
Abstract
The purpose of this study was to quantify D2 receptors density and affinity in living rats using [11C]raclopride and to validate the multiinjection modelling approach. To this aim, we used an intracerebral β+-sensitive probe as a highly sensitive system to quantify the radioligand activity using a single three-injection experimental paradigm. The study was divided into three main parts: (i) [11C]raclopride catabolism evaluation without and with cimetidine pretreatment (cytochrome P450 inhibitor); (ii) quantification of kinetics parameters in the striatum, enthorinal cortex, and cerebellum of living rats using a three-compartment model with an arterial input function; (iii) correlation study of in vivo and in vitro binding density and affinity values in the same striatal tissues. (i) raclopride catabolism was very reproducible between individuals; cimetidine pre-treatment resulted in a 30% reduction of raclopride metabolites. (ii) D2 striatal B'max and KdVr estimates obtained by compartmental modelling were 19.87 ± 6.45 and 6.2 ± 3.3 nmol/L, respectively. Cerebellum is the best candidate as a reference region with no specific binding detectable in vivo. (iii) When comparing density (Bmax/B'max) and affinity (Kd/KdVr) values in vivo and in vitro for each striatum, a high strict correlation was found (r2 = 0.90 and 0.72, for density and affinity, respectively). These results validate the multi-injection modelling approach coupled to β-microprobe acquisitions as a mean to provide accurate and separate estimates of dopamine D2-receptor density and affinity, in the living rodent striatum.
Introduction
Positron emission tomography (PET) imaging is a widely used method for the in vivo diagnosis and monitoring of neuroreceptor changes in neurological and psychiatric diseases. In this context, [11C]raclopride is a widely used radiotracer for dopamine receptor quantification because of its high selectivity for the D2 receptor sub-type (Kohler et al, 1985).
The main strategies for quantifying [11C]raclopride binding with PET rely on the use of a region of reference such as the cerebellum (assumed to be devoid of D2-receptors) to calculate a binding potential (BP, Lammertsma and Hume, 1996), which is defined as the ratio between B'max (the available receptor density) and Kd (the receptor affinity) values. Determining a BP may be sufficient for many studies. However, a separate estimation of the two parameters could be very interesting to distinguish between changes in receptor density or affinity that would occur independently in the course of pathological or recovery processes. To estimate these two parameters separately, experimental PET protocols require multiple radiotracer injections at different specific activity; either performed separately (Nikolaus et al, 2003) or within one single experiment called a multi-injection protocol (Delforge et al, 1990). Using an appropriate compartmental model, the latter approach identifies all the compartments and parameters, including receptor density (B'max) and ligand—receptor affinity (KdVr). A major interest of this modelling approach is that, knowing the values of all model parameters, it becomes also possible to design by computer simulation, the best and simplest protocol providing separate quantification of B'max and KdVr values (Delforge et al, 1993). Up to now, such protocols could only be performed on humans and large animals such as non-human primates because of PET imaging constraints (resolution, sensitivity). To meet the time-resolution constraints of the multi-injection protocol, we used the β-Microprobe system, a highly sensitive beta radioactivity counter which allows in vivo measurements of radioactivity concentrations to be obtained from a detection volume restricted to few millimetre around the stereotaxically implanted probes (Pain et al, 2000).
Our goal was to quantity B'max and KdVr separately with [11C]raclopride using a β-microprobe. In that context, we investigated the feasibility and established the validity of the multiinjection protocol in the rat.
The entire study was designed as a series of three complementary experiments: (i) quantification of nonmetabolised [11C]raclopride concentration in arterial plasma under experimental conditions that would increase the amount of radiotracer available for specific binding (cimetidine pretreatment); (ii) separate quantification of B'max and KdVr values for [11C]raclopride in various brain regions of cimetidine pretreated rats using a three-compartment model and the multiinjection modelling approach; (iii) systematic comparison of B'max and KdVr estimates in vivo with in vitro binding values (Bmax and Kd) obtained in the same striatal tissues post mortem.
Methods
Animals
All experimental procedures were in strict compliance with the directives of the EEC (86/609/EEC) and the French National Committee (87/848). On arrival at the laboratory, animals were housed three per cages in a temperature-controlled room under reversed light/dark conditions using a 12h on/off schedule and allowed 1 week of adaptation before being used in experiments. Owing to a shortage in animal production during this study, the various experiments had to be performed on two strains of Lewis rats issued from two different breeding colonies. For experiments 1 and 2, Lewis rats (male, average weight: 340g) were purchased from IFFA CREDO (L'Arbreles, France). For experiment 3, Lewis rats (males, average weight 370g) were obtained from JANVIER (Le Genest-St-Isle, France).
Radiochemistry
[11C]raclopride ((S)-(–)-3,5-dichloro-N-((1-ethyl-2-pyrrolidinyl)methyl)-2-hydroxy-6 methoxy benzamide) was labelled with carbon-11 on its methylether function from the corresponding desmethyl phenolic precursor ((S)-(–)-3,5-dichloro-N-((1-ethyl-2-pyrrolidinyl)methyl)-2,6-dihydroxy benzamide) and the highly efficient methylating reagent [11C]methyl triflate according to Langer et al (1999). Typically, 2.96 to 5.55 GBq of [11C]raclopride (radiochemical and chemical purity >95%) was obtained in routine within 40 to 45mins of radiosynthesis with specific radioactivities ranging from 18.5 to 55.5 GBq/μmol.
[3H]Raclopride (2.96 GBq/μmol) used for in vitro binding assay was purchased from Perkin-Elmer Life Science (Boston, MA, USA).
Drugs
Cimetidine (100 mg/mL) was purchased from Enteris (France) as an injectable solution. Alpha-chloralose anhydro-
β-Microprobe Measurements
The β-microprobe is a local positron detector that can be directly implanted into the rat brain under stereotaxic guidance. The system, which is sensitive to the β+ particles, takes advantage of the limited range of β+ particles within biological tissues to define a restricted detection volume around the probe (typically, a sphere of 2.01mm radius around the probe for 11C; Pain et al, 2000). Two probes (2 mm long, 0.5mm diameter) of plastic scintillating fibre (BCF-12; Bicron, Newbury, OH, USA) fused to an optic fibre of 7mm long (BCF-98; Bicron, Newbury, OH, USA) were used for this study. A detailed report on the operating protocol can be found in a previous report (Pain et al, 2002). Briefly, the signal emitted by the scintillating plastic of the probe is guided by means of an optic fibre to a photodetector (Hamamatsu R7400P; Hamamatsu Photonics, Hamamatsu City, Japan), which delivers a count rate proportional to the concentration of radioligand in the detection volume. In the present study, frequency of detection set to 1sec counting rate was displayed online on the computer screen using the Labview Software (National Instruments, Austin, TX, USA). The β-microprobe system used, allowed simultaneous recordings from two probes. At least 30 mins before each experiment, photodetectors were turned on to allow stabilisation of the electronics (Ginovart et al, 2004). Each detection line (comprising the probe, the associated optical guide and the photodetector) was carefully calibrated after each experiment with 10 mL of an homogeneous aqueous solution of a known amount of [11C]raclopride taken from the same radiosynthesis. The measured sensitivities of the two lines were always comprised between 0.7 and 0.9 cps/kBq mL. This calibration was essential for the full quantification used in this study as fluctuations in the detection rate of the detection lines were observed from one experiment to another.
Surgical Interventions
Surgery was performed under isoflurane anaesthesia (induction: 5%; maintenance 2.5%) delivered in a gas mixture of 30% O2 and 70% N2O. Animals were mounted in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA), the scalp was incised along the midline and a hole was made in the skull to allow insertion of two β+-sensitive microprobes either bilaterally in the striatum, or one in the striatum and the other either in the cerebellum or in the entorhinal cortex, depending on the planned experiment. The two probes were implanted into the rat brain under stereotaxic guidance using micromanipulators (David Kopf Instruments, Tujunga, CA, USA) that also helped to maintain the devices in place during the entire course of the experiment. The following coordinates were selected according to the Paxinos and Watson Atlas (1986) striatum: AP; +0.5 mm; ML; 3 mm; DV; −5 mm; cerebellum: AP; −3 mm; ML; 3 mm; DV; −3 mm; entorhinal cortex: AP; +0.3 mm; ML; 4.3 mm; DV; −4.5 mm. For all regions, DV reference was dura matter, AP and ML reference was lambda, except the striatum for which AP and ML coordinates are given from bregma. In addition to the brain surgery, venous and arterial catheters (polyethylene catheters internal diameter = 0.58 mm, outer diameter = 0.96 mm) were placed for radioligand injections and blood samplings.
For experiment 1, one catheter was inserted in the femoral artery to allow timed arterial blood sampling and a second catheter was placed in the femoral vein to allow [11C]raclopride injection(s). For experiments 2 and 3, three catheters had to be inserted: two in the femoral veins (one for [11C]raclopride injections, one for unlabelled raclopride injection) and the third one in the femoral artery to allow timed arterial blood sampling.
Immediately following surgery, isoflurane anaesthesia was progressively replaced by a continuous α-chloralose anaesthesia (given in a cyclodextrin complex) delivered as a bolus injection of 60mg/kg, intraperitoneally, followed by a chronic intraperitoneal infusion of 40 mg/kg h. Animals were then placed under mechanical ventilation (rodent ventilator 683, Harvard Apparatus) and their body temperature was maintained at 37°C using a thermo-regulated electric blanket (rodent home-othermic blanket control unit, Harvard Apparatus). The physiologic status of the animals was monitored during the entire course of the experiment by assessing various physiologic parameters such as PCO2, PO2 and pH at regular timepoints before, during, and after the β-microprobe recordings and haematocrit at the end of the experiment.
Experimental Paradigms
Experiment 1 (catabolism study): As an attempt to increase the β+ signal-to-noise ratio, we first tested the ability of cimetidine, a selective histamine H2-receptor antagonist and a cytochrome P450 inhibitor, to decrease the amount of [11C]raclopride metabolised during the study (Levine et al, 1998). Thus, without interfering with D2 receptors (Nowak, 1980) but by slowing down [11C]raclopride peripheral catabolism, cimetidine increases the availability of plasmatic [11C]raclopride and consequently the tissular [11C]raclopride concentration. A total of 14 male Lewis rats, divided into two groups, were used in this experiment: seven received cimetidine as a single injection (150 mg/kg intraperitoneally) and seven animals were not treated with cimetidine to constitute the control group. In each group, all animals received a first [11C]raclopride injection (45.88 ± 16.28 MBq corresponding to 3.20 ± 1.86 nmol), 50 mins after cimetidine. After 30 mins, three animals of each group received another injection of [11C]raclopride (40.33 ± 1.85 MBq corresponding to 5.38 ± 1.26 nmol) to mimic the situation that will be encountered in the multiinjection protocol (Figure 1A). Arterial blood samples (0.5 mL) were also collected at 2, 5, 20, 40, and 60 mins in both groups of rats after the last radioligand injection to establish the arterial time—activity curve and determine the fraction of nonmetabolised [11C]raclopride present at these timepoints. Two other arterial samples were collected at 80 mins, one on a cimetidine-treated rat and one on a control rat. Arterial blood samples were immediately centrifuged (5 mins, 2000g, 4°C) to obtain cell-free plasma. For deproteinization, 0.5 mL plasma was mixed with 0.7 mL acetonitrile containing raclopride (5 μg/mL) as a reference compound. After centrifugation at 2000g for 5 mins the supernatant was directly used for HPLC analysis. The HPLC system consisted of two LC-10AS pumps (Shimadzu, Kyoto, Japan), a 2.6 mL mixing chamber, a Valco injector (model C6W; Valco, Schenkon, Switzerland) with a 1 mL loop and a reverse-phase Waters μ Bondapak C18 column (300 × 7.8 mm, 10 μm; Waters, Milford, MA, USA) connected to an ultraviolet detector (Shimadzu SPD-10A; Shimadzu, Kyoto, Japan) operated at 254 nm followed by a radioisotope detector (model LB 506, 2500-4MX cell; Berthold, Wildbad, Germany). Data acquisition and handling were performed with the software Winflow (version 1.21; JMBS Developpements, Grenoble, France). The column was eluted applying a gradient from 10% acetonitrile in 0.01 mol/mL phosphoric acid increasing to 70% in 6.5 mins, increasing to 90% acetonitrile in 8.5 mins, decreasing to 10% acetonitrile in 8.6 mins with a total run length of 15 mins. The flow rate of the eluant was maintained at 5 mL/min.
Data were expressed as a percentage of nonmetabolised [11C]raclopride over the total plasma concentration.
Experiment 2 (Multiinjection Study)
In vivo acquisition protocol: In vivo β-microprobe experiments were performed on cimetidine-treated male Lewis rats. Four experiments were performed with a β-microprobe implanted in one striatum. Two other experiments were performed with the two probes implanted in the left striatum and in the cerebellum. Two experiments corresponded to rats implanted with one probe in the right entorhinal cortex and one in the left striatum. In all cases, the multiinjection protocol consisted of three successive injections of labelled and/or unlabelled ligand timed as follows:
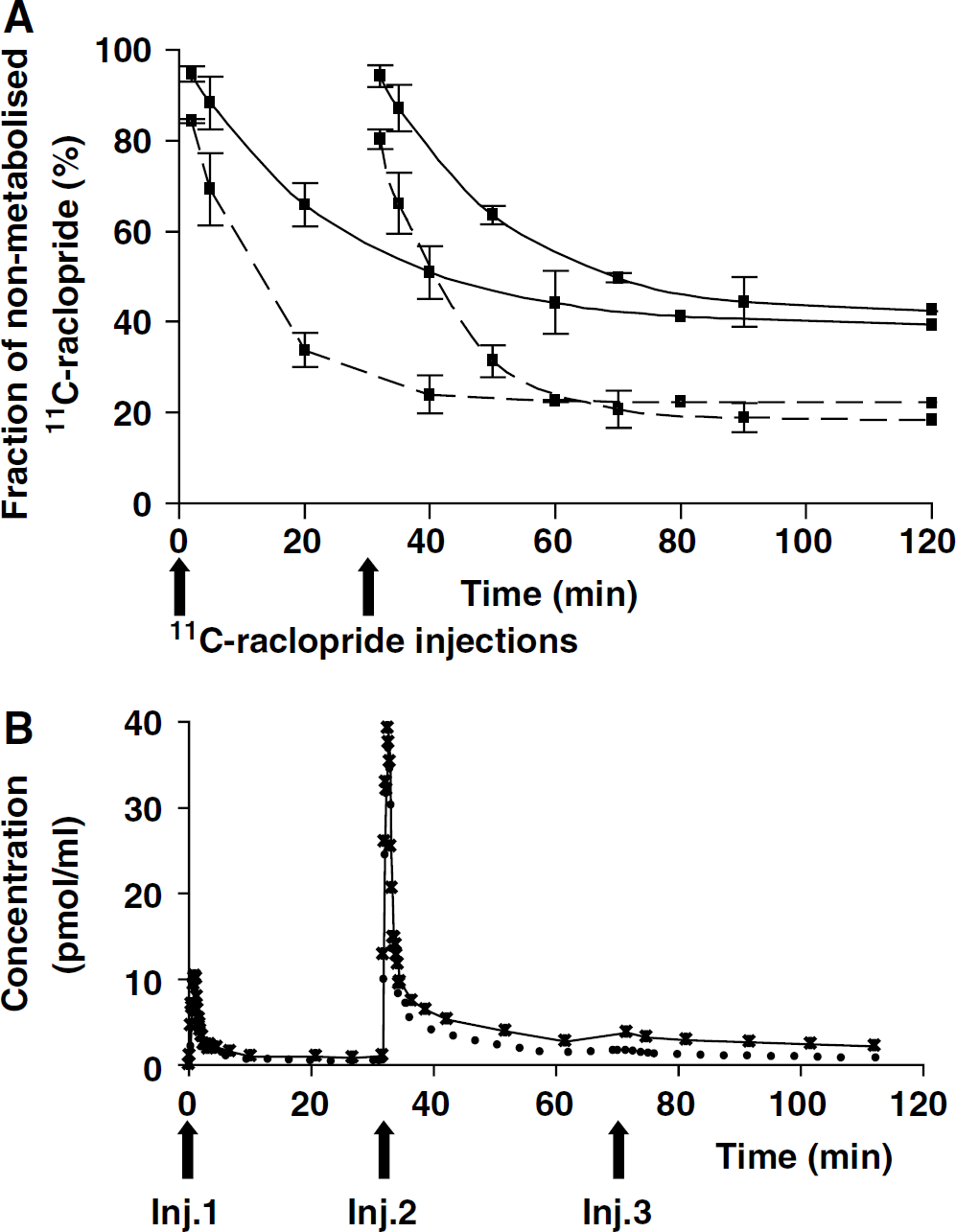
(1) At T=0 mins, a tracer dose (17.39±4.07 MBq; 0.88±0.22 nmol) of [11C]raclopride; (2) at T =30 mins, a second injection of labelled [11C]raclopride (18.5±5.55 MBq; 2.63±1.02 nmol); (3) at T=50 mins, a single injection of unlabelled raclopride (7 μmol).
Arterial blood samples (around 30 μL each) were collected continuously (every 4 to 5 secs) during the first 2 mins after each injection and then at increasing intervals up to the end of the protocol (Figure 1B). About 50 samples (a total blood volume of around 1.5 mL) were withdrawn during the entire experiment, which had no significant effect on haematocrit (data not shown).
To establish the [11C]raclopride time—activity plasmatic curves, blood samples were centrifuged for 5 mins and 10 μL aliquots of plasma were taken for counting in a cross-calibrated gamma counter (Packard, France). The plasmatic curves were then corrected for physical decay of 11C activity and specific radioactivity to establish the time-concentration curves of [11C]raclopride (in pmol/mL).
The correct positioning of the β-microprobes was verified post mortem in each animal. To this aim, shortly after the in vivo protocol completion, rats were killed by decapitation and their brains quickly removed, frozen at −80°C and sectioned (coronal sections, 20 μm thick) in a cryostat at −20°C. Slices were then reacted and stained with Cresyl Violet to determine the actual probe track locations by comparison with the Paxinos and Watson Atlas (1986).
Data analysis: The compartmental model used in this study is based on the usual nonequilibrium nonlinear ligand-receptor model developed in house (Syrota et al, 1984; Delforge et al, 1993). The model structure (Figure 2A) was composed of three compartments (plasma, free ligand, and specifically bound ligand) and six parameters. Because the multiinjection protocol includes the injection of unlabelled raclopride, the kinetics of the unlabelled ligand affects the local concentration of receptor sites accessible to the ligand. As a consequence, the model describing unlabelled ligand behaviour had to be included, assuming that all parameters are identical to that of the labelled ligand model.
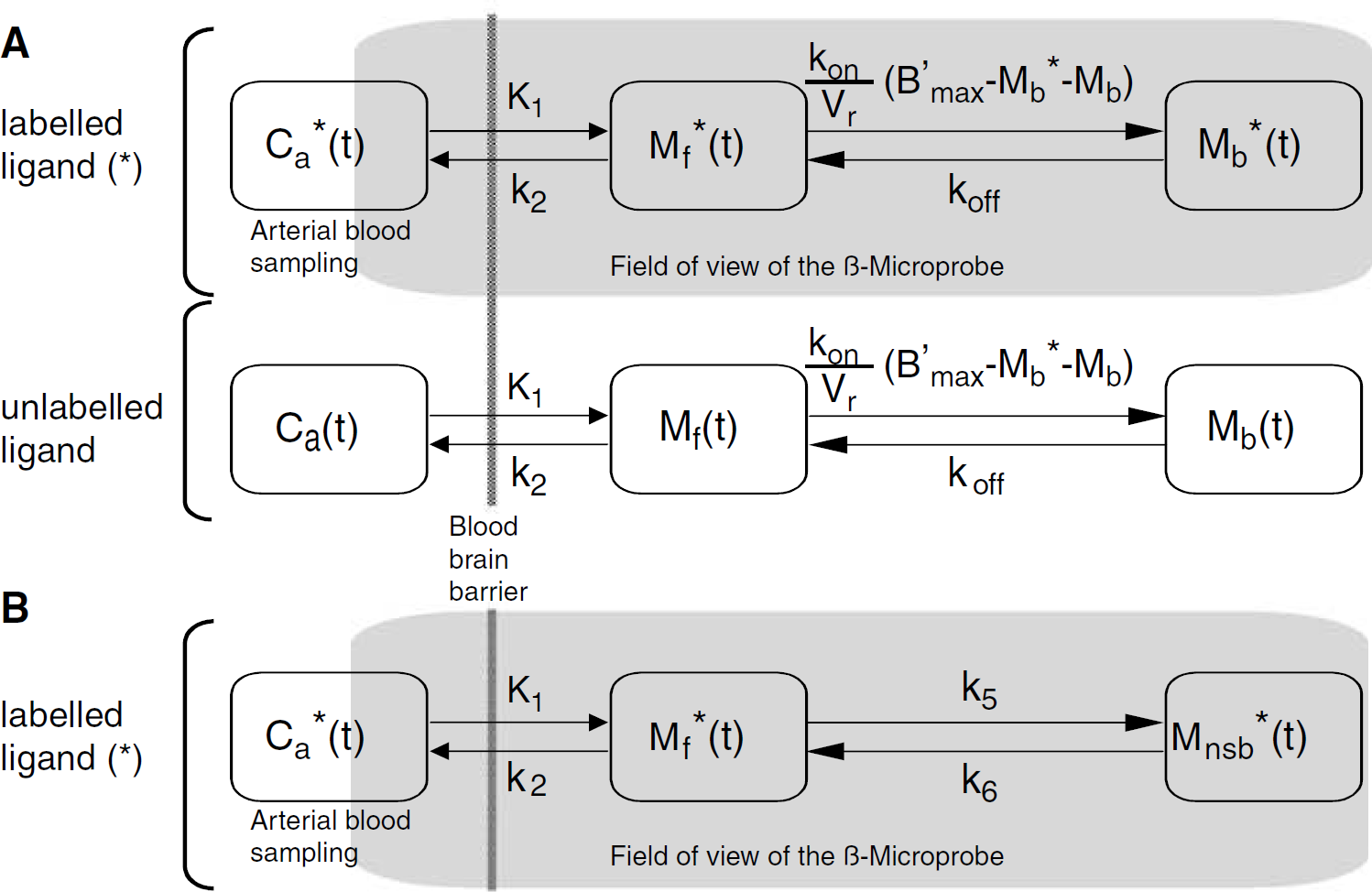
Three-compartment ligand—receptor model (Delforge et al, 1999) used in the analysis of raclopride kinetics obtained with the β-microprobe. The measured data correspond to the labelled ligand in tissular compartments plus a fraction Fv of the vascular labelled ligand concentration. C*a(t) is the plasma concentration of nonmetabolised [11C]raclopride measured by arterial blood sampling.
The flux of radioligand moving from the plasma compartment to the brain compartment is given by K1 C*a(t), where C*a(t) is the plasma concentration of non-metabolised [11C]raclopride derived from the standard curve defined in the first experiment (Figure 1B). The concentration of free labelled ligand is denoted by M*f(t). In the model considered, the free ligand can either bind directly to the free receptor sites or escape back to the blood circulation with a rate constant k2. The specific binding is a saturable reaction which depends on (i) the bimolecular association rate constant kon, (ii) the free ligand concentration at the vicinity of the receptor sites M*f(t)/Vr, Vr representing the reaction volume of the radioligand (Delforge et al, 1996) and (iii) the concentration of free receptor sites, which is equal to B'max – M*b(t)–Mb(t), M*b(t), and Mb(t), respectively, are the concentration of labelled and unlabelled ligand bound to the D2-receptors in the detection volume. The rate constant for the dissociation of the specifically bound ligand is denoted koff. The model also takes into account the vascular fraction in the field of view of the probe, denoted Fv. The plasma concentration of unlabelled ligand (Ca(t)) was derived from the labelled ligand plasmatic concentration C*a(t).
In summary, six parameters have to be identified in the striatum (B'max, K1, k2, kon/Vr, koff, and Fv). However, in brain regions displaying low D2 receptor densities (i.e., the cerebellum and the entorhinal cortex), we also tested a non-saturable model, including or not, a nonspecific compartment (M*nsb). This model (Figure 2B) involves a pool of non-specifically bound ligand which is driven by association and dissociation rate constants denoted k5 and k6, respectively. Five parameters (K1, k2, k5, k6, and Fv) are then required to describe the radioligand kinetics in the entorhinal cortex and the cerebellum.
Compartmental kinetics were calculated by integrating the nonlinear differential equations with a 4th order (classical) Runge—Kutta method (GSL library). Owing to the arterial catheter length, a systematic delay was introduced between C*a(t) and the actual cerebral input function (calibrated, then fixed at 10 secs).
For all regions, the β-microprobe measurements were simulated as the sum of the labelled compartments kinetics and the vascular activity included in the field-of-view. The vascular fraction Fv was estimated, along with all the model parameters by means of minimization of a weighted least-square cost function using a Levenberg—arquardt least-squares minimization procedure. Weights of the cost function were calculated based on the assumption that measurements followed a Gaussian distribution derived from a Poisson emission process, and were expressed as follows:
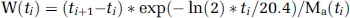
where Ma(ti) is the activity measured by the β-microprobe between ti and ti+1.
The maximum iteration number was twelve, and we used two stopping criteria: one on the fractional improvement (sum-of-squares, stol = 0.0001), the other on parameter variations (Xtol = 0.00001). We ensured that all the minimization process converged, and used different initial parameter values to check for multiple solutions existence. Estimation of the standard errors on each parameter was based on the use of a sensitivity analysis and of the covariance matrix, as previously described (Delforge et al, 1999).
Experiment 3 (Correlative Study)
In vivo acquisition protocol: In the second series of experiments, the doses of injected radiolabelled ligand and the timing of the third injection were slightly adapted according to results of the first series of experiments. Injections were defined as follows:
(1) At T=0 mins, 37.37 ± 10.73 MBq (0.54 ± 0.07 nmoles) of [11C]raclopride; (2) at T=30 mins, 68.45 ± 17.39 MBq (2.96 ± 0.52 nmol) of [11C]raclopride; (3) at T=75 mins, 7 μmol of unlabelled raclopride.
A group of five male Lewis rats was used and implanted bilaterally in the striatum. All animals first underwent a complete multiinjection study according to the paradigm described above. After the end of the acquisition (75 mins after the last injection of unlabelled raclopride), animals were killed by decapitation, their brain removed and left and right striatum dissected, weighted and frozen at −80°C until further processing for binding assay.
In vitro binding protocol: Bmax and Kd values for [3H]raclopride were then determined in each striatum separately, using a binding assay as described by Seeman et al (1992). Briefly, each striatum was homogenized separately in cold buffer (final concentration was 1 mg/mL) 15 up-and-down strokes, at 500 rpm, in a glass Teflon homogenizer. The buffer contained 50 mmol/L Tris-HCl (pH 7.4 at 20°C), 1 mmol/L EDTA, 5 mmol/L KCl, 1.5 mmol/L CaCl2, 4 mmol/L MgCl2, and 120 mmol/L NaCl. Nonspecific binding was determined in the presence of unlabelled raclopride (2 μmol/L).
The assays were performed in duplicate. They were initiated by adding, in the following order, 0.25 mL buffer, 0.25 [3H]raclopride (eight concentrations between 0.312 and 30 nmol/L) and 0.5 mL tissue homogenate. Incubation lasted 2 h at room temperature. Assays were terminated by immediate filtration using Whatman GFC filter presoaked in cold buffer containing 1% Polyethyleneimine (Sigma-Aldrich, France). The filters were washed two times with 4 mL of ice-cold buffer. The filters were then dissolved in 8 mL of liquid scintillation (Optiphase, Hisafe 2, PerkinElmer Life Science, France) and the radioactivity was counted 12 h later (radioactivity counter analyzer, Packard, France). Bmax and Kd values were determined by a Scatchard analysis (eight points).
Results
Probe Placements
All probes were correctly located in the aimed regions (striatum, lateral cerebellum, and the entorhinal cortex). As shown in Figure 3A, the trace resulting from the probe implantation was hardly detectable. Furthermore, the volume of detection of the probe, a sphere of 2.01 mm radius for 11C (Pain et al, 2000), corresponds to a major part of the striatum and encompasses the entire entorhinal cortex (Figure 3B).
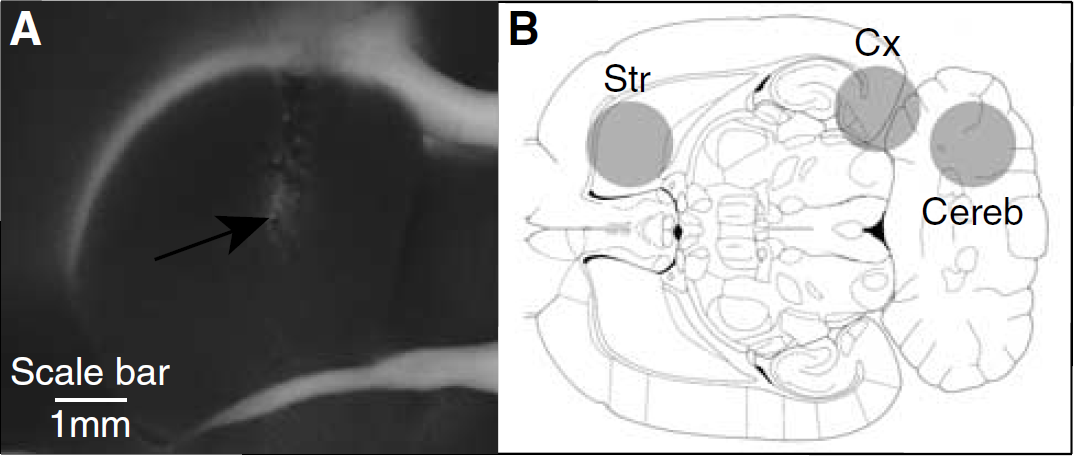
Probe location in the striatum and detection volume of the β-microprobe (90% of measured signal).
Catabolism of [11C]raclopride
Figure 1A shows that the fraction of nonmetabolised [11C]raclopride in the group of control rats (nontreated with cimetidine) decreases rapidly to reach a plateau around 20% by 40 mins after radiotracer injection. Pretreatment with cimetidine significantly increased the fraction of nonmetabolised [11C]raclopride to 55% at 40 mins. As observed in Figure 1A, increasing the injected mass by a second injection of [11C]raclopride did not modify the metabolism of the radiotracer.
Therefore, a pretreatment by cimetidine, 50 mins before the first injection of [11C]raclopride was used in experiments 2 and 3.
Moreover, given the small interindividual variations of these measurements, an exponential fit was performed and a standard curve was extrapolated up to 120 mins.
Time—Concentration Curves of [11C]raclopride in Striatum, Entorhinal Cortex, and Cerebellum
Figure 4 shows representative examples of [11C]raclopride binding kinetics in the striatum, the entorhinal cortex, and the cerebellum obtained with the multiinjection protocol. All kinetics are rather slow, compared with other experiments (Ginovart et al, 2004), a phenomenon related to the cimetidine pretreatment. The uptake of [11C]raclopride could be observed for at least 20 to 30 mins after each injection of labelled and unlabelled raclopride.
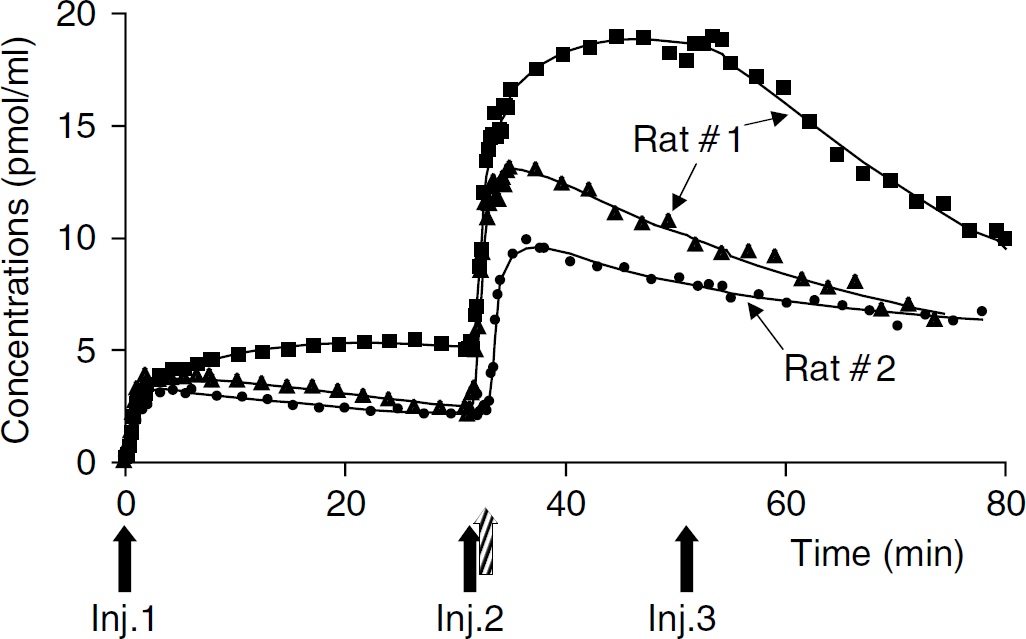
Representative [11C]raclopride time—activity curves recorded in various regions of the living rat brain (experiment 2). Solid lines correspond to the best fitted curve for data recorded in: striatum (squares), entorhinal cortex (triangles), and cerebellum (circles). Labelled and unlabelled doses of raclopride are injected subsequently (↑): a first injection of [11C]raclopride at tracer dose, followed 30 to 35 mins later by a second [11C]raclopride injection at low specific radioactivity and 52 mins later by a large amount of unlabelled raclopride. These data are from two different rats.
After the first injection (tracer dose), the radiotracer concentration in the striatum increased rapidly reaching a plateau, around 5 mins after [11C]raclopride injection. In contrast, the cerebellar time—concentration curve reached a maximum within the first 3 mins after the radioligand injection and then decreased slowly until the second [11C]raclopride injection. Cortical time—activity curve showed a shape comparable to that of the cerebellum increasing rapidly after radiotracer injection then slowly decreasing. At 30 mins, radioactive concentrations present in the entorhinal cortex and cerebellum were approximately 2.5 times smaller than that observed in the striatum.
The last injection of unlabelled ligand produced a displacement of the [11C]raclopride binding in the striatum, whereas cerebellar and cortical kinetics remained largely unaffected.
Identification of the Model Parameters
Striatal binding kinetics: In the striatum, the quality of the fits obtained with the model described in Figure 2A was satisfactory as it can be seen in the example of Figure 5 (best fit: solid line, experimental data: squares). Taking into account the percentage of radioligand reaching the detection volume of the probe (about 0.8%), [11C] raclopride occupied 5% to 10% to 60% to 80% of the D2 striatal available receptors, after the first (high specific radioactivity) and the second (low specific radioactivity) injections, respectively.
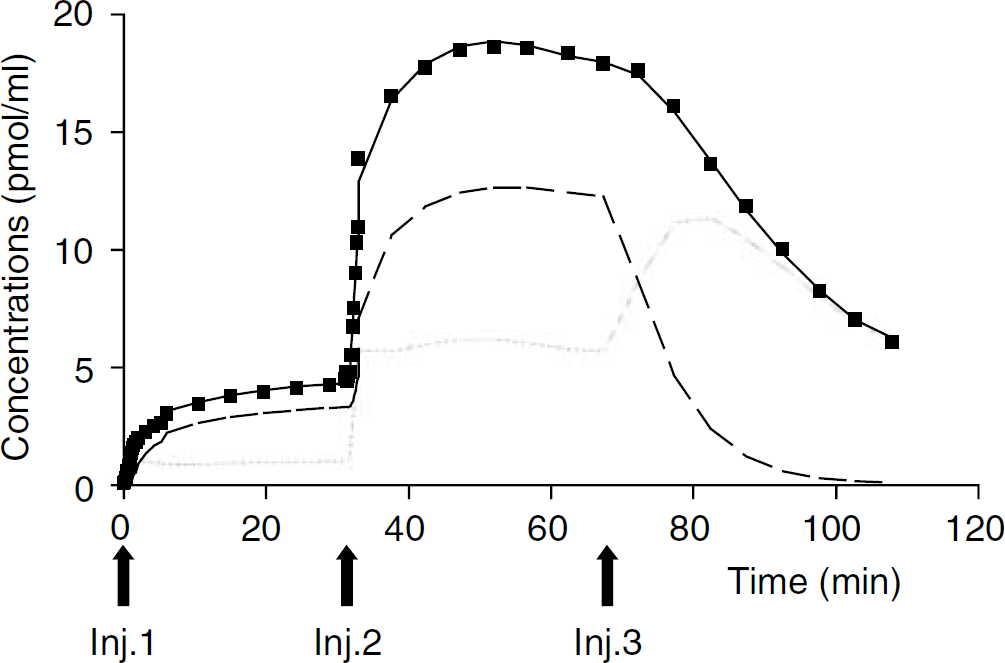
Striatal kinetics of [11C]raclopride (squares) and best fit (plain line) predicted by the three-compartment—six-parameter model of Figure 2A. Arrows point to the time of labelled and unlabelled raclopride injections (experiment 3). The figure displays the radioactive concentrations of [11C]raclopride predicted by the model for the free ligand compartment (dotted line) and the bound ligand compartment (dashed line). The vascular fraction is not represented.
Table 1 summarizes the values of the estimated parameters, two relative standard deviations (RSD = s.d./mean, expressed in percent) were derived: one on the studied population (RSDp), one on the estimators (RSDe).
Mean and standard deviations of model parameters in experiment 2
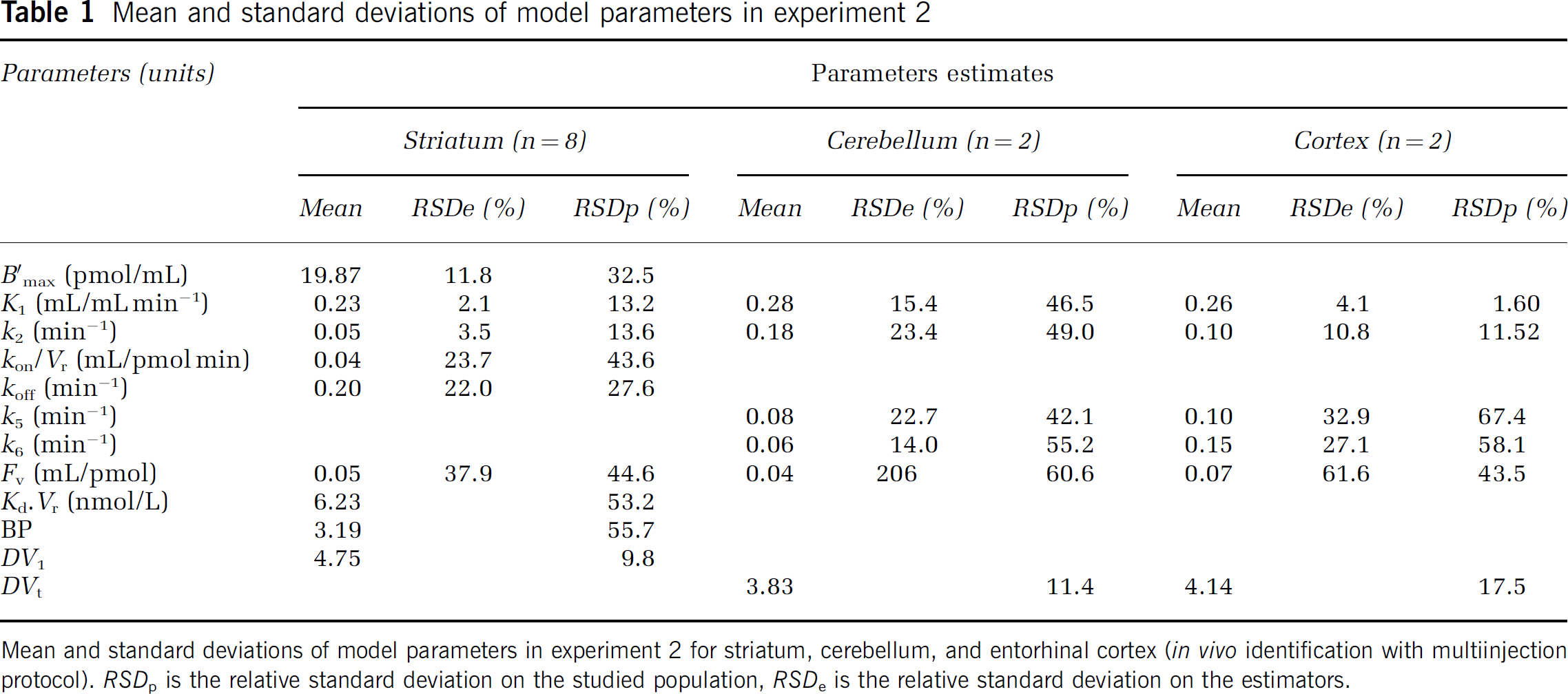
Mean and standard deviations of model parameters in experiment 2 for striatum, cerebellum, and entorhinal cortex (in vivo identification with multiinjection protocol). RSDp is the relative standard deviation on the studied population, RSDe is the relative standard deviation on the estimators.
Two additional parameters were calculated from the estimated parameters: the free volume of distribution DV1 = K1/k2, and the binding potential BP = Bmax/KdVr.
All the model parameters were always correctly identified (RSDe from 2% to 24%). One can notice that the mean percentage of covariance (RSDe) was lower than the interindividual variations (RSDp) for all parameters.
Cerebellar and cortical binding kinetics: In these two nonstriatal regions, we tested the model with one (free only) or two (free and non specific) tissular compartments. For all curves, the model including only one compartment was not sufficient to describe correctly the measured kinetics. Adding a nonspecific compartment (Figure 2B) led to significantly better fits. RSDe of the parameters were ranging from 4% and 10% (for K1 and k2, respectively) to 20% and 25% (for k5 and k6, respectively). In Table 1, the total volume of distribution was calculated as DVt = K*1(1 + k5/k6)/k2. In both structures, although parameters were poorly identified, the tissue volume of distribution was stable (RSDp = 11% and 17.5%).
Comparison Between In vivo and In vitro Data in the Same Striatum
The in vivo striatal D2 receptor density B'max, determined by the multiinjection modelling approach with [11C]raclopride was highly correlated with in vitro Bmax determined with [3H]raclopride in the same striata (Figure 6A; r2 = 0.91, P<0.0001). Similarly, a highly significant correlation was observed between in vivo KdVr and in vitro Kd (Figure 6B; r2 = 0.75; P<0.001). Average striatal in vivo B'max was 34.75 pmol/mL (RSDp = 35.9%) and KdVr was 9.15 nmol/L (RSDp = 31%). In vitro, mean Bmax and Kd values were 67.96 pmol/mL (RSDp = 34.7%) and 39.85 nmol/L (RSDp = 43.5%), respectively. The ratio in vitro Bmax/in vivo B'max was 1.94 and the ratio in vitro Kd/in vivo KdVr was 4.4.
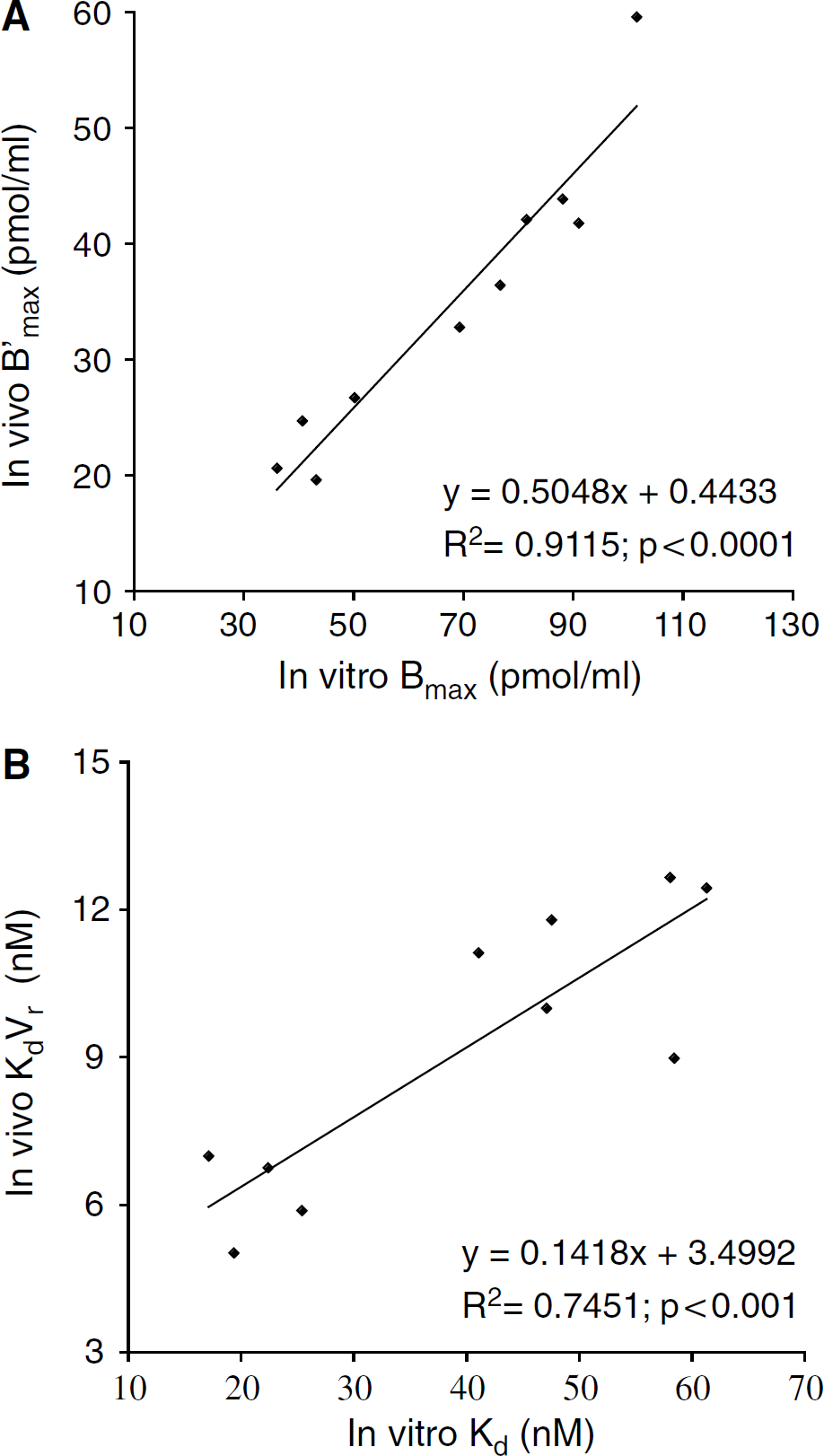
Relationships between D2-receptor density and affinity determined in the same rat striatal tissues by in vivo [11C]raclopride multiinjection protocol and in vitro [3H]raclopride binding assay.
Discussion
The aim of this study was to quantify in rodents, striatal D2 receptors specific binding in vivo and, if possible, to estimate receptor affinities and densities separately. To this aim a multiinjection approach had to be used to allow the identification of each of the six parameters (including B'max and KdVr) of a three-compartment ligand—receptor model. This method requires that the regional brain kinetics of the tracer can be determined at a sampling rate high enough to allow proper identification of the kinetics and accurate compartmental modelling. This prerequisite was fulfilled by the use of the β-microprobe, an implantable β-sensitive system, which proved to be capable of quantifying the radiotracer concentrations, locally, with a sensitivity and temporal resolution compatible with that needed by the multiinjection experimental protocol.
Cimetidine Pretreatment
Although the sensitivity of the probe was high, the combination of tracer decay and washout from the tissue resulted in a fast decrease of the count rate, especially during the displacement phase. Pretreatment with cimetidine, a cytochrome P450 inhibitor, reduced the catabolism of [11C]raclopride, without altering major physiologic parameters such pH, PCO2, PO2, and haematocrit. Interestingly, the cimetidine pretreatment significantly increased the signal-to-noise ratio of the tissue activity. This allowed a better characterisation of the radiotracer washout during the displacement phase, which appeared essential for accurate estimation of the Koff value (see below). Moreover, the average BP value calculated in this study (3.19, see Table 1) is very similar to the value obtained (2.71) without cimetidine pretreatment in a study performed with Sprague—Dawley rats (Ginovart et al, 2004). This confirms that cimetidine does not interfere with D2 dopamine receptors (Nowak, 1980).
Probe Implantation
Two kinds of effects may occur after probe implantation: the blood—brain barrier may be damaged, or there could be a variation of local extra—cellular dopamine concentration. A disruption of the blood—brain barrier would lead to very high values of K1 and k2 (transfer rates between vascular and the free compartments), a phenomenon that was never observed in these experiments. A study specifically addressed this point by performing microPET [18F]FDG and [11C]raclopride after a 500 μm micro-dialysis probe implantation (Schiffer et al, 2003). The authors reported modifications of [18F]FDG metabolism, but no effect on transport, nor on [11C]raclopride binding could be detected.
As probe implantation consequence is a large increase in dopamine concentration levels, the effect of shortening the delay between probe implantation and measurements should result in a decrease in the BP estimation. However, we found a BP mean value (BP = 3.19) that is higher than the value of 1.2 obtained in a noninvasive study with the RATPET (Houston et al, 2004).
The Multiinjection Protocol
The aim of the multiinjection modelling approach is to estimate all the model parameters from a single experiment. A difficulty of this approach for the study of brain dopamine receptors is that the various brain regions studied (striatum, entorhinal cortex, cerebellum) present very different receptor site concentrations. As the injected masses of [11C]raclopride were selected to occupy less than 5% to 10% of the striatal D2 receptors after the first injection (tracer injection), and up to 60% to 80% of the same receptors after the second injection, these doses lead to a complete saturation of the receptor sites in the cerebellar and cortical regions in which densities of D2 receptors represent, respectively, 1% and 10% of that of striatum (Martres et al, 1985). Thus, even after the first tracer injection, all receptors were fully occupied in these extrastriatal regions, which resulted in a lack of displacement after the second [11C]raclopride injection. As the slope of this displacement is used by the model to identify the apparent dissociation constant of the ligand with the receptors, this explain that the masses of [11C]raclopride injected in the present study could not allow an accurate estimation of specific binding parameters (B'max, KdVr) in these extrastriatal regions. Despite such limitations, the experimental conditions allowed us to measure the total volume of distribution of the radiotracer in the entorhinal cortex and in the cerebellum and to show, as expected, that the volume of distribution in these regions was close to that of the free compartment measured in the striatum.
Identification of Compartmental Model Parameters
Being able to run a multiinjection compartmental modelling in the living rat allowed us to gain access to each of the six parameters (B'max, K1, k2, kon/Vr, koff, Fv), which fully describe raclopride striatal kinetics in the living rat. One way to evaluate the reliability of parameter identification is to calculate the uncertainty (standard deviations) on the estimate of each parameter within each separate experiment. Such analysis showed that most parameters were correctly identified. K1 values were the best identified parameters with the smallest relative standard deviation (around 4%) whereas koff and KdVr values were identified with the largest relative standard deviation of 22 to 24%. B'max estimates were relatively well identified with intermediate relative standard deviations (around 12%).
As we mentioned above, a previous work was performed with [11C]raclopride on rodents (Ginovart et al, 2004). The authors used a reference-region quantification method (Lammertsma and Hume, 1996) with the cerebellum as the reference region. This method avoids arterial blood sampling, but only allows estimation of R1, k2, and BP. Binding potential is a valuable macro-parameter, however the purpose of our work was to quantify all the model parameters separately, including both Bmax and KdVr. These parameters cannot be estimated separately with the reference-region method, and the compartments kinetics cannot be derived as well. In the same study, the authors derived a koff value of 0.025 mins−1 from data measured in the striatum and cerebellum, with a double-injection protocol: tracer dose followed by a massive injection of unlabelled raclopride (10 mins later). They calculated koff as the exponential rate of the ‘specific’ kinetic calculated as ‘striatum – cerebellum’ (we derive the same exponential rate from our kinetics with the same method). However, Ginovart's method does not take into account the impact on the free compartment of a massive dissociation phenomenon, because of the displacement injection. This effect is nonnegligible, as the clearance from the free to the vascular compartment (k2) is rather slow and the free compartment concentration represents at least 30% of the total activity. In our identification procedure, forcing the koff value to 0.025 mins−1 resulted in a very strong underestimation of the free compartment (inconsistent with presaturation kinetics, data not shown) and poor quality fits. Ginovart's koff estimate was in agreement with the in vitro koff (0.024 min−1) previously reported by Kapur (Kapur and Seeman, 2000). However, we have to keep in mind that this study was performed at room temperature. Finally, the koff value found in our study (0.198 min−1) is comparable to the value obtained (0.137 min−1) in another study with [11C]raclopride in healthy humans (Gregoire et al, 2000).
Another way to assess the reproducibility of parameter estimates is to study the inter-individual variability through standard deviations on the studied population. In this respect, B'max and KdVr values were surprisingly estimated with the highest relative standard deviations (30% to 50%, respectively). Two phenomena could explain such a scattering of these estimators. First, this could reflect the various errors because of the deployment of a complex protocol such as probe misplacements or lack of standardisation in the anaesthetic protocol, which could greatly influence the radiotracer biodisponibility. Alternatively, actual interindividual differences could be responsible for this variability. Systematic postoperative histologic assessment of probe placement confirmed that all probes were correctly located in the target regions, ruling out surgical variability as a factor of data scattering.
Similarly, registration of various physiologic parameters (PO2, PCO2, rectal temperature, pH) at regular timepoints during the entire experiment confirmed the stability of each animal's physiologic status. Therefore, actual interindividual variability in D2-receptor densities in the studied rat population may have contributed to the large standard deviation on the B'max estimates observed here. Interestingly, in a previous study performed on 20 humans, Farde et al (1995) quantified both B'max and KdVr, using five separate PET acquisitions and a Scatchard-like approach in vivo. The relative standard deviations on these parameters were close to 25%, similar to the one reported here in the living rat striatum. In addition, the direct comparison between in vivo B'max and in vitro Bmax values performed in the present study confirms that large interindividual as well as intraindividual (see Table 2) variations in D2-receptor density accounts for the large standard deviation observed.
Comparison Between In Vivo and In Vitro D2-Receptor Density Estimates
The average density of receptor sites available for in vivo binding in the striatum was 34.75 pmol/mL (RSDp 35.9%). When comparing these in vivo estimates to in vitro values (67.96 pmol/mL, RSDp = 34.7%), determined on the very same striatal tissues using [3H]raclopride and classical in vitro binding techniques, a ratio of 1.94 was observed, suggesting that B'max values are systematically underestimated in vivo. It is interesting to note that (Ginovart et al, 2004) recently reported a similar ratio between [11C]raclopride radioactive concentrations measured in vivo by β-microprobe recordings and post mortem after striatal dissection. These authors explained such a ratio by a partial volume effect because of a detection volume of the probe (a sphere of about 4 mm diameter; Pain et al, 2000) exceeding the size of the striatum (AP: 2.5 mm; ML: 3 mm; DV: 4 mm) and resulting in a true underestimation of actual striatal radioactive concentrations. It is worth noting that a 50% underestimation of actual radioactive striatal concentrations was also observed under similar experimental conditions with the active stereoisomer of another dopamine receptor ligand but not with the nonactive stereoisomer of the same compound (Besret et al, unpublished data). Provided that one corrects in vivo B'max values for this partial volume effect, the strong correlation observed between in vivo and in vitro binding density validates our experimental approach and shows feasibility of accurately quantifying striatal D2-receptor density in vivo using a multiinjection approach.
Comparison Between In Vivo and In Vitro D2-Receptor Affinity Estimates
As for B'max values, a strong correlation was found between in vitro and in vivo estimates of receptor affinities (Kd versus KdVr). However, Kd values obtained in vitro in the same striatal tissues (average 41.80 nmol/L) were considerably higher compared with other in vitro binding data (between 1.2 and 1.9 nmol/L) previously published in rodents (Kohler et al, 1985; Sun et al, 2003). One explanation to such a discrepancy can be related to the use of cimetidine pretreatment to slow-down radiotracer catabolism in the living rat. Indeed, reducing metabolism of raclopride lead to a higher signal when injecting [11C]raclopride, but also reduces the washout rate of the unlabelled raclopride injected for displacement. Consequently, it is likely that an excess of unlabelled raclopride remained in the tissue at the moment of killing and thereafter in the striatal homogenates. In that case, a competition of unlabelled raclopride with the tritiated radiotracer should occur, resulting in a systematic bias on the estimation of Kd. To test this hypothesis, a specific study was performed out on one rat pretreated with cimetidine. At 2 h after cimetidine administration, the rat received an intravenous injection of 7 μmol of raclopride. After a survival time of 2 h to allow raclopride washout, the rat was sacrificed and an in vitro binding study was performed to measure Bmax and Kd. This Scatchard analysis yielded Bmax and Kd values within the range of previously published in vitro estimates (36.6 pmol/mL and 1.15 nmol/L, respectively).
As the delay between displacement and killing was always 75 mins, and the effect of cimetidine is very reproducible between animals (see study 1), it is very likely that the occupancy of D2 receptors by remaining raclopride at the time of killing was similar for every rat. Thus, the correlation between in vivo and in vitro measured affinities is shifted but still significant.
Conclusion
The present study shows that the β-Microprobe recordings combined with a multiinjection protocol of [11C]raclopride is a well-adapted tool for the in vivo quantification of D2-receptors density and affinity in the striatum of rats. Highly significant linear correlations were found between in vivo and in vitro estimates for D2-receptor density (B'max versus Bmax) and receptor affinity (KdVr versus Kd). The method allows accurate and separate measurements of D2-receptor affinity and density in living rats, within one experiment. The complete characterisation of the model should allow us to derive a single-injection protocol which aims at quantifying separately B'max and KdVr.
This approach can be used to address a wide range of issues relevant to in vivo mechanisms of receptor changes occurring in various psychiatric or neurodegenerative diseases and in system repair or protection in animal models of diseases involving the dopaminergic neurotransmission system.
Footnotes
Acknowledgements
The authors thank Dr Héric Valette and Lucette Garreau for their helpful comments and technical assistance.