
Abstract
Transient cerebral ischemia kills CA1 pyramidal cells of the hippocampus, whereas most CA1 interneurons survive. It has been proposed that calcium-binding proteins, neurotrophins, and/or inhibitory neuropeptides protect interneurons from ischemia. However, different synaptic responses early after reperfusion could also underlie the relative vulnerabilities to ischemia of pyramidal cells and interneurons. In this study, we used gramicidin perforated patch recording in ex vivo slices to investigate γ-aminobutyric acid (GABA) synaptic function in CA1 pyramidal cells and interneurons 4 h after a bilateral carotid occlusion accompanied by hypovolemic hypotension. At this survival time, the amplitudes of both miniature inhibitory postsynaptic currents (mIPSCs) and GABA-evoked currents were reduced in CA1 pyramidal cells, but not in CA1 interneurons. In addition, the mean rise time of mIPSCs was reduced in pyramidal cells. The reversal potential for the GABA current (EGABA) did not shift toward depolarizing values in either cell type, indicating that the driving force for chloride was unchanged at this survival time. We conclude that early during reperfusion GABAergic neurotransmission is attenuated exclusively in pyramidal neurons. This is likely explained by reduced GABAA receptor sensitivity or clustering and possibly also reduced GABA release, rather than by an elevation of intracellular chloride. Impaired GABA function may contribute to ischemic neuronal death by enhancing the excitability of CA1 pyramidal cells and facilitating N-methyl-
Introduction
Transient cerebral ischemia kills neurons in vulnerable regions of the brain, including the hippocampus, striatum, and somatosensory cortex (Pulsinelli et al, 1982; Crain et al, 1988). Within the hippocampus of both humans and rodents, area CA1 is particularly sensitive to cerebral ischemia (Pulsinelli et al, 1982; Kirino, 1982; Petito et al, 1987). The pyramidal cells degenerate within a few days after cerebral ischemia, whereas most of the interneurons remain intact (Johansen et al, 1983; Schlander et al, 1988; Nitsch et al, 1989; Tortosa and Ferrer, 1993). However, some interneurons do exhibit degenerative changes and morphological abnormalities within weeks or months after ischemia (Fukuda et al, 1993; Inglefield et al, 1997; Arabadzisz and Freund, 1999). Various mechanisms have been proposed to account for the relative resistance of CA1 interneurons to transient ischemia, including (1) the presence of Ca2+-buffering proteins (e.g., parvalbumin) within interneurons (Freund et al, 1990), (2) the expression of inhibitory neuropeptides such as somatostatin (Bering et al, 1997) and (3) increased neurotrophin signaling within interneurons (Larsson et al, 2001). Although these factors might be involved, they do not explain interneuron resistance completely. For example, parvalbumin-containing interneurons within the striatum degenerate after transient cerebral ischemia (Larsson et al, 2001).
Changes in synaptic responses early after an ischemic insult may explain, at least partially, the difference in vulnerability between CA1 pyramidal cells and interneurons. Enhanced excitatory transmission in area CA1 has been observed within a few hours after ischemia, along with impairment of CA1b pyramidal cell excitability (Urban et al, 1989; Shinno et al, 1997; Gao et al, 1998; Mitani et al, 1998). Decreases in inhibitory transmission have been observed in neocortex 10 months after cerebral ischemia (Luhmann et al, 1995), but, to our knowledge, inhibitory synaptic responses have not been examined in hippocampal pyramidal cells early after ischemia nor have either the excitatory or inhibitory synaptic responses of CA1 interneurons. Synaptic responses in area CA1 interneurons have been measured during anoxia in vitro; both excitatory and inhibitory synaptic responses are depressed, but recover within 10mins (Khazipov et al, 1995).
Interneurons within hippocampal area CA1 provide inhibitory GABA innervation to pyramidal cells and other interneurons. It has been hypothesized that GABA neurotransmission in hippocampus is reduced early after cerebral ischemia (Li et al, 1993; for a review, see Schwartz-Bloom and Sah, 2001). In support of this idea, we showed that GABAA receptors are downregulated in hippocampal area CA1 within 30 mins after cerebral ischemia (Alicke and Schwartz-Bloom, 1995). Consistent with this finding, GABA-stimulated chloride influx is reduced in isolated rat forebrain synaptoneurosomes within hours after ischemia (Verheul et al, 1993). Similarly, GABA-stimulated chloride influx is reduced after oxygen-glucose deprivation of hippocampal slices, an in vitro model of cerebral ischemia (Inglefield and Schwartz-Bloom, 1998; Galeffi et al, 2004b). This impairment of GABA function in hippocampal pyramidal cells was associated with an increase in intracellular chloride. If intracellular chloride increases after cerebral ischemia in vivo, one would expect to see a positive shift in the reversal potential for GABA-evoked currents (EGABA).
The objective of this study was to determine if cerebral ischemia alters GABA function in pyramidal neurons and/or interneurons of area CA1 within a few hours after transient cerebral ischemia. We also determined whether any impairment of GABAA receptor-mediated responses could be explained by a reduced driving force for chloride. To preserve the chloride gradient across the plasma membrane, recordings were made with the gramicidin-based perforated patch technique (Akaike, 1996).
Materials and methods
Forebrain Ischemia
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Duke University Institutional Animal Care and Use Committee.
Transient forebrain ischemia was produced using two-vessel occlusion, as described by Zhan et al (2001). Briefly, adult male Sprague—Dawley rats (Charles River, Raleigh, NC, USA) that weighed 160 to 200 g (42 to 47 days old) were fasted for 7 to 10 h before surgery, but allowed free access to water. Animals were anesthetized with 2.5% halothane delivered through a face mask and intubated tracheally; the lungs were ventilated mechanically with 30% O2/70% N2O. The inspired concentration of halothane was adjusted to maintain a mean arterial pressure between 80 and 100 mm Hg. A digital thermistor probe was inserted into the rectum to record core temperature and another one was placed under the left temporalis muscle to monitor pericranial temperature. Cortical surface electroencephalograph (EEG) was monitored continuously during the surgery with active subdermal electrodes positioned over the parietal cortex bilaterally and a ground lead inserted into the tail. The left femoral artery was cannulated for continuous monitoring of blood pressure and for blood sampling. The right jugular vein was cannulated with a soft catheter for drug infusion and blood withdrawal. Both common carotid arteries were isolated from the carotid sheaths via a ventral midline incision. At 15 min before occlusion of the common carotid arteries, a sample of arterial blood (200 μL) was withdrawn for blood gas analysis and plasma glucose assay. Mean blood pressure was lowered to 30 to 40 mm Hg by injecting 0.4 mg of phentolamine into the arterial catheter, followed by withdrawal of blood from the jugular vein with a heparinized syringe. Subsequently, both common carotid arteries were occluded with small vascular clips for 8 mins. During the period of occlusion, the core temperature was controlled at 37.2°C ± 0.2°C, whereas the pericranial temperature was allowed to decrease gradually from 37.2°C ± 0.2°C at the beginning of the occlusion to 36.8°C ± 0.2°C at the end of the occlusion. The clips were then removed and the withdrawn blood was reinfused. Reperfusion in each artery was verified visually. Metabolic acidosis caused by hypoperfusion was corrected by intravenous administration of 8.4% (w/v) sodium bicarbonate (0.3 ml) immediately after reperfusion. Before discontinuation of anesthesia, the vascular catheters were removed and the wounds were infiltrated with 1% lidocaine and sutured. The endotracheal tube was removed after recovery of spontaneous respiration and the righting reflex. The animal was then maintained in a warm (30°C to 32°C) humidified chamber for another 3 h before being returned to its cage at room temperature. Rats accepted for further study met the following criteria: (1) blood gas parameters were within the normal range (pH 7.35 to 7.45, pCO2 35 to 45 mm Hg, pO2 100 to 180 mm Hg); (2) plasma glucose concentration ranged between 90 and 150 mg/dL; and (3) the EEG flattened immediately after occlusion and remained flattened throughout the entire occlusion period. Rats were killed 4 h later for electrophysiological studies or 7 days later for cell counting and immunofluorescence studies.
Cell Counting and Immunofluorescence
To determine whether hippocampal CA1 interneurons were more resistant to ischemic damage than pyramidal cells in the relatively young rats used in this study, hippocampal sections were double-stained with antibodies against microtubule-associated protein-2 (MAP-2) and parvalbumin, a Ca2+-binding protein that is expressed in a large population of interneurons in stratum pyramidale of area CA1 (Aika et al, 1994; Shetty and Turner, 1998). Adjacent sections were stained with cresyl violet for counting intact neuronal cell bodies.
At 7 days after reperfusion, rats (4 sham-operated controls and 4 rats subjected to transient cerebral ischemia) were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneally) and perfused through the left ventricle, first with heparinized 0.1 mol/L phosphate-buffered saline (PBS) and then with a fixative that contained 2% (w/v) paraformaldehyde, 0.075 mol/L lysine, 0.01 mol/L sodium peroxidate and 0.0375 mol/L sodium phosphate, pH 6.8. Brains were removed and tissue blocks that contained the neocortex and hippocampus were cut. Tissue blocks were postfixed with the same fixative for 4 h. After the blocks were washed in gradually increasing concentrations of phosphate-buffered sucrose, they were frozen rapidly in ethanol cooled with solid CO2. A cryostat was used to cut coronal sections of 14 µm (for immunofluorescence) or 30 µm (for cresyl violet staining) thickness from the rostral hippocampus as far as 4.16 mm posterior to bregma. The protocol for double immunofluorescence staining was as follows. After sections were exposed to 100% acetone at 4°C for 60 secs, they were washed with 0.1 mol/L PBS 3 times for 5 mins each. Sections were incubated with 2% (w/v) IgG-free bovine serum albumin in 0.1 mol/L PBS for 90 mins, followed by an incubation with primary antibodies at 4°C overnight. The concentrations of rabbit polyclonal anti-MAP-2 (Chemicon, Temecula, CA, USA) and mouse monoclonal antiparvalbumin (Sigma, St Louis, MO, USA) used were 12.5 and 5 µg/mL, respectively. Sections were washed 3 times with 0.1 mol/L PBS (10 mins each) and then incubated with a mixture of Alexa Fluor 594 goat anti-rabbit IgG (1:400) and Alexa Fluor 488 goat anti-mouse IgG (1:400) for 150mins at 4°C. The two secondary antibodies were obtained from Molecular Probes (Eugene, OR, USA). After washing, sections were mounted with 75% glycerol in 0.1 mol/L PBS and visualized under a fluorescence microscope. Images were captured with a digital camera controlled by Adobe Photoshop (Adobe, San Jose, CA, USA).
Cell counting was performed in cresyl violet-stained sections adjacent to those used for immunofluorescence. Intact neuronal cell bodies in stratum pyramidale of the whole CA1 area were counted in both hemispheres from at least three sections per rat. The values, expressed as the number of cells per millimeter length of the stratum pyramidale, were averaged for each animal.
Preparation of Acute Brain Slices (‘Ex Vivo’)
At 4 h after reperfusion, the animal was reanesthetized with halothane and decapitated. The whole head was immersed immediately in cold (5°C to 6°C) oxygenated artificial cerebrospinal fluid (ACSF) and the forebrain was removed. Coronal hippocampal slices (400-µm thickness) were prepared with a Vibratome in oxygenated (95% O2/5% CO2) ACSF at 5°C to 6°C. Slices were cut from the rostral hippocampus as far as 4.16 mm posterior to bregma (Paxinos and Watson, 1986). The ACSF consisted of 125 mmol/L NaCl, 2.5 mmol/L KCl, 2 mmol/L CaCl2, 1.0 mmol/L MgCl2, 1.25 mmol/L NaH2PO4, 26 mmol/L NaHCO3, 20 mmol/L
Electrophysiological Recording
A slice was transferred to a plexiglas recording chamber and held in place with platinum wires. The slice was superfused continuously (2.5 mL/min) with ACSF at room temperature (22°C to 24°C). Pyramidal cells and interneurons located within the stratum pyramidale of area CA1b were identified visually based on their size, shape, location, and dendritic morphology under a Nikon Eclipse E600FN microscope equipped with infrared-differential interference contrast (IR-DIC) optics, a ± 40 water immersion objective, and epifluorescence illumination (Nikon Inc., Melville, NY, USA). Images were captured with an infrared charge-coupled device (CCD) video camera (IR-1000; DAGE MTI, Michigan City, IN, USA) and displayed on a monitor.
Patch electrodes were pulled from borosilicate glass (OD: 1.5 mm; ID: 1.1 mm; Sutter Instruments, Novato, CA, USA) with a Flaming/Brown electrode puller (Sutter Instruments, Novato, CA, USA). Pipettes with resistances of 4 to 6 MΩ were filled with an internal solution that contained 136 mmol/L CsCl, 1 mmol/L MgCl2, 0.1 mmol/L CaCl2, 1 mmol/L Ethylene glycol-bis(2-aminoethylether)-N,N,N‘,N’-tetraacetic acid (EGTA), 10 mmol/L N-(2-Hydroxyethyl) piperazine-N‘-(2-ethanesulfonic acid) (HEPES), 2 mmol/L adenosine 5‘-triphosphate (ATP) tris salt, and 0.4 mmol/L guanosine 5‘-triphosphate (GTP) tris salt, pH 7.30. The osmolality ranged between 294 and 297 mosm. To visualize the recorded cells, Alexa Fluor 488 hydrazide (Molecular Probes, Eugene, OR, USA) was added to the internal solution at a concentration of 0.003% (w/v). For perforated-patch recordings, patch electrodes were immersed in the gramicidin-free internal solution for 1 min and then back-filled with the same solution that contained 40 µg/mL of gramicidin. Gramicidin was dissolved in methanol (5 mg/mL), mixed for 1 min, and sonicated for 2 mins before being diluted into the internal solution. The gramicidin-containing solutions were used within 90 mins after preparation.
Signals were recorded with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). After a gigaseal was formed, the progress of perforation was monitored until the access resistance dropped below 100 MΩ. Series resistance (20 to 30 MΩ) was compensated to 90%. Series resistance was monitored throughout the experiment to ensure its constancy (± 15%). The resting membrane potential was recorded when the access resistance was ∼ 100 MΩ and was corrected for a 4 mV liquid junction potential. Recordings were made only from neurons with a resting membrane potential that exceeded −50 mV. Miniature inhibitory postsynaptic currents (mIPSCs) and responses to applied GABA were recorded at a holding potential of 0 mV in the presence of 0.5 µmol/L tetrodotoxin (TTX), 100 µmol/L
In another set of experiments, EGABA was determined by applying GABA to the recorded cells at holding potentials from −100 to 0 mV in 10 mV increments. Each holding potential was maintained for 4 secs, during which GABA (100 µmol/L) was delivered with a Picospritzer (General Valve, Fairfield, NJ, USA) for 10 ms beginning 250 ms after the change of holding potential. Pressure ejection pipettes had a tip resistance of 3 to 5 MΩ and were placed 20 to 30 µm from the somata of recorded cells.
At the end of each recording, the membrane was ruptured to allow Alexa Fluor® 488 access to the cytoplasm, and the recorded cell was visualized at an emission wavelength of 535 nm to confirm its identity 15 to 20 mins later. Cells were discarded if the patch broke during the recording, as indicated by the appearance of intracellular fluorescence or a sudden drop in EGABA. Additionally, data were not included from cells that could not be identified morphologically as either a pyramidal cell or an interneuron.
Data Acquisition
Data were acquired using an IBM-compatible computer equipped with a Digidata 1322 interface and pClamp 8.1 software (Axon Instruments, Union City, CA, USA). Whole-cell currents were filtered at 1 to 2 kHz and digitized at 20 kHz for off-line analysis with pClamp 8.1 and/or MiniAnalysis 5.2.8 (Synaptosoft Inc., Decatur, GA, USA). Miniature inhibitory postsynaptic currents were first screened automatically with a set of prespecified parameters and then accepted or rejected manually with an event detection amplitude threshold at 5pA (Molnár and Nadler, 2001). Current amplitudes were measured at their absolute maximum after subtraction of baseline noise. The frequency of mIPSCs in a given cell was calculated from the number of events over a 2.5-mins period. To construct cumulative probability plots, mIPSC recordings within groups were pooled together and then reanalyzed with MiniAnalysis. EGABA was determined in individual cells from the I/V curve by a least-squares regression. The intracellular chloride concentration was calculated from the Nernst equation.
Statistical Analyses
Data are presented as means ± s.e.m. unless otherwise specified. Data were analyzed with two-way and one-way analyses of variance (ANOVAs) with repeated measures, followed by Fisher's protected-least-significant-difference (PLSD) test when interactions of main effects were significant. When only two groups of means were compared, a Student's t-test was used. The Kolmogorov—Smirnov test was used to compare the cumulative probability curves for amplitude, charge transfer, decay time constant, 10% to 90% rise time, and interevent interval. Values were considered statistically significant at P > 0.05.
Materials
γ-Aminobutyric acid, IgG-free bovine serum albumin, gramicidin,
Results
Relative Sensitivity to Ischemic Damage of Pyramidal Cells and Interneurons in Area CA1
In the present study, rats younger than those normally used in studies of cerebral ischemia (42 to 47 days postnatal) were required so that gramicidin-based perforated patch recordings could be made. Similar to the extent of neuronal damage caused by 2-vessel occlusion for 8mins in older rats (Zhan et al, 2002), cresyl violet staining revealed that approximately 90% of neurons in the stratum pyramidale of area CA1 had degenerated 7 days after reperfusion. The number of intact neurons per mm length of stratum pyramidale was reduced significantly from 261 ± 4 in 4 sham-operated animals to 25 ± 2 in rats subjected to transient cerebral ischemia (P > 0.001, unpaired Student's t-test). To confirm that interneurons were more resistant to ischemic damage than pyramidal cells in area CA1, we used a double-labeling approach with MAP-2 and parvalbumin antibodies to distinguish the loss of pyramidal neurons from the loss of interneurons, respectively. As shown in Figure 1, both pyramidal cells and interneurons were stained with an anti-MAP-2 antibody. At 7 days after reperfusion, there were only few MAP-2-positive cells remaining in area CA1 stratum pyramidale. Most of the MAP-2 immunolabeled cells were also parvalbumin-positive, indicating that interneurons are more resistant to ischemia than the neighboring pyramidal cells.
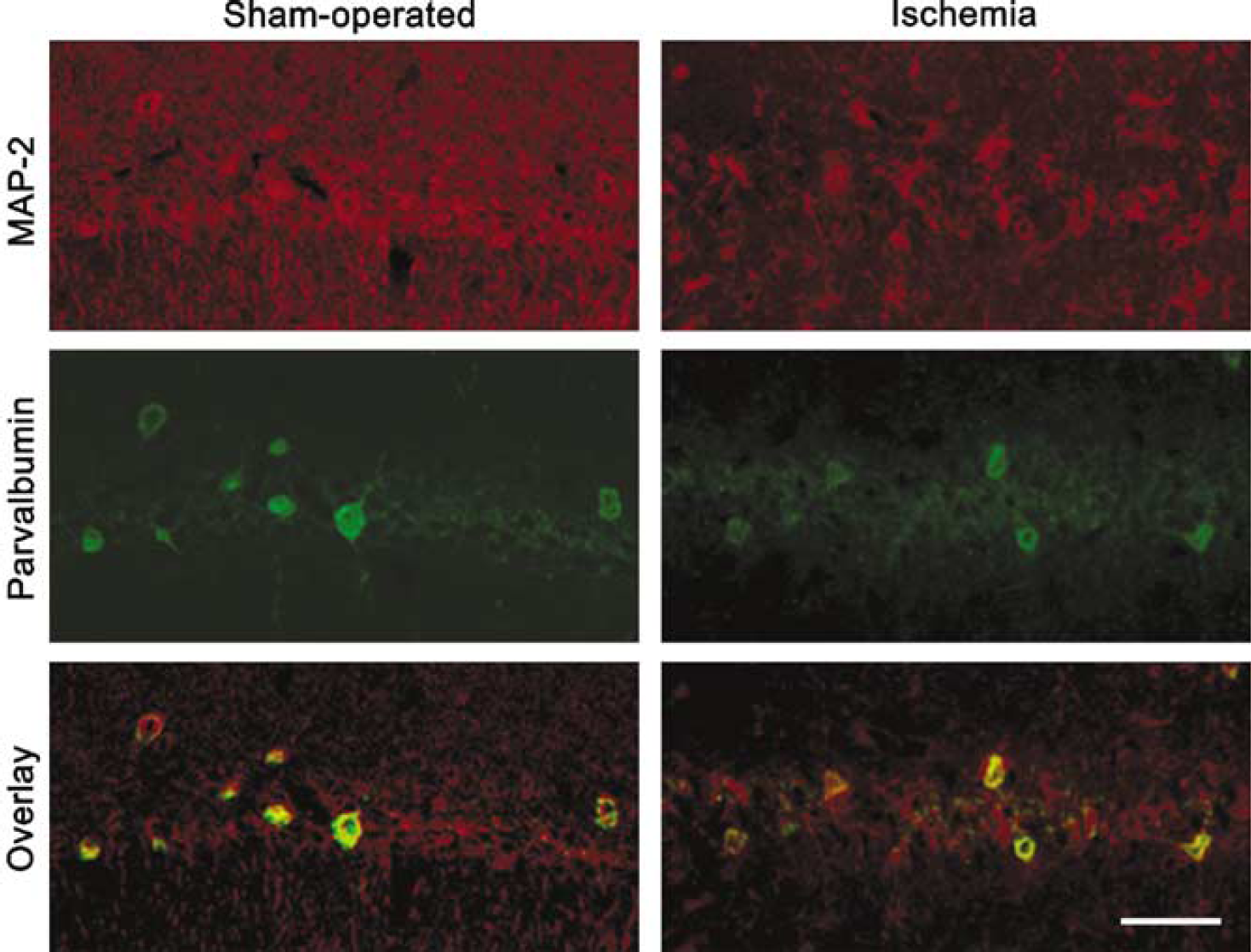
CA1 interneurons are more resistant to ischemic damage than surrounding CA1 pyramidal cells. Sections were prepared 7 days after reperfusion. Anti-MAP-2 and antiparvalbumin antibodies were used to label all neurons and interneurons, respectively. Note that a large percentage of interneurons survived transient cerebral ischemia, whereas most pyramidal cells were lost. Scale bar = 50 µm.
GABAergic Responses in CA1 Pyramidal Cells and Interneurons 4 h after Reperfusion
With the use of IR-DIC visualization and fluorescent imaging, most cells recorded in hippocampal slices prepared 4 h after ischemia could be identified easily as pyramidal cells or interneurons. Individual pyramidal cells had a triangular soma and a thick apical dendrite that extended into the stratum radiatum (Figure 2). In general, pyramidal cells recorded in slices prepared from rats 4 h after ischemia or sham operation exhibited similar cellular morphology. Area CA1 interneurons were diverse in their somatic shape and size and in the appearance of major dendrites. The morphology of interneurons obtained from animals subjected to transient ischemia was indistinguishable from that of interneurons in sham-operated rats.
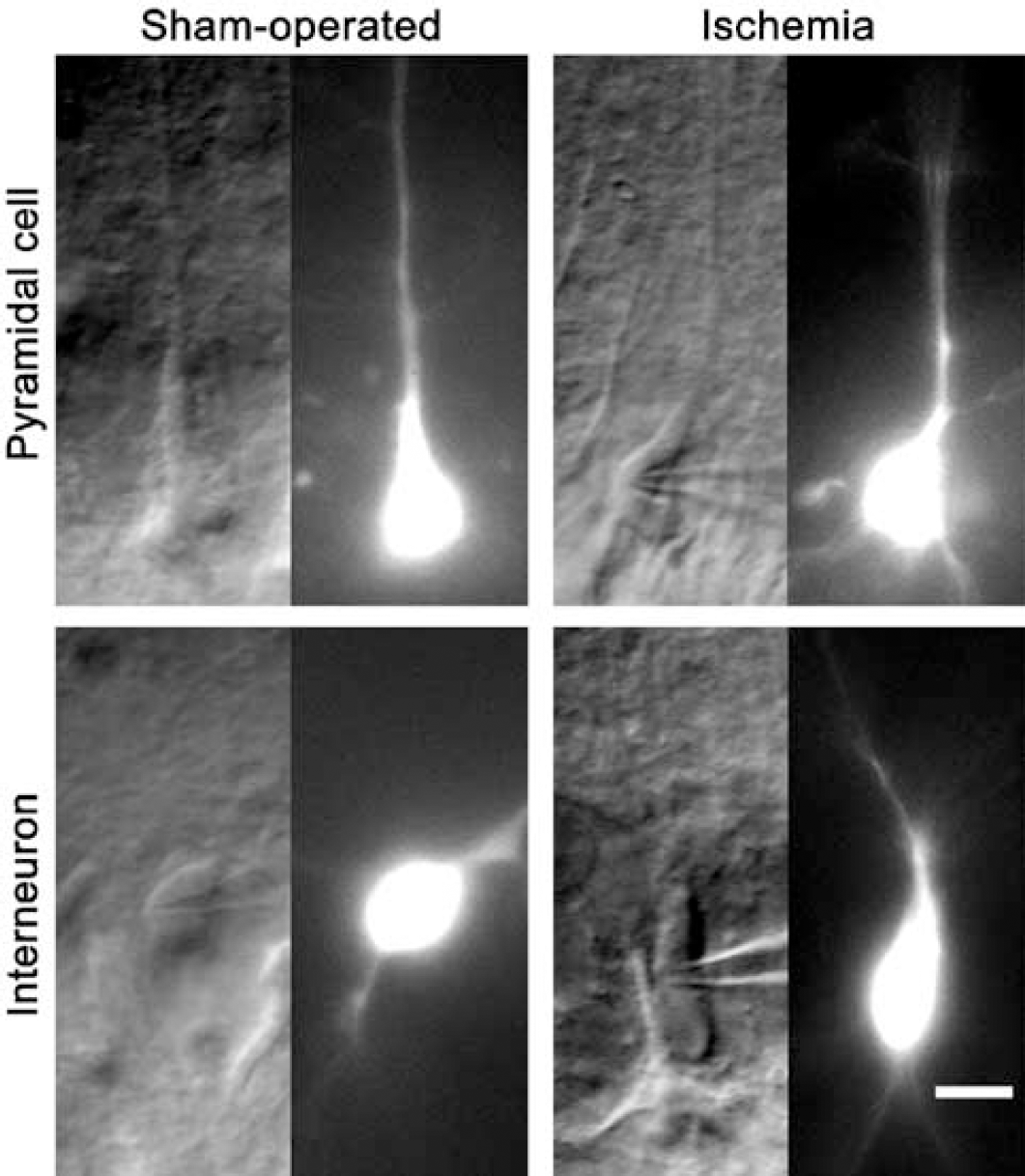
Morphological identification of recorded cells with IR-DIC visualization and Alexa Fluor 488 fluorescent imaging. Infrared-differential interference contrast images (the left in each panel) were taken within a few minutes after seal formation and fluorescent images were obtained 15 to 20 mins after membrane rupture. Upper panels: pyramidal cells were identified by their triangular soma and thick apical dendrite that penetrated the stratum radiatum. Lower panels: interneurons were identified by their multiple major dendrites and the varied shape and size of their somata. Scale bar = 10 µm.
To record GABAergic currents without disrupting the chloride gradient across the plasma membrane, we made gramicidin perforated patch recordings from the somata of pyramidal cells and interneurons in area CA1 stratum pyramidale. When the membrane potential (Vm) was changed from −60 to 0 mV, a potential considerably more positive than ECl, the outwardly directed mIPSCs became substantially larger (Figure 3). Transient cerebral ischemia altered mIPSCs in several respects (Figures 3–5, Table 1). First, there was a significant reduction in the mean frequency (∼60%) and mean peak amplitude (∼50%) of mIPSCs in pyramidal cells, but not in interneurons (Table 1). The mean frequency and peak amplitude differences in pyramidal neurons are supported by the corresponding cumulative probability curves for the interevent interval (a measure of frequency) and the mIPSC amplitude (Figures 4 and 5; Kolmogorov—Smirnov test, P > 0.01). The reduced number of mIPSCs in pyramidal cells might be explained either by reduced probability of GABA release or by the accompanying reduction in amplitude of mIPSCs. A reduction in amplitude would likely cause some of the smaller mIPSCs to become lost in the background noise. To discriminate these possibilities, we recalculated mIPSC frequency in rats subjected to ischemia on the assumption that ischemia reduced all mIPSC amplitudes by ∼50%. By increasing the detection threshold to 10 pA (twice the standard threshold), the frequency of mIPSCs fell by only ∼30%. This reduction was substantially less than the ∼60% expected, if reduced amplitude accounted completely for the lower mIPSC frequency observed after ischemia. These considerations led us to conclude that GABA release, in addition to postsynaptic GABAA receptor function, was probably impaired by transient cerebral ischemia.
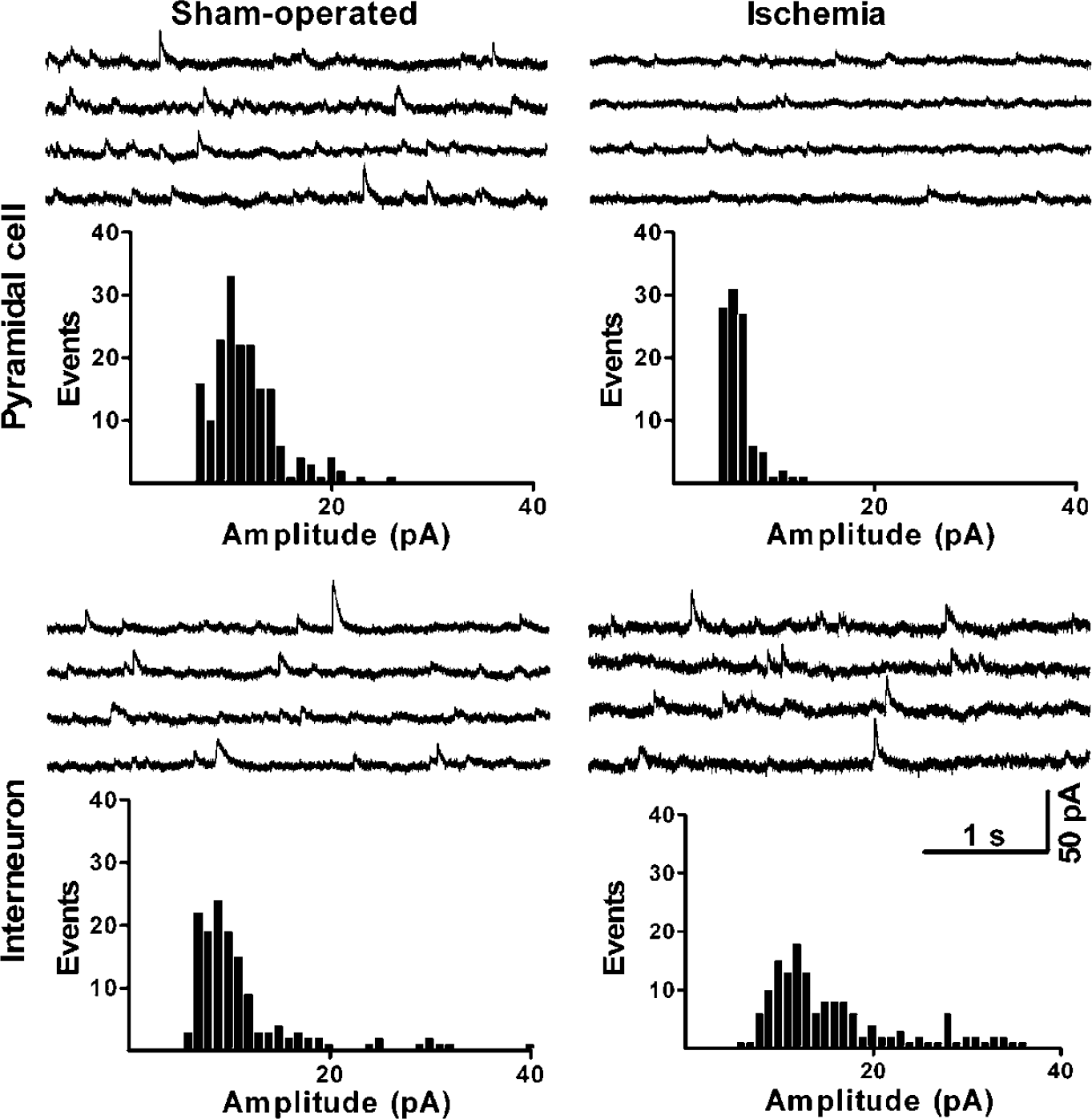
Transient cerebral ischemia alters the properties of mIPSCs in CA1 hippocampal pyramidal cells, but not in CA1 interneurons, 4 h after reperfusion. Gramicidin perforated patch recordings were made in the presence of 0.5 µmol/L TTX, 20 µmol/L CNQX, 100 µmol/L DL-AP5, and 1 µmol/L CGP 55845 at a holding potential of 0 mV. Larger events of mIPSCs were frequently seen in pyramidal cells from sham-operated control rats, but rarely seen in pyramidal cells subjected to transient cerebral ischemia. Ischemia had no such effect in CA1 interneurons. Spontaneous events were recorded from each cell for a 2.5-mins period. Results shown were obtained from representative experiments.
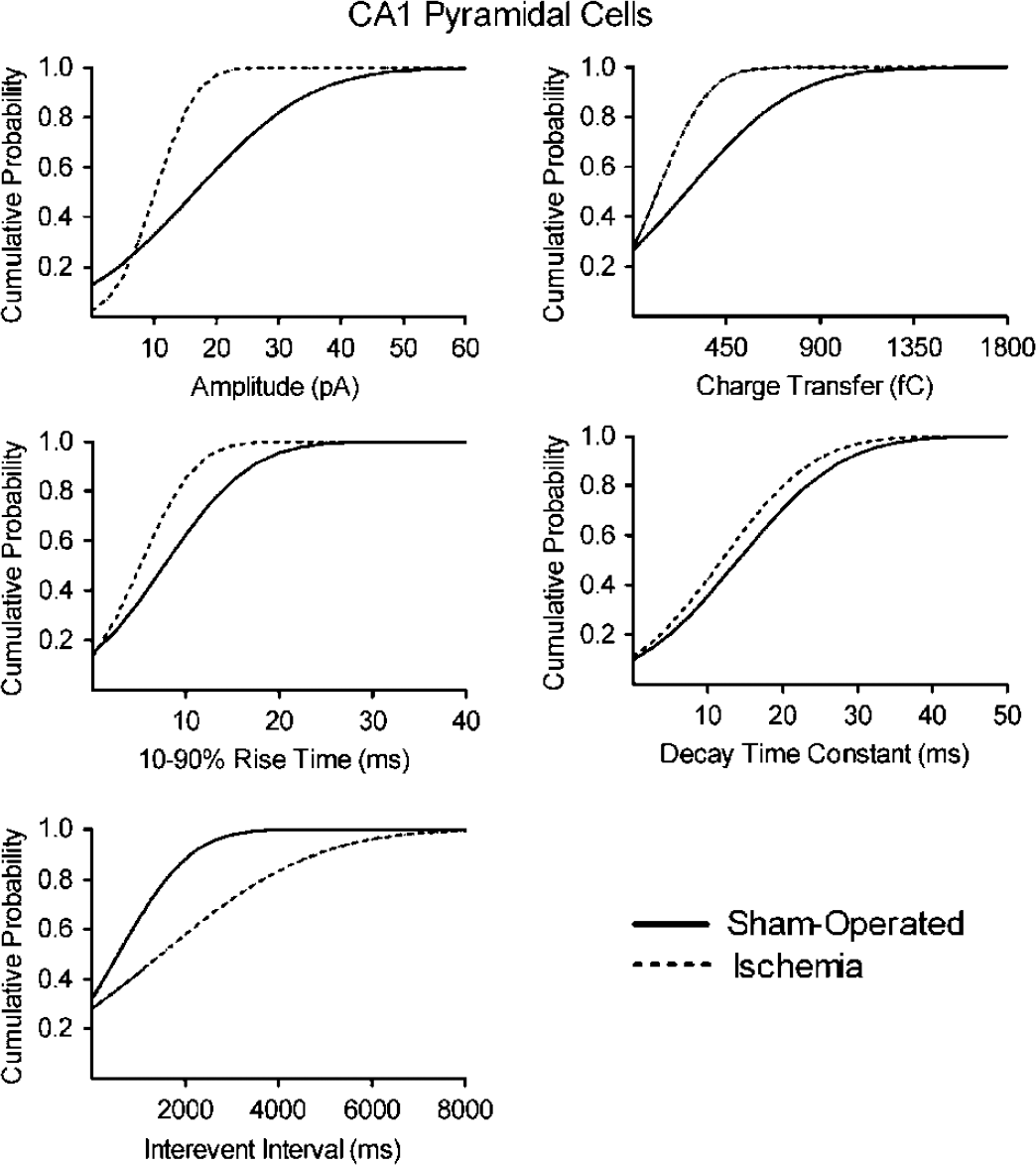
Cumulative probability plots of amplitude, charge transfer area, 10% to 90% rise time, and decay time constant reveal that ischemia had profound effects on miniature IPSCs in pyramidal cells. Recordings in pyramidal cells from sham-operated rats (solid line, n = 6) and from ischemic rats (dotted lines, n = 6) were pooled together and analyzed with MiniAnalysis. Note that ischemia produced a considerable left shift of the amplitude, charge transfer area, 10% to 90% rise time, and interevent interval probability curves (Kolmogorov—Smirnov test, P > 0.01).
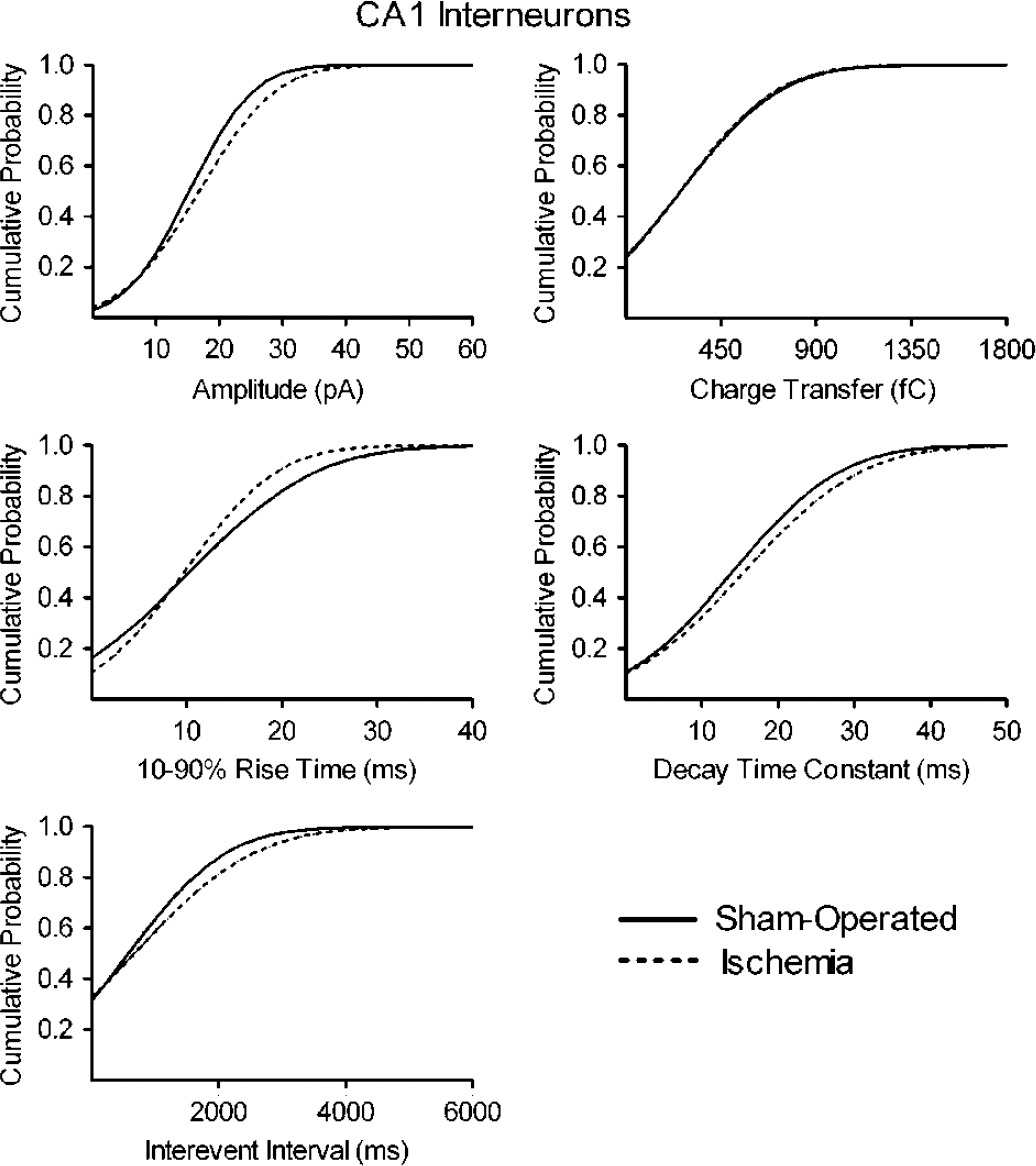
Cumulative probability plots show that cerebral ischemia had negligible effects on all electrophysiologic parameters measured in interneurons. Recordings from interneurons within stratum pyramidale (area CA1) from sham-operated rats (solid line, n = 6) and from ischemic rats (dotted lines, n = 6) were pooled together and analyzed with MiniAnalysis.
Effects of transient cerebral ischemia on the properties of mIPSCs in CA1 hippocampal pyramidal cells and CA1 interneurons
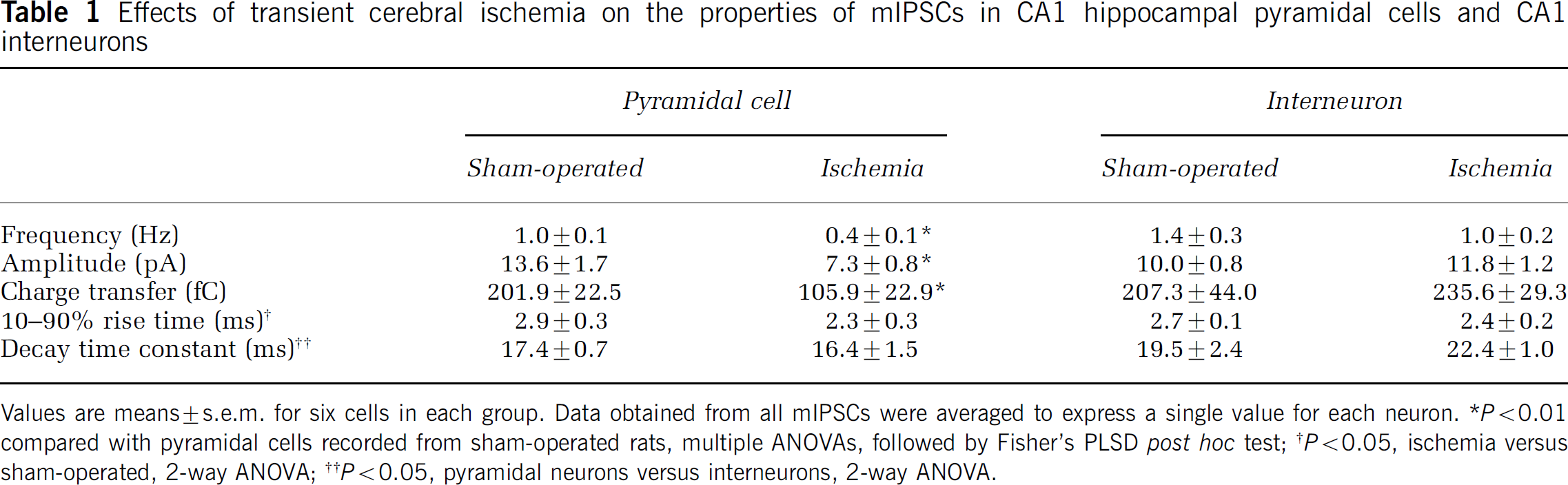
Values are means ± s.e.m. for six cells in each group. Data obtained from all mIPSCs were averaged to express a single value for each neuron.
P > 0.01 compared with pyramidal cells recorded from sham-operated rats, multiple ANOVAs, followed by Fisher's PLSD post hoc test;
P > 0.05, ischemia versus sham-operated, 2-way ANOVA;
P > 0.05, pyramidal neurons versus interneurons, 2-way ANOVA.
Second, consistent with the reduced amplitude of the mIPSCs, the charge transfer per event was significantly smaller in pyramidal cells recorded from animals subjected to ischemia compared with sham-operated rats (Table 1, Figure 4; Kolmogorov—Smirnov test, P > 0.01). Third, 10% to 90% rise times of mIPSCs in both pyramidal cells and interneurons were shorter after ischemia (Table 1), but the magnitude of the effect was substantially greater in pyramidal cells (Figures 4 and 5; Kolmogorov—Smirnov test, P > 0.01). Fourth, the average decay time constant was insensitive to ischemia in both pyramidal cells and interneurons, although the cumulative probability distributions for the decay time constant were slightly different in sham-operated rats versus rats subjected to ischemia (Figures 4 and 5; Kolmogorov—Smirnov test, P > 0.01).
The reduced mean mIPSC amplitude in pyramidal cells suggested impairment of the postsynaptic response to GABA (Cossart et al, 2001). To test this possibility, we measured GABA-evoked currents at a holding potential of 0 mV. GABA (100 µmol/L) was applied to pyramidal cells by addition to the superfusion medium for 90 secs. The peak current decreased significantly from 138 ± 23 pA in sham-operated animals to 57 ± 5 pA in animals subjected to transient cerebral ischemia (Figure 6).
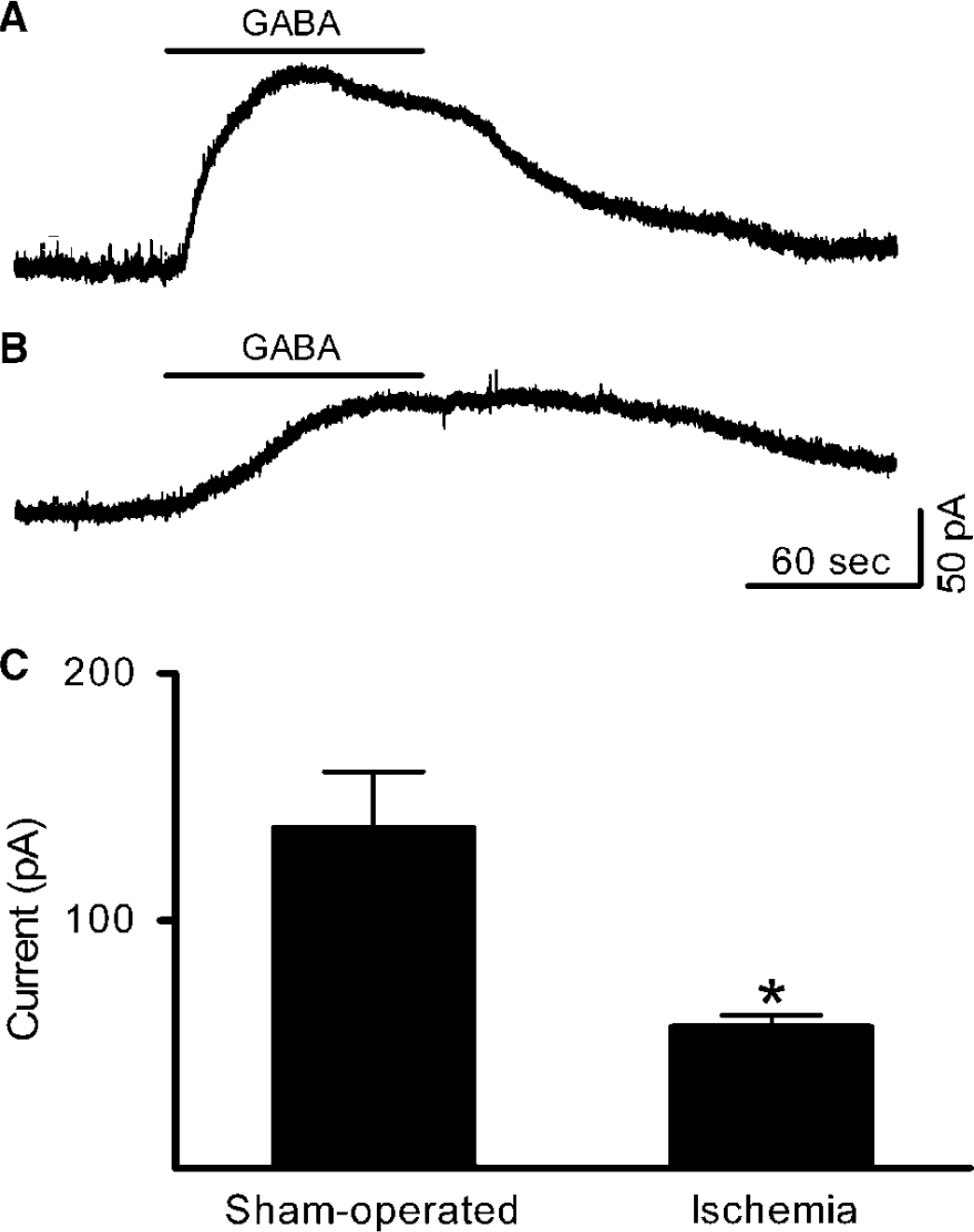
Transient cerebral ischemia reduces the size of currents evoked by the application of GABA to CA1 pyramidal cells 4 h after reperfusion. Recording conditions were the same as described in Figure 2. GABA (100 µmol/L) was applied for the 90-sec period indicated by the horizontal bar above each trace. (
Determination of EGABA
To determine if the reduced GABAergic responses could be due to a reduced driving force for chloride, we measured EGABA, which reflects the chloride gradient across the plasma membrane. In sham-operated animals, EGABA did not differ between pyramidal cells and interneurons (Figure 7). Similarly, there was no significant difference in EGABA between pyramidal cells and interneurons from animals subjected to transient cerebral ischemia. There were no significant differences in the intracellular chloride concentration estimated from the Nernst equation (Figure 8).
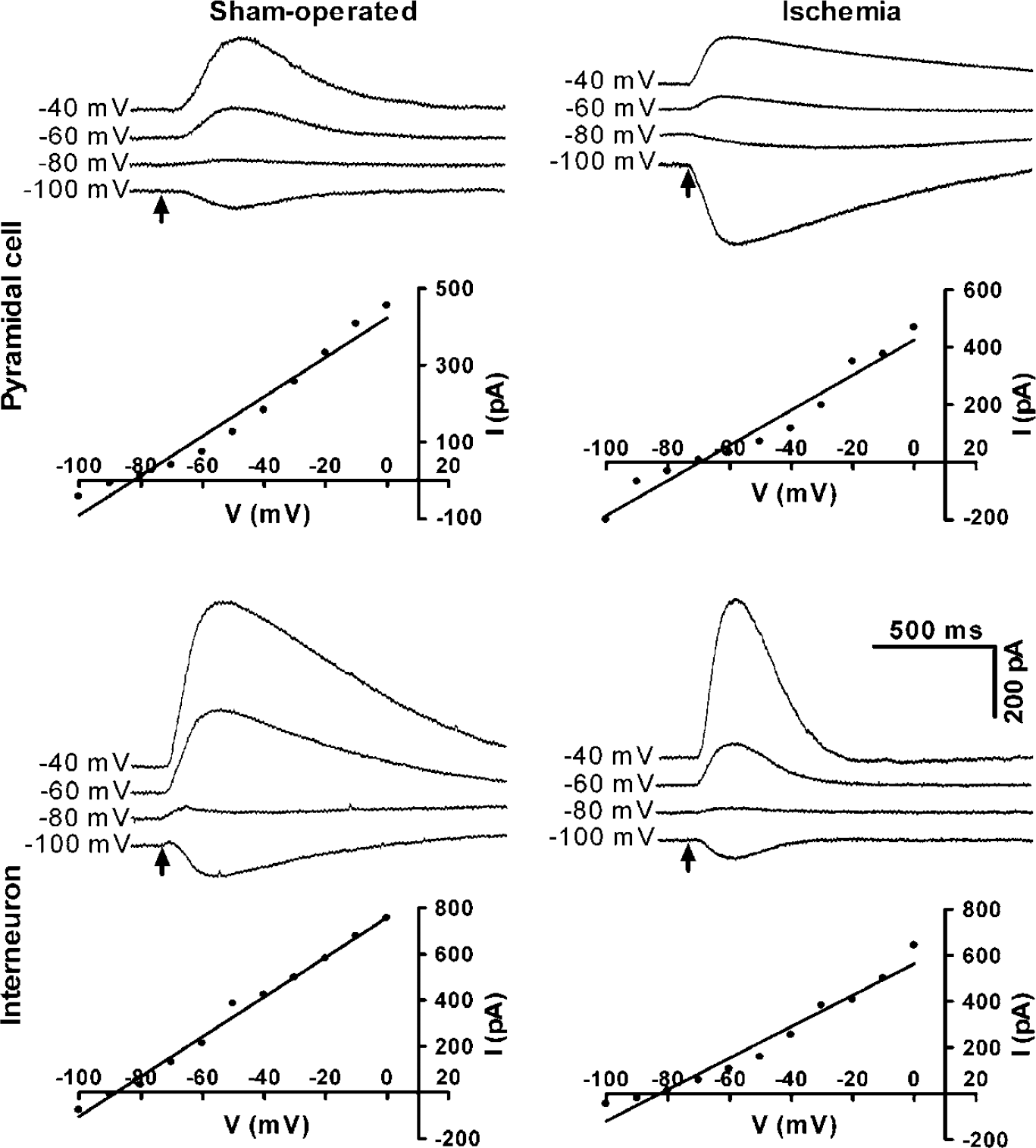
Transient cerebral ischemia does not change EGABA in either CA1 pyramidal cells or CA1 interneurons 4 h after reperfusion. Traces show the response to somatic GABA application at different holding potentials in the same cell. GABA was applied for 10 ms at each holding potential beginning at the arrow. The corresponding current—voltage (I–V) relationship is shown beneath each trace. Results shown were obtained from representative experiments.
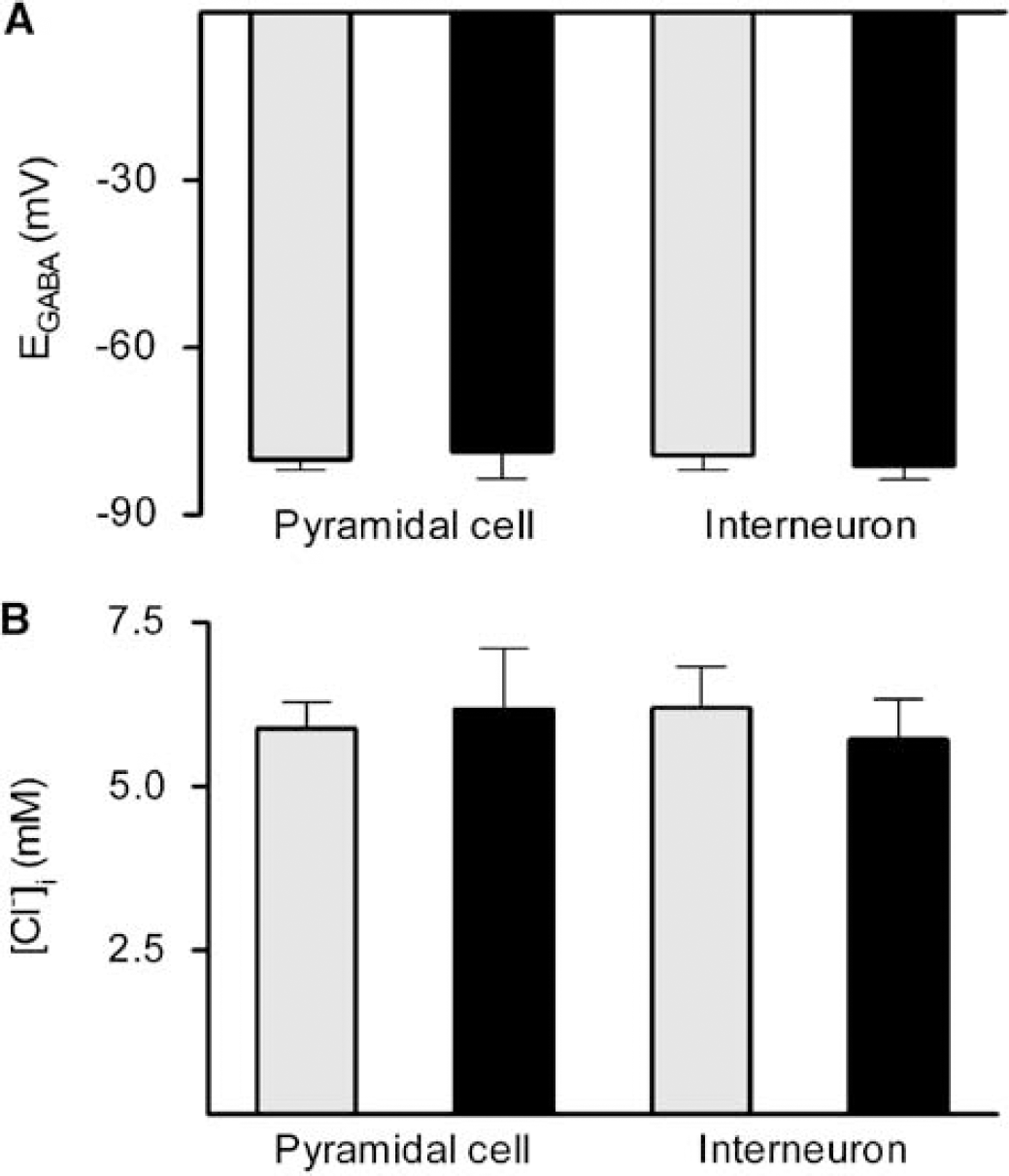
Neither EGABA nor the calculated intracellular chloride concentration ([Cl−]i) was altered by transient cerebral ischemia in either cell type 4 h after reperfusion. (
Discussion
Here we show that transient forebrain ischemia reduces GABA-evoked currents and the mean frequency, amplitude, and rise time of mIPSCs in CA1 hippocampal pyramidal cells, but not in CA1 interneurons, 4 h after reperfusion. The selective reduction in GABAergic responses parallels the differential vulnerability of these neurons to ischemia-induced cell death. With use of the gramicidin perforated patch technique to measure EGABA, we determined that our findings could not be explained by a reduction in the driving force for chloride influx, at least at the level of the soma and at a survival time of 4 h. We reported previously that oxygen-glucose deprivation in hippocampal slices reduces GABAA receptor-mediated responses of CA1 pyramidal cells to applied GABA, coincident with an increase in somatic chloride concentration (Inglefield and Schwartz-Bloom, 1998; Galeffi et al, 2004b). The difference between our ex vivo and in vitro findings may relate to temporal issues. Oxygenglucose deprivation of hippocampal slices kills CA1 pyramidal cells within hours, whereas death of these cells is delayed by several days after transient cerebral ischemia. The delayed onset of degenerative cellular events in vivo allows time for chloride transporters to normalize intracellular chloride concentration. In addition, elements of the physiological milieu present in the intact brain, but not in vitro, may promote restoration of the chloride gradient.
Many investigators have used acutely prepared brain slices (ex vivo) to examine neuronal electrophysiological properties and intracellular ionic changes caused by a previous episode of cerebral ischemia (Urban et al, 1989, 1990; Kirino et al, 1992; Tsubokawa et al, 1992; Shinno et al, 1997; Taga et al, 2000). Compared with in vivo recordings, the ex vivo slice offers experimental advantages, especially the ability to use patch-clamp techniques. However, the ex vivo approach does have some limitations. First, the process of slice preparation may render the slices temporarily ischemic, and this could affect neurons from the sham-control and ischemia groups differently. Second, some neuronal connections are disrupted by slicing. Third, superfusion with ACSF alters the environment of both damaged and intact neurons compared with the natural environment of neurons in vivo. Despite these limitations, several studies showed that electrophysiological changes produced by cerebral ischemia observed in the ex vivo slice were comparable to those determined from in vivo recordings. For example, Urban et al (1989, 1990) first showed that transient ischemia-enhanced excitatory transmission in CA1 pyramidal cells of ex vivo slices is an N-methyl-
The reduced mean mIPSC amplitude observed in the ex vivo slice might be explained either by a smaller postsynaptic response to released GABA or by a reduced vesicular concentration of GABA. However, reduction of the membrane current evoked by application of GABA to CA1 pyramidal cells implies the involvement of a postsynaptic mechanism, reflecting a reduced GABAA receptor conductance and/or activation of fewer GABAA receptors. These results are consistent with previous reports. GABAA receptors are downregulated (possibly by internalization) in area CA1 within 30 mins after global cerebral ischemia in the gerbil (Alicke and Schwartz-Bloom, 1995) and in cultured cortical neurons within hours after oxygen-glucose deprivation (Mielke and Wang, 2005). Furthermore, specific GABAA receptor subunits, including α1, α2, α5 and γ, decline in vulnerable brain regions after focal cerebral ischemia in the rat (Redecker et al, 2002). The shorter mean 10% to 90% rise time indicates that the onset kinetics of pyramidal cell mIPSCs are accelerated. This result might be explained by a change in the relative expression of GABAA receptor subunits or declustering of GABAA receptors. With respect to the latter possibility, destabilization of microtubules in cultured hippocampal neurons resulted in the declustering of GABAA receptors associated with a reduced rise time (Petrini et al, 2003). Cerebral ischemia causes rapid degradation of microtubule-associated proteins (Kitakawa et al, 1989; Matesic and Lin, 1994), which are important in stabilizing microtubules (for a review, see Hirokawa, 1994). Thus, transient cerebral ischemia may lead to declustering of postsynaptic GABAA receptors, reducing the number of receptors in close proximity to presynaptic active zones.
Other factors that depress GABAA receptor function include elevated intracellular calcium, arachidonic acid, and oxygen-free radicals (for a review, see Schwartz-Bloom and Sah, 2001; Sah et al, 2002). Transient cerebral ischemia increases all of these. However, it is unclear whether these metabolic events persist in the ex vivo preparation or reverse once the hippocampus is removed from the post-ischemic milieu and glutamate receptors are blocked. Further studies are required to assess these possibilities.
EGABA was unchanged 4 h after ischemia, suggesting that the transmembrane chloride gradient was preserved in both CA1 pyramidal cells and interneurons at or near the soma. Thus, responses to GABA would be expected to remain hyperpolarizing. In contrast, it has been suggested that the chloride gradient might be dissipated or reversed. As stated above, the somatic chloride concentration increases after oxygen-glucose deprivation in vitro (Inglefield and Schwartz-Bloom, 1998; Galeffi et al, 2004b). Increases in extracellular K+ concentration or glutamate receptor activation, conditions that occur after ischemia, stimulate Na+/K+/2Cl− cotransporter (NKCC-1) activity (Sun and Murali, 1998; Su et al, 2000) or reverse the direction of the K+/Cl− cotransporter (KCC2) to increase intracellular chloride (Payne et al, 1996; Jarolimek et al, 1999; Kakazu et al, 1999; DeFazio et al, 2000). NKCC-1 protein expression and NKCC-1 activity increase in infarcted brain regions from rats subjected to focal cerebral ischemia (Yan et al, 2001, 2003). Finally, downregulation of KCC2 is associated with increased intracellular chloride concentration after oxygen-glucose deprivation (Galeffi et al, 2004b) and other pathological conditions (Coull et al, 2003; Nabekura et al, 2002; Toyota et al, 2003). We suggest that an initial postischemic rise in intracellular chloride concentration reverses within 4 h of reperfusion. At least two factors may contribute to the reversal of high intracellular chloride. Immediately after ischemia, extracellular GABA levels are high enough to saturate GABAA receptors (Globus et al, 1988; Schwartz et al, 1995). However, within 1 h of reperfusion, extracellular GABA levels normalize due to restoration of GABA transport. Thus, 4 h after the onset of reperfusion, GABAA receptors are probably unsaturated with agonist, allowing restoration of the normal chloride gradient. It is not technically feasible to evaluate this hypothesis by measuring EGABA earlier than 4 h after ischemia because CA1 neurons remain leaky and depolarized for some time. It is extremely difficult to form and maintain a gigaohm seal under these conditions. Another possibility is that the expression/activity of chloride transporters changes in a direction that restores the previously disrupted chloride gradient. In this regard, Kang et al (2002) reported that NKCC-1 immunoreactivity in CA1 pyramidal cells is downregulated within 12 h after cerebral ischemia in the gerbil.
The lack of change we observed in EGABA does not preclude the possibility that dendritic chloride concentration remained elevated at the 4 h survival time, because the membrane potential of distal dendrites is not well controlled by somatic current injection. Interestingly, there is a high density of KCC2 expressed in the vicinity of excitatory synapses on the distal dendrites in area CA1 (Gulyás et al, 2001). This extrusion mechanism is proposed to counteract the rise in intracellular chloride associated with excitation-induced dendritic swelling. Recently, we showed that KCC2 is downregulated in the hippocampus after oxygen-glucose deprivation in vitro and after cerebral ischemia (Galeffi et al, 2004a, b ), possibly limiting its ability to restore the dendritic chloride gradient. Thus, an attenuated chloride gradient in distal dendrites could explain the reduced amplitude of some mIPSCs 4 h into reperfusion. Theoretically, this possibility could be investigated by recording directly from the dendrites. However, it is technically very difficult to make gramicidin perforated patch recordings from dendrites in acutely prepared hippocampal slices. An alternative approach is required.
One of the most interesting findings in the present study was that CA1 interneurons retained normal GABA responses at a time when CA1 pyramidal cells responded poorly to GABA. This difference may contribute to the differing vulnerability of these neuronal populations to ischemic damage. Both glutamate neurotransmission and calcium influx may play a role. Although some interneurons express more calcium-permeable AMPA receptors than pyramidal cells (He et al, 1998; Catania et al, 1998), the increase in intracellular calcium induced by AMPA is smaller in hippocampal interneurons than in pyramidal cells (Segal and Greenberger, 1992). In addition, pyramidal cells express more NMDA receptors (Nyíri et al, 2003), and both NMDA currents and calcium influx through the NMDA channel are larger in pyramidal cells than in interneurons (Avignone et al, 2003). Furthermore, interneurons buffer intracellular calcium more effectively than do pyramidal cells, due to their high expression of calcium-binding proteins (for a review, see Baimbridge et al, 1992). An increase in intracellular calcium might be important in mediating the reduced GABA response. First, an ischemia-induced increase in intracellular calcium may reduce GABAA responses via a G protein-dependent mechanism (Chen and Wong, 1990). Second, calcium influx through NMDA receptor channels is important for the loss of microtubule-associated proteins (Buddle et al, 2003), resulting in declustering of GABAA receptors in postischemic brain. Notably, application of MK-801, a high-affinity blocker of the NMDA channel, can attenuate the downregulation of GABAA receptor subunits after focal cerebral ischemia (Redecker et al, 2002) and the suppression of GABA currents in vitro (Allen et al, 2004).
Depressed GABAA receptor-mediated synaptic transmission may, in turn, account for part of the postischemic enhancement of the NMDA receptor contribution to excitatory synaptic responses in CA1 pyramidal cells. The latter phenomenon has been attributed to increased phosphorylation of the NMDA receptor by cyclin-dependent kinase 5 (Wang et al, 2003). However, synaptically driven NMDA receptor-mediated responses are limited in both amplitude and duration by the hyperpolarizing feedforward IPSP (Wu et al, 2004). Reduction of feedforward IPSPs is expected to enlarge and prolong the NMDA receptor contribution to EPSPs in CA1 pyramidal cells and to promote calcium influx through NMDA channels. Thus, depressed GABA inhibition may promote excitotoxicity in two ways: by enhancing the overall excitability of area CA1, which would increase calcium influx by several routes, and specifically by enhancing calcium influx through NMDA channels.
In addition to the phasic inhibition (IPSCs) mediated by synaptically released GABA, there is another type of GABAergic inhibition called tonic inhibition. The average charge carried by the tonically active GABA receptors is several times larger than the charge carried by all of the IPSCs, even when IPSCs occur at high frequencies (Nusser and Mody, 2002). Tonic inhibition is present in hippocampal interneurons under physiological conditions and in pyramidal cells upon disturbance of GABA uptake (Semyanov et al, 2003). It is considered to be important in regulating neuronal gain and signal-to-noise ratio (Mitchell and Silver, 2003; Chadderton et al, 2004; for a review, see Semyanov et al, 2004). Given that the concentration of extracellular GABA is increased during and within the first hour after ischemia, further studies are required to determine if tonic inhibition is altered by cerebral ischemia and if the effect differs between CA1 pyramidal cells and interneurons.
We conclude that GABA responses are reduced in CA1 hippocampal pyramidal cells, but not in interneurons, within a few hours after reperfusion. This deficit in synaptic inhibition may contribute to the selective vulnerability of pyramidal cells and to the enhancement of NMDA receptor-mediated synaptic responses observed at the same survival time. It may also explain the substantial neuroprotective efficacy of benzodiazepines and other GABA-enhancing drugs observed in animal models of global cerebral ischemia (for a review, see Schwartz-Bloom and Sah, 2001). Thus, normalizing GABA synaptic function might be a useful approach to counteract excessive glutamatergic excitation during the postischemic period, thereby attenuating pyramidal cell death.