO2 decreased slightly to 40.3 mm Hg (P < 0.05), and blood pH increased slightly to 7.524 (P < 0.001). Global glucose and oxygen metabolism remained constant ((61 versus 62 μmiol/100 g min), P = 0.941) and (375 versus 361 μmol/100 g min), P = 0.673), respectively. The arteriovenous differences for glucose and oxygen decreased significantly approximately 36% due to the CBF increase and the unchanged CMRs, (a – v) = CMR × CBF−1. The OGI was unaffected and remained constant, 109% versus 101% (P = 0.582) (Table 1).
Cerebral Blood Flow and Metabolism in the Awake Animal during Bicarbonate Infusion
The pH of the blood increased during the bicarbonate infusion from 7.415 to 7.566 (P < 0.001). Cerebral blood flow, CMRO2, CMRglc, and the OGI remained constant. The arterial lactate increased and a slight reduction in (a – v)O2 was observed (Table 2).
Physiological variables, global CBF, global CMR and arteriovenous differences for oxygen, glucose, and lactate before (control) and during infusion with sodium bicarbonate (12.5 mg/kg body weight-min) (bicarbonate infusion)
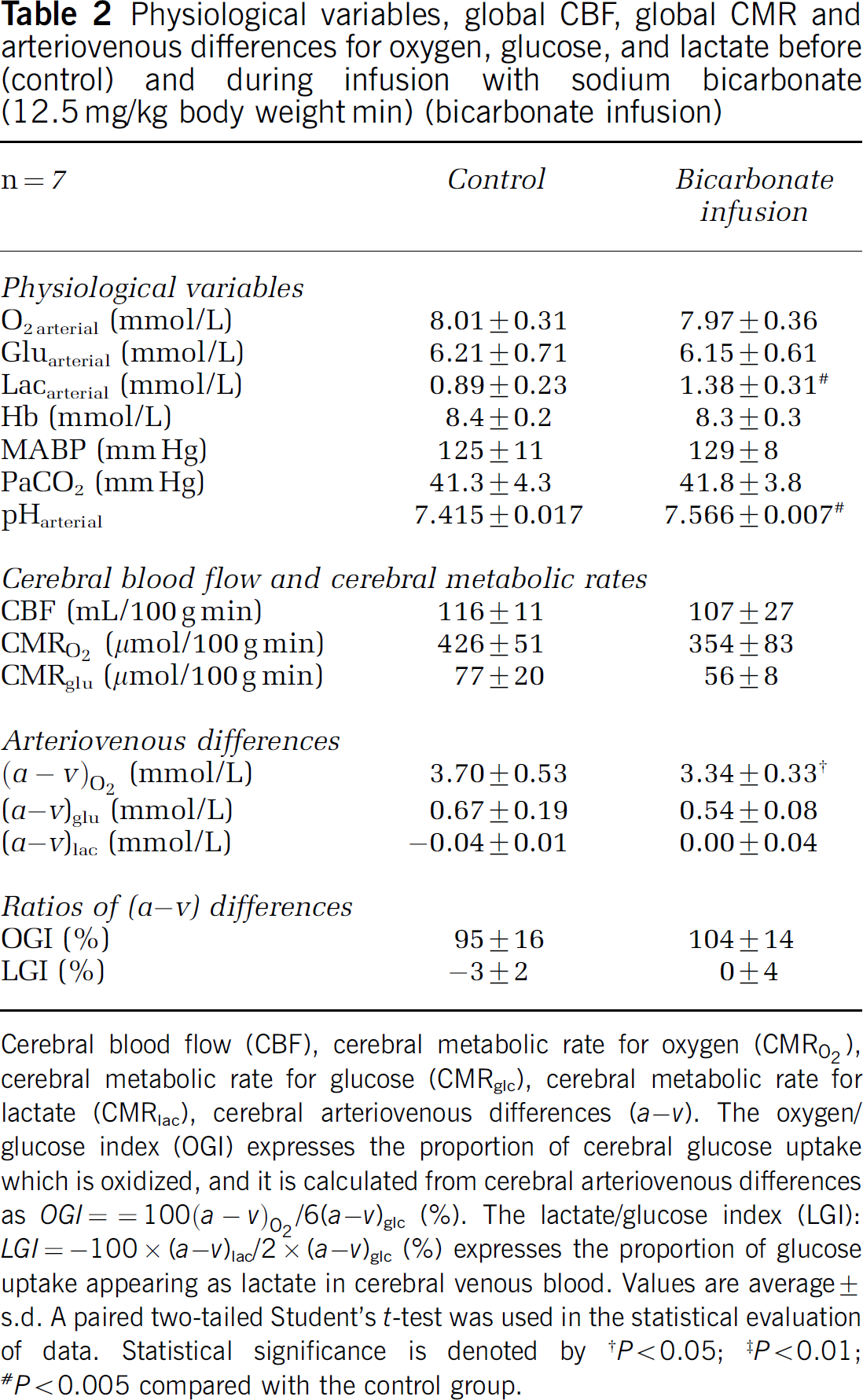
Cerebral blood flow (CBF), cerebral metabolic rate for oxygen (CMRO2), cerebral metabolic rate for glucose (CMRglc), cerebral metabolic rate for lactate (CMRlac), cerebral arteriovenous differences (a – v). The oxygen/glucose index (OGI) expresses the proportion of cerebral glucose uptake which is oxidized, and it is calculated from cerebral arteriovenous differences as OGI = 100(a – v)02/6(a –v)glc (%). The lactate/glucose index (LGI): LGI = −100 × (a –v)lac/2 × (a – v)glc (%) expresses the proportion of glucose uptake appearing as lactate in cerebral venous blood. Values are average ± s.d. A paired two-tailed Student's t-test was used in the statistical evaluation of data. Statistical significance is denoted by
P < 0.05
‡ P < 0.01
P < 0.005 compared with the control group.
Cerebral Blood Flow and Metabolism in the Anesthetized Animal during Hyperketonemia
The overall picture of the changes in the anesthetized animals is equal to the results in the awake animals (Tables 3 and 4). In the halothane-anesthetized animals, the global CBF increased by 213% to 360 mL/100 g min (s.d.: 133 mL/100 g min) during acute hyperketonemia (P < 0.05). In the pentobarbital anesthetized animals, the global CBF increased by 54% to 103 mL/100 g min (s.d.: 27 mL/100 g min) during acute hyperketonemia. The cerebral metabolism and the OGI were unaffected and remained constant, while the arteriovenous differences for glucose and oxygen decreased significantly due to the CBF increase and the unchanged CMRs, (a – v) = CMR × CBF−1.
Physiological variables, global CBF, global CMR and arteriovenous differences for oxygen, glucose and lactate before (control) and during infusion of β-hydroxybutyrate (37.5 mg β-hydroxybutyrate/kg body weight min) (acute hyperketonemia) in the halothane-anesthetized animal
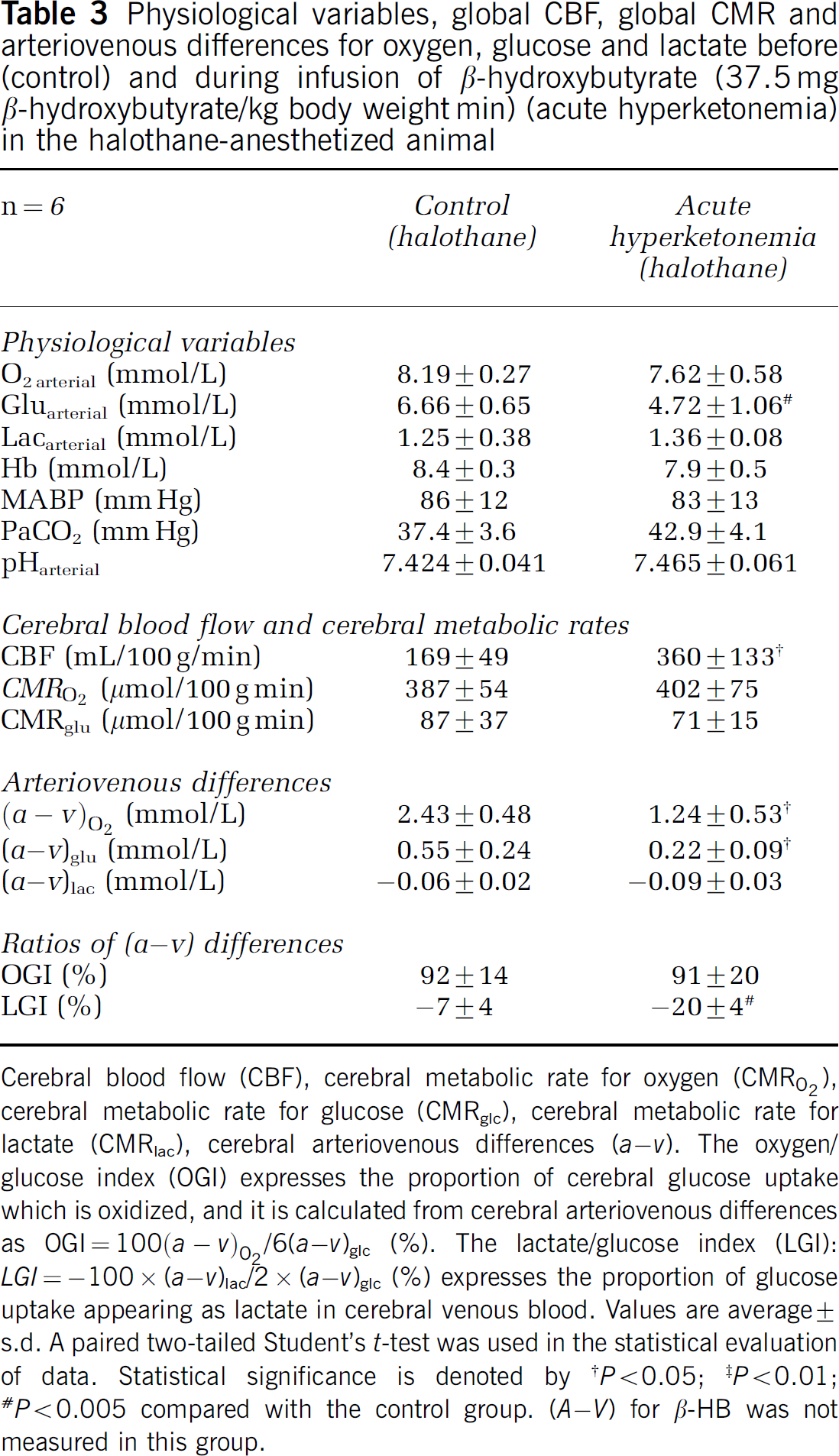
Cerebral blood flow (CBF), cerebral metabolic rate for oxygen (CMRO2), cerebral metabolic rate for glucose (CMRglc), cerebral metabolic rate for lactate (CMRlac), cerebral arteriovenous differences (a – v). The oxygen/glucose index (OGI) expresses the proportion of cerebral glucose uptake which is oxidized, and it is calculated from cerebral arteriovenous differences as OGI = 100(a – v)O2/6(a – v)glc (%). The lactate/glucose index (LGI): LGI = −100 × (a – v)lac/2 × (a – v)glc (%) expresses the proportion of glucose uptake appearing as lactate in cerebral venous blood. Values are average ± s.d. A paired two-tailed Student's t-test was used in the statistical evaluation of data. Statistical significance is denoted by
P < 0.05
‡ P < 0.01
P < 0.005 compared with the control group. (A – V) for β-HB was not measured in this group.
Physiological variables, global CBF, global cerebral metabolic rates (CMR) and arteriovenous differences for oxygen, glucose and lactate before (control) and during infusion of β-hydroxybutyrate (37.5 mg β-HB/kg body weight min) (acute hyperketonemia) in the pentobarbital anesthetized animal
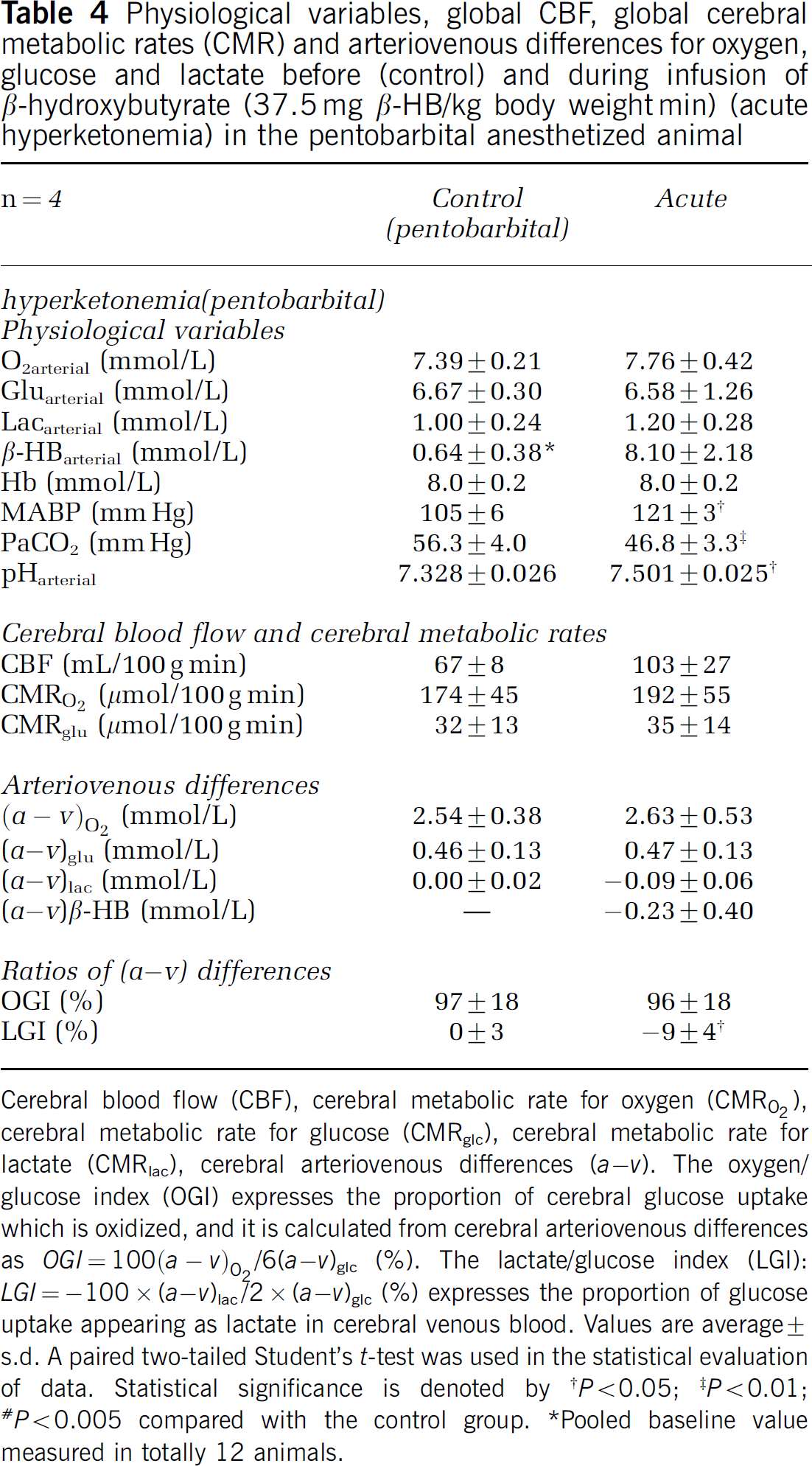
Cerebral blood flow (CBF), cerebral metabolic rate for oxygen (CMRO2), cerebral metabolic rate for glucose (CMRglc), cerebral metabolic rate for lactate (CMRlac), cerebral arteriovenous differences (a – v). The oxygen/glucose index (OGI) expresses the proportion of cerebral glucose uptake which is oxidized, and it is calculated from cerebral arteriovenous differences as OGI = 100(a – v)O2/6(a – v)glc (%). The lactate/glucose index (LGI): LGI = −100 × (a –v)lac/2 × (a – v)glc (%) expresses the proportion of glucose uptake appearing as lactate in cerebral venous blood. Values are average ± s.d. A paired two-tailed Student's t-test was used in the statistical evaluation of data. Statistical significance is denoted by
P < 0.05
P < 0.01
# P < 0.005 compared with the control group.
Pooled baseline value measured in totally 12 animals.
31P Nuclear Magnetic Resonance
The average intracellular pH of the brain did not change after β-hydroxybutyrate infusion, 7.08 (s.d.: 0.09) and 7.14 (s.d.: 0.04), before and after infusion, respectively. A similar result was obtained after bicarbonate infusion, 7.09 (s.d.: 0.04) and 7.11 (s.d.: 0.04), before and after infusion respectively. Both infusion of β-hydroxybutyrate and infusion of bicarbonate increased the blood pH, as observed in the other series (Table 5).
Average intracellular pH of brain, blood pH, PaCO2 and arterial concentration of β-hydroxybutyrate before (control) and during infusion of β-hydroxybutyrate (37.5 mg β-hydroxybutyrate/kg body weight min) or bicarbonate (12.5 mg/kg body weight min) in the halothane-anesthetized animal
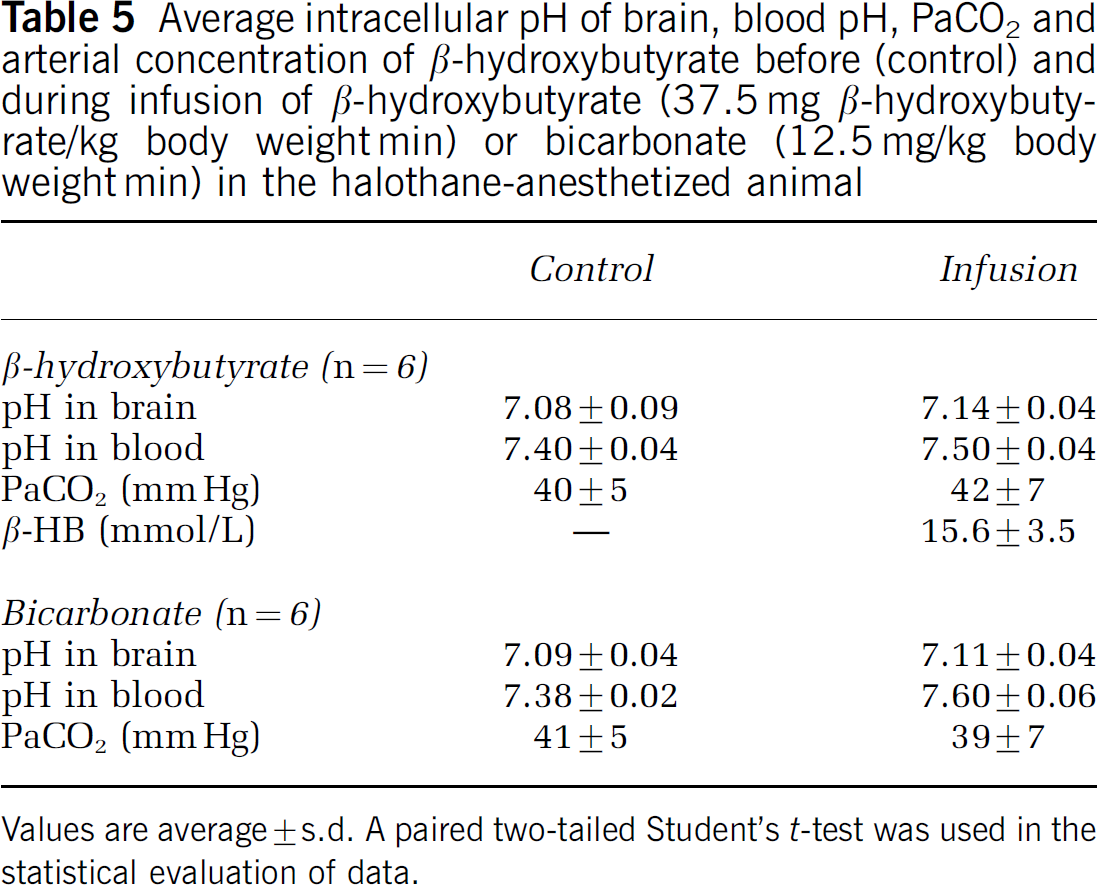
Values are average ± s.d. A paired two-tailed Student's t-test was used in the statistical evaluation of data.
Discussion
Technical Considerations
The applied Kety–Schmidt technique for determination of global CBF and global CMRs are validated for repeated determinations of global values for CBF, CMRO2, and CMRglc in the awake rat in a previous study (Linde et al, 1999). The applied method provides accurate and reproducible values and provides simultaneous determinations of global CBF and global CMRs (Linde et al, 1999).
Figure 1 shows the changes in the arterial concentration of β-hydroxybutyrate and in blood pH from infusion start to 120 mins. After 45 mins, the β-hydroxybutyrate concentration remains relatively constant and has reached a steady-state level at approximately 6 mmol/L. All blood sampling and determinations are started 45 mins after onset of the β-hydroxybutyrate infusion to ensure a high and constant level of β-hydroxybutyrate in the blood, and the infusion of β-hydroxybutyrate was performed in the same way in all the study series.
The β-hydroxybutyrate concentration in arterial blood of 6 mmol/L obtained during ketone body infusion was higher than values obtained in diabetic rats (Ikeda et al, 1991; Ferreira et al, 2001; Okuda et al, 2002), or in humans during short-term fasting (Pan et al, 2000; Hasselbalch et al, 1994), whereas similar values can be seen in long-term fasting (Owen et al, 1967) and in severe diabetic ketoacidosis (Taboulet et al, 2004). However, the aim of the present study was to study the possible mechanisms behind the CBF-hyperketonemia coupling, and therefore we aimed for a high blood ketone body concentration to ensure a sufficient CBF response. One may speculate that the still moderate increase in blood osmolality after β-hydroxybutyrate per se may influence CBF. However, this hypothesis does not find support in the literature where other forms of increased osmolality have been used (Arvidsson et al, 1981; Burgess et al, 1985).
Measurement of brain pH by the 31P NMR methodology is performed during halothane/N2O anesthesia, and therefore we determined CBF and CMRs before and during infusion of β-hydroxybutyrate in a series of halothane-anesthetized rats. This series showed that the increase in CBF during infusion of β-hydroxybutyrate is independent of halothane anesthesia. The CBF increased from 169 to 360 mL/100 g min and the CMRO2 and CMRglc remained constant, excluding the effects of anesthesia on these results.
Alterations in Cerebral Blood Flow
Global CBF increased by 65% from a control value of 108 to 178 mL/100 g min during infusion of β-hydroxybutyrate. The control value is in good accordance with results obtained in a previous study (Linde et al, 1999), measuring global CBF in the awake rat. The increase in global CBF is similar to results obtained by Hasselbalch et al (1996) in the human setting, showing a 39% increase in global CBF during infusion of β-hydroxybutyrate. In the anesthetized animals, we observed increases of CBF at 54% and 213% for pentobarbital and halothane, respectively. We assume that it is the same mechanism that is in function in both the awake and anaesthetized animals. The higher increase in the present study can be explained by infusate concentration differences and species differences.
The observed increase in global CBF was not accompanied by any changes in the classic modulators of CBF. The CO2 tension in the arterial blood remained almost constant, the PaCO2 decreased 2 mm Hg from 42.3 mm Hg (s.d.: 4.4 mm Hg) to 40.3 mm Hg (s.d.: 5.2 mm Hg), which, if anything, should lead to a decrease in CBF. The O2 content in the arterial blood remained constant and, due to the CBF increase, the oxygen availability increased; so the oxygen content could not explain the increase in CBF.
The CBF increase was not accompanied by increases in the CMR for oxygen and glucose that remained unchanged before and during infusion of β-hydroxybutyrate. The CBF increase can therefore not be explained by a general increase in neuronal activity, as this is accompanied by an increased cerebral glucose consumption, and further the rat did not show any behavioral signs of activation or epileptic seizures during the infusion of β-hydroxybutyrate.
A very important factor for regulating the CBF is the H+ concentration. In this study, special care was taken to check if the β-hydroxybutyrate infusion changed the average intracellular pH of the brain. By the 31P NMR experiments, we observed a minor nonsignificant alkalizing effect of the brain during β-hydroxybutyrate infusion, probably because areas with no blood–brain barrier were affected by the significant increase in blood pH. The plasma volume in the brain is only approximately 0.5% and cannot explain the measured pH increase of the brain. Very similar results are obtained in the control group during bicarbonate infusion. The result obtained for the baseline pH of the brain at 7.08 is in good accordance with results obtained by other groups by 31P NMR methodology (Petroff et al, 1985).
The fact that the brain is not acidified during β-hydroxybutyrate infusion excludes the possibility that the CBF increase is caused by changes in brain pH. The blood pH is changed during infusion of β-hydroxybutyrate because the β-hydroxybutyrate is infused as a solution of the salt of the acid, thereby introducing metabolic alkalosis. Changing the blood pH by bicarbonate infusion, in the control group, did not affect CBF and metabolism.
Both the β-hydroxybutyrate infusion and the bicarbonate infusion resulted in an increased arterial concentration of lactate. This effect has previously been observed and has been explained by a lactate production by the muscles and other tissues due to stimulation of the glycolysis (Delcher and Shipp, 1966) when the blood is alkalized. Different groups have shown that β-hydroxybutyrate increases the production of lactate by the brain due to an inhibition of the pyruvate dehydrogenase enzyme. This might also be true in our study while we measured a significant decrease (P < 0.005) of CMRlac from −3.24 μmol/100 g min (s.d.: 3.54 μmol/100 g min) to −12.83 μmol/100 g min (s.d.: 7.27 μmol/100 g min) during infusion of β-hydroxybutyrate in the awake animal. This production of lactate by the brain is not big enough to be responsible for the increase of the concentration of lactate in the arterial blood and this significant increase must have been caused by the production of lactate by muscles. It has been shown that an increase in arterial lactate concentration does not affect the overall CBF and metabolism (Mintun et al, 2004), and the increase in arterial lactate concentration seems not to explain the CBF increase.
The infusion of β-hydroxybutyrate in the form of the two isomers (D and L forms) may induce a different physiological response as compared with fasting-induced ketosis in which the primary ketone body is D-β-hydroxybutyrate. As the pharmacological effect of the L-form is unknown, except for a possible NMDA-blocking effect (Donevan et al, 2003), we cannot exclude that part of the discrepancy between CBF changes during ketone infusion and during fasting could be ascribed to this isoform.
In conclusion, we observed a 65% increase in global CBF during acute, β-hydroxybutyrate infusion in the awake fed rat without any changes in the classic parameters of CBF control, that is, arterial tension of CO2, arterial content of O2, neuronal activity and intracellular pH of the brain.
Alterations in Cerebral Metabolism
The CMR for oxygen, CMRO2, remained constant at a level of 375 μmol/100 g min versus 361 μmol/100 g min before and during infusion of β-hydroxybutyrate. The CMR for glucose, CMRglc, remained constant at a level of 61 μmol/100 g min versus 62 μmol/100 g min before and during infusion of β-hydroxybutyrate. The β-hydroxybutyrate infusion therefore caused a resetting between the CBF and the cerebral metabolism of oxygen and glucose. We observed no decrease in glucose metabolism and no substitution of glucose by ketone bodies for the oxidative metabolism, as has been shown in humans both during short-term fasting (Hasselbalch et al, 1994) as well as during ketone body infusion (Hasselbalch et al, 1996; Pan et al, 2002), indicating a possible species difference. The arteriovenous differences for β-hydroxybutyrate during infusion of β-hydroxybutyrate were negligible — 0.02 mmol/L (s.d.: 0.33 mmol/L), and not different from zero, indicating that β-hydroxybutyrate is not being used by the brain of the adult-fed rat when the β-hydroxybutyrate concentration is acutely raised to approximately 6 mmol/L. However, the variability in the measurements was very high, and the uptake of β-hydroxybutyrate might have been more than 120 μmol/100 g min (and thus more than twice the glucose uptake) without detecting it at a 5% significance level. Although a type II error thus cannot be excluded, OGI, remained constant during infusion, indicating that glucose was the only substrate for the oxidative metabolism of the brain during β-hydroxybutyrate infusion. A substitution of glucose by ketone bodies or other substances would have increased the OGI significantly. A study in rats by Corddry et al (1982) and by Hawkins et al (1986) is supporting our results by showing no effect on regional glucose consumption during ketone body infusion. Thus, acknowledging that the variability of arteriovenous differences for β-hydroxybutyrate might have obscured an increase in the uptake of ketone bodies, the constant OGI does not suggest that this occurred. There was, however, a small, but significant increase in the efflux of lactate (LGI changed from −3 to −11%, Table 3). Thus, we cannot exclude a small decrease in the oxidation of glucose (corresponding to a small increase in the production of lactate) and a similar increase in the oxidation of ketone bodies that was not detected in the arteriovenous differences for β-hydroxybutyrate, because of variability in the data set. In line with this, in fasting-induced ketosis in humans, Pan et al (2000) suggested that ketone body oxidation displaced lactate oxidation without altering glucose uptake. Other studies in sheep and rats are in contrast to our findings showing that uptake of ketone bodies by the brain is only limited by the concentration of ketone bodies in the blood (Kammula, 1976; Hawkins et al, 1971). It should be noted, however, that the present observations only concern acute changes in ketone levels, and adaptational processes that may occur under prolonged hyperketonemia are not likely to take place within the time frame of the present study. Further, although we tried to exclude possible confounders, the infusion itself might have prevented a shift towards ketone body utilization that would otherwise have occurred under more physiological circumstances.
Possible Mechanisms for the CBF Increase
Various explanations of the mechanism by which the acute hyperketonemia increases CBF can be considered. A few of these possibilities are presented here.
Nitric oxide (NO) is constitutively produced in vascular endothelial cells and in some scattered neurons by either endothelial NO synthetase (eNOS), neuronal NOS (nNOS) or inducible NOS (iNOS) (Moncada et al, 1989; Nozaki et al, 1993). Nitric oxide is a highly diffusible and a potent vasodilator, and it has been shown that NO is of major importance in regulating CBF in both neuronal activation (Dirnagl et al, 1994) and limbic seizures (Pinard et al, 1987). Nitric oxide has been proposed to be a part of the coupling between CBF and neuronal activity in a complex system involving adenosine (Dirnagl et al, 1994). Due to the very potent effect of NO, NO could be a reasonable candidate to explain the very dramatic increase in global CBF observed during infusion of β-hydroxybutyrate. The fact that we did not observe any net uptake of ketone bodies during the infusion and that the metabolism was unchanged indicates that the ketone bodies may not enter the brain with the possibility of influencing the neuronal NOS or iNOS. Ketone bodies may act via vascular endothelial cells without crossing the blood–brain barrier, and in future studies it should be clarified whether NO is involved in the CBF response.
Adenosine is another chemical factor known to be involved in control of CBF. During energy consumption in the brain, dephosphorylation of the energy-rich adenosine nucleotides leads to rising adenosine concentrations within the cells and in the extracellular space, and to increase in CBF. If ketone bodies influenced the energy metabolism of the cells, the adenosine concentration within the cells or in the extracellular space may increase. This study, however, indicates that the energy metabolism of the brain is unchanged and changes in the adenosine system are not the most probable explanation of the CBF increase shown in this study.
Cerebral arteries have an intense supply of adrenergic nerves as well as cholinergic nerves. Under normal circumstances, CBF changes only little when the activities of the nerves are altered either by activation or blockade. In a pilot study, we observed no effect of an unspecific blockade of the β-adrenoreceptors, by propranolol (2 mg/kg), on the increase in CBF during β-hydroxybutyrate infusion (data not shown). Thus, there is little evidence for changes in nervous control in acute hyperketonemia.
In the study by Hasselbalch et al (1996) in man, it was hypothesized that the ketone bodies alter the glycolytic pathways in a way that increases the concentration of some CBF-regulating intermediate substances. This hypothesis was based on findings in the study where the glucose metabolism was depressed by 33% and the CBF increased by 39%. This inverse relationship between glucose metabolism and CBF could be similar to the situation observed during hypoglycemia (Tallroth et al, 1992; Bryan et al, 1987) or by blocking glycolysis (Breier et al, 1993), which in both instances decreased the glycolytic flux. This hypothesis would also fit recent data that suggest an astrocyte-mediated control of CBF (Zonta et al, 2003), since blocking of glycolysis in astrocytes by ketone body infusion may elicit a CBF response despite the fact that neighboring neurons are not energy depleted, since they may use ketone bodies as an alternative fuel.
The mechanisms accounting for the CBF increase in hypoglycemia do, however, not need to be the same, because in the present study normoglycemia was maintained and oxygen metabolism was constant. In a study by Randle et al (1966), it was shown in the perfused heart that, when ketone bodies were added to the perfusate, the pyruvate oxidation was inhibited. An inhibition like that could then lead to a decrease in oxidation of glucose. Metabolism of acetoacetate in the mitochondria generates acetyl-CoA, resulting in increased formation of citrate that is transported into cytosol, where it inhibits phosphofructokinase activity. Consequently, fructose 6-phosphate concentrations rise, and the resulting increase in the concentration of glucose-6-phosphate inhibits hexokinase activity, thus decreasing glucose uptake. Experimental studies in the rat brain have found similar metabolite changes compatible with inhibition of hexokinase, and phosphofructokinase, and thus inhibition of glycolysis (Thurston et al, 1986), possibly due to an increase in citrate production and a decrease in ADP concentration induced by the increased β-hydroxybutyrate metabolism through the Krebs cycle (Ruderman et al, 1974). Another in vitro study using a purified enzyme obtained from pig brain indicates inhibition of pyruvate dehydrogenase (Siess et al, 1971). This inhibition of the pyruvate oxidation was proposed to be a result of the competition between acetyl-CoA derived from acetoacetate metabolism and free CoA for the pyruvate-oxidation system.
In the present study, we did not observe any changes in cerebral metabolism; both the CMRO2 and the CMRglc remained constant. Thus, a mechanism for upregulation of CBF where the metabolism is playing a key role is not very likely. As there seems to be no net uptake of ketone bodies, it is not likely that ketone bodies exert their effect on CBF through changes in brain tissue environment. Thus, the data in the present study indicate that the mechanism responsible for the CBF increase in the fed rat could be directly on the endothelial cells rather than via changes in glycolytic metabolic pathways, but the mechanisms for this effect remain to be determined.
Concluding Remarks
In this study, we found that, during acute hyperketonemia, the brain of the fed rat showed no net uptake of ketone bodies and CMRO2 and CMRglu remained unchanged. Further, we have confirmed the observation by Hasselbalch et al (1996): CBF increases dramatically during infusion with ketone bodies. Thus, in contrast to humans, the rat brain does not switch to ketone body oxidation in favor of glucose oxidation, when ketone bodies are introduced acutely into the blood in normoglycemia. None of the classic modulators of CBF: PaO2, PaCO2 and pH of blood and brain, could explain the increase in CBF. These findings keep the question open: what is the mechanism of the observed CBF increase? We speculate that the action of the ketone bodies is probably directly on the endothelial cells rather than via changes in metabolic pathways inside the brain. Further studies are needed to clarify these issues.
Footnotes
Acknowledgements
The authors would like to thank Dr Jens Kondrup and the technical staff at the Department of Medicine A, Rigshospitalet, for help with analysis of the blood samples.