
Abstract
Stroke patients have increased levels of endothelin-1 (ET-1), a strong vasoconstrictor, in their plasma or cerebrospinal fluid. Previously, we showed high level of ET-1 mRNA expression in astrocytes after hypoxia/ischemia. It is unclear whether the contribution of ET-1 induction in astrocytes is protective or destructive in cerebral ischemia. Here, we generated a transgenic mouse model that overexpress ET-1 in astrocytes (GET-1) using the glial fibrillary acidic protein promoter to examine the role of astrocytic ET-1 in ischemic stroke by challenging these mice with transient middle cerebral artery occlusion (MCAO). Under normal condition, GET-1 mice showed no abnormality in brain morphology, cerebrovasculature, absolute cerebral blood flow, blood-brain barrier (BBB) integrity, and mean arterial blood pressure. Yet, GET-1 mice subjected to transient MCAO showed more severe neurologic deficits and increased infarct, which were partially normalized by administration of ABT-627 (ETA antagonist) 5 mins after MCAO. In addition, GET-1 brains exhibited more Evans blue extravasation and showed decreased endothelial occludin expression after MCAO, correlating with higher brain water content and increased cerebral edema. Aquaporin 4 expression was also more pronounced in astrocytic end-feet on blood vessels in GET-1 ipsilateral brains. Our current data suggest that astrocytic ET-1 has deleterious effects on water homeostasis, cerebral edema and BBB integrity, which contribute to more severe ischemic brain injury.
Introduction
Ischemia and reperfusion injury causes blood-brain barrier (BBB) disruption, which accelerates development of abnormal vascular permeability and exacerbates postischemic edema (Cole et al, 1991; Yang and Betz, 1994). Blood-brain barrier is formed by endothelial cells of cerebral blood vessels under the influence of astrocytes (Abbott, 2002). Humoral agents released by astrocytes have been shown to increase BBB permeability, one of which is endothelin-1 (ET-1) (Abbott, 2002), suggesting a role of ET-1 in BBB disruption in brain injury. ET-1, a 21-amino-acid potent vasoconstrictor originally isolated from aortic endothelial cells (Yanagisawa et al, 1988), is expressed in neuronal groups and involved in central autonomic control (Kuwaki et al, 1997). The distribution of its receptors, ETA and ETB, in central nervous system (CNS) suggests its role in CNS functions. Indeed, ET-1 level was significantly elevated in cerebrospinal fluid and plasma of stroke patients (Volpe and Cosentina, 2000; Franceschini et al, 2001) and in rat brain tissue after cerebral ischemia (Barone et al, 1994; Bian et al, 1994), suggesting a role of ET-1 in pathogenesis of cerebral ischemic injury. For example, increased ET-1 level was observed in experimental stroke models such as 24-h permanent middle cerebral artery occlusion (MCAO) and 80-mins MCAO with 24-h reperfusion (Barone et al, 1994). In addition, levels of ETA and ETB receptors are upregulated after 2 h of MCAO with 46 h of reperfusion (Stenman et al, 2002), suggesting that the endothelin system is affected even at 2 days after 2 h of MCAO.
Moreover, intense ET-like immunoreactivity was detected in astrocytes of damaged brain areas under several CNS pathologic conditions (Kuwaki et al, 1997). Astrocytes in hypoxia/ischemia (H/I)-damaged region of neonatal mouse brain also expressed high level of ET-1 mRNA (Tsang et al, 2001), indicating that increased ET-like immunoreactivity in astrocytes seen in other studies was because of increased ET-1 gene transcription. However, whether increased ET-1 protects or hastens H/I-induced brain damage is unclear. Contradictory results were also obtained from in vivo studies on effects of ET receptor antagonists in ischemic brain injury. Some studies showed beneficial effects (Feuerstein et al, 1994; Barone et al, 1995, 2000; Patel et al, 1996; Tatlisumak et al, 1998; Dawson et al, 1999; Matsuo et al, 2001) while some showed no protection (McAuley et al, 1996; Patel and McCulloch, 1996; Bhardwaj et al, 2000).
Because the in vivo efficacy of ET receptor antagonists are not so clear, mice deficient in ET-1, ETA, or ETB receptor would be useful models for understanding the role of ET-1 in ischemic brain damage and BBB maintenance. Unfortunately, these mice have lethal phenotypes (Baynash et al, 1994; Kurihara et al, 1994; Clouthier et al, 1998). Nevertheless, ET-1 knockout mouse embryos have provided a source of ET-1-deficient astrocytes that are more sensitive to H/I stress, indicating that ET-1 is essential for their survival under H/I conditions (Ho et al, 2001).
To further understand the role of astrocytic ET-1 in ischemia-induced brain injury, BBB integrity and cerebral edema formation, we generated transgenic mice overexpressing ET-1 in astrocytes (GET-1) to exaggerate the effects of ET-1 during ischemia. As mentioned above, previous results suggest that the endothelin system is affected even at 2 days after 2 h of MCAO while infarct size could be reduced by administration of ETA receptor antagonist at similar time points (Matsuo et al, 2001). In addition, the findings that various agents can decrease cerebral infarct size after 2 h of MCAO with 22-h reperfusion (Endres et al, 1998; Ellsworth et al, 2003) suggest that such ischemia-induced brain injury can still be reversed. Cerebral edema is also present 24 h after 2 h MCAO (Yang et al, 1992). Moreover, transient MCAO with reperfusion is proposed to more closely mimic the clinical situation (Hunter et al, 1995). Therefore, GET-1 mice were subjected to similar transient MCAO model (Hara et al, 1996; Bonventre et al, 1997; Endres et al, 1997, Endres et al, 1998; Kondo et al, 1997; Asahi et al, 2001). Here, GET-1 mice showed more severe neurologic deficits and larger infarct size after transient MCAO. GET-1 mice also showed more BBB disruption with increased water accumulation and brain edema possibly because of elevated aquaporin (AQP)4 expression in astrocytic end-feet. Our results suggest that under ischemic condition, ET-1 overexpression in astrocytes has adverse effects on neurons.
Methods
Mice were housed under diurnal lighting condition and allowed free access to food and water. The protocol of this study was reviewed and approved by the Committee on the Use of Live Animals in Teaching and Research in The University of Hong Kong.
GFAP-ET-1 (GET-1) Transgenic Mice
These mice were generated using the 15-kb SfiI fragment of pGFAP-ET1 (pGET-1) plasmid (mouse ET-1 complementary DNA (cDNA) (Chan et al, 1995) with SV40 polyA inserted into exon 1 of modified mouse glial fibrillary acidic protein (GFAP) gene, pGFAP-445(-)) (Toggas et al, 1994)) (Figure 1A) for microinjection into pronuclei of F1 (CBA ϗ C57BL/6) mouse embryos. GET-1 transgenic mice were identified by PCR and Southern blots on genomic DNA extracted from the tail (data not shown). Mice used were age-matched nontransgenic (NTg) and homozygous male littermates unless otherwise stated and all procedures were performed blinded to the genotype.
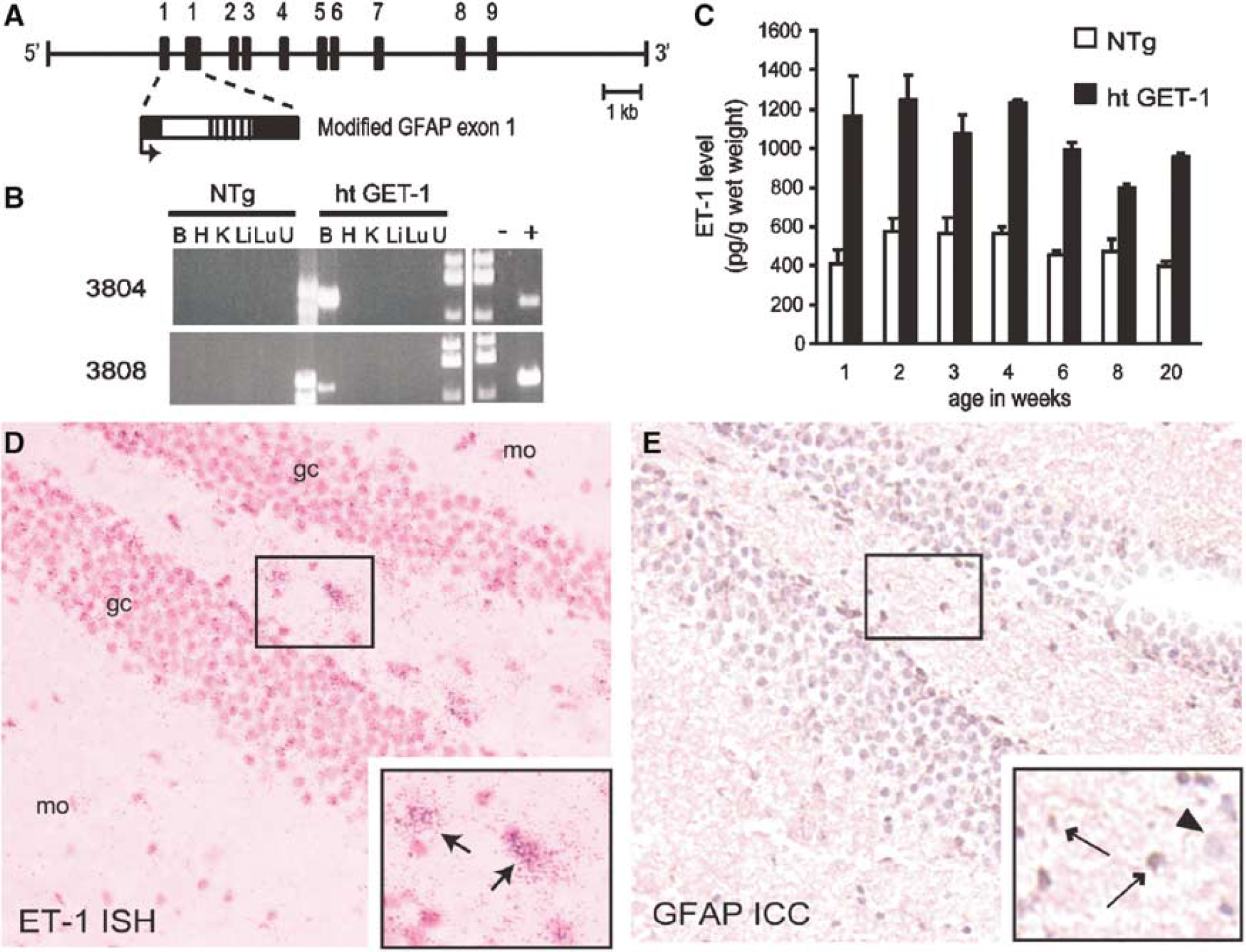
Brain-specific expression of transgenic endothelin-1 (ET-1) in GET-1 mice. (
RT-PCR Analysis
RT-PCR analysis was performed on total RNA isolated from various mouse tissues. Primers ET4U2 (5'-AGTGTGTCTACTTCTGCCACCTGGAC-3') and CAT1L (5'-GGCATCAGCACCTTGTCGCCTT-3') recognize a fragment from transgenic ET-1 mRNA (1.2 kb, 60°C annealing) while primers ET4U2 and ET8L (5'-TCAAGATTCAATC***TAACCTCTTCC-3') were used for PCR-amplifying endogenous ET-1 in separate reactions (55°C annealing, data not shown). Controls were rt-PCR products from C6 (astrocytic cell line transfected with pGFAP-ET-1, +; Figure 1B) or W28 (wildtype, –; Figure 1B) cells.
Quantitative Real-Time PCR
This technique was used to quantitate ET-1 mRNA level in ipsilateral cerebral hemisphere after MCAO using primers mET1-1252F (5'-CCTTTAATTATTGCCTCCCTGT***ATTC-3') and mET1-1331R (5'-TGTGAGGCAAGGATT***CAGTATAGG-3') and TaqMan probe mET1-1281T (5'-CCTCTCCGCTATCCGGGTTCCCA-3') (Primer Express software version 1.6, Applied Biosystems, Foster City, CA, USA) with TaqMan Universal PCR Master Mix (Roche, Branchburg, NJ, USA) and ABI PRISM 7700 Sequence
Endothelin-1 ELISA
For active ET-1 (not inactive precursor big ET-1), ET-1 ELISA was performed on whole-brain homogenate (Sakai et al, 1996) of 2-month-old mice. Briefly, whole brains that were frozen in liquid nitrogen and stored at –80°C was homogenized with a Polytron homogenizer for 2 × 60 sec in 10 vol of 1 mol/L glacial acetic acid containing 10 μg/mL pepstatin and immediately boiled for 10 mins. After being chilled, the homogenate was centrifuged at 20,000 g for 30 mins at 4°C and the supernatant was first concentrated by passing through C2 columns (Amersham, Piscataway, NJ, USA) and then assayed for ET-1 levels using the Biotrak ET-1 ELISA system (Amersham) according to the manufacturer's instructions.
In Situ Hybridization and Immunocytochemical Analyses
In situ hybridization (ISH) using a riboprobe hybridizing to both endogenous and transgenic ET-1 mRNA (Chan et al, 1995) was performed on cryostat sections of 2-month-old mouse brains (Tsang et al, 2001). For histology, 7-μm paraffin brain sections fixed with 4% paraformaldehyde were stained with hematoxylin and eosin (H&E) to assess the cytoarchitecture.
For immunohistochemical (ICC) analyses, brain sections were incubated with antibodies against GFAP (1:500, Z0334, DAKO, Glostrup, Denmark), occludin (1:50, Santa Cruz, CA, USA), and AQP4 (1:25, a generous gift of Dr Mark Knepper) (Terris et al, 1995). Signals were visualized by Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) with 3,3'-diaminobenzidine tetrahydrochloride (Zymed, South San Francisco, CA, USA). Control included omission of primary antibody on adjacent sections. For quantitative GFAP ICC analyses, digitized images of brain sections (~1.34 mm interaural line; Franklin and Paxinos, 1997) were collected from 3 mice of each genotype and the number of GFAP-positive astrocytes (with a center nucleus and stellate morphology) in the hippocampal area was counted.
Cerebrovasculature and Water Content Measurement
Cerebrovasculature (circle of Willis and its major branches) was evaluated by black ink injection into anesthetized mouse until it turned visibly black (Huang et al, 1994). Water content in the mouse brain tissue was calculated from measured wet and dry weight by the formula water content = 100%(wet weight-dry weight)/wet weight (Hara et al, 1996). For water content measurement under normal conditions, whole brains were used whereas cerebral hemispheres (ipsilateral and contralateral) were used in the determination of cerebral water content after transient MCAO in both NTg and GET-1 mice.
Mean Arterial Blood Pressure
Mean arterial blood pressure (mABP) was measured via cannulation of the right femoral artery (Chen et al, 1999) in halothane-anesthetized mice (see below) using a blood pressure transducer (MLT1050, AD Instruments, NSW, Australia) connected to PowerLab 8 s data acquisition system (AD Instruments) and analyzed by Chart for Windows v4.0.4 (AD Instruments).
Cerebral Blood Flow
Absolute cerebral blood flow (CBF) was measured in halothane-anesthetized mice (see below) by an indicator fractionation technique using N-isopropyl-[methyl, 1,3-14C]-p-iodoamphetamine (American Radiolabeled Chemicals Inc, St Louis, MO, USA) (Fujii et al, 1997). Relative regional CBF (rCBF) was monitored by laser Doppler flowmetry (Perimed, Järfälla, Sweden) using an optic fiber (with probe 418-2) placed directly on the skull (2 mm posterior and 6 mm lateral to bregma) and expressed as percentage relative to baseline values (Huang et al, 1994). Before each measurement, the probe was calibrated with a motility standard (Perimed).
Transient Focal Cerebral Ischemia and Drug Treatment
Transient focal cerebral ischemia was induced by intraluminal occlusion of the right middle cerebral artery as previously described (Hara et al, 1996; Bonventre et al, 1997; Endres et al, 1997; Kondo et al, 1997; Endres et al, 1998; Asahi et al, 2001). Here, adult mice (24 to 28 g) were subjected to MCAO by the filament method under gas anesthesia (2% halothane in 70% N2O/30% O2 for induction and 1% halothane in 70% N2O/30% O2 for maintenance) (Huang et al, 1994). Briefly, a nylon monofilament (Johnson & Johnson, Brussels, Belgium) coated with impression material (3 mol/L Dental Products, St Paul, MN, USA) was inserted into the right internal carotid artery to block the origin of right middle cerebral artery. Regional CBF was monitored during the whole surgical procedure to confirm appropriate suture placement while rectal temperature was maintained at 37°C±0.5°C with a temperature control system (FHC, Brunswick, ME, USA). Two hours later, the filament was pulled out to allow for reperfusion and the mouse was kept in an intensive care system (ThermoCare Inc., Incline Village, NV, USA) at 32°C for 4 h (Chen et al, 1999). Sham-operated animals underwent the same anesthesia and surgical procedures without MCAO. For drug treatment, GET-1 mice were administered with ABT-627 (10 mg/kg body weight, intraperitoneal, a generous gift from Dr Ruth Wu-Wong) or BQ788 (1 mg/kg body weight, intraperitoneal, Novabiochem, Germany) 5 mins after the onset of ischemia. Vehicle-treated mice were also included as controls (0.25 mol/L NaHCO3 for ABT-627 or saline for BQ788).
Neurologic Deficits, Infarct Size and Hemispheric Brain Swelling
Mice were evaluated for neurologic deficits 22 h after reperfusion (Huang et al, 1994) as follows: 0, no observable neurologic deficits (normal); 1, failure to extend opposite forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking and righting reflex (severe). The rater was naïve to the animal identity and to the treatment protocol.
Immediately after scoring of neurologic deficits, the mouse brain was cut into 2-mm coronal slices, stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma, St Louis, MO, USA) and fixed in 10% buffered formalin overnight. The posterior surface of each brain slice was photographed and analyzed using a digital image analysis system (NeuroLucida, MicroBrightfield Inc, Colchester, VT, USA). Infarct area and volume percentages were calculated using the indirect method while hemispheric brain swelling was assessed as follows: hemispheric brain swelling = 100%(ipsilateral volume-contralateral volume)/contralateral volume (Huang et al, 1994).
Evans Blue Extravasation
Right after reperfusion, 0.1 mL of 4% Evans blue (EB) (Sigma) in saline was injected intravenously into another group of NTg and GET-1 mice in separate transient MCAO experiments. Mice were killed by transcardial perfusion of heparinized saline (10 U/mL in saline) 22 h after reperfusion. Evans blue content was then assayed quantitatively (Kondo et al, 1997).
Western Blot Analyses
Separate transient MCAO experiment was performed to collect protein samples from NTg and GET-1 brains. Proteins were isolated from ipsilateral and contralateral cerebral hemispheres homogenized in lysis buffer (50 mmol/L Tris-HCL, pH 6.8, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.5% sodium deoxycholate, 0.5% NP-40, plus protease inhibitor cocktail) (Lo et al, 1994). Homogenate was centrifuged (3000 g, 5 mins, 4°C) and the resulting supernatant was further centrifuged (200,000 g, 30 mins, 4°C) to obtain tissue membrane fractions for AQP4 and AQP 9 (AQP9) analyses (Neely et al, 1999). Blots were incubated with antibodies against AQP4 (1:750, AB3594, Chemicon, Temecula, CA, USA), AQP9 (1:1,000, AB3079, Chemicon), GFAP (1:1,000), occludin (1:1,000, sc-5562, Santa Cruz, Santa Cruz, CA, USA), and α-tubulin (1:5,000, sc-5286, Santa Cruz). Signals were visualized by ECL (Amersham) and quantitated using PhosphoImager (Molecular Dynamics). Values for protein levels were given as percentage of NTg sham-operated group after normalization with individual α-tubulin levels for equal loading except in AQP4 and AQP9 Western analyses where Coomassie Blue staining was used to ensure equivalent sample applications.
Statistical Analyses
Data are presented as means±s.e.m. and statistical tests were performed using the GraphPad Prism software (San Diego, CA, USA) as specified. The neurologic score in individual experiments as well as infarct and hemispheric swelling data comparison between NTg and GET-1 mice were analyzed using the Mann-Whitney test. Unpaired t-test was used to analyze infarct and hemispheric swelling data obtained in the drug studies with GET-1 mice. All other measurements were analyzed statistically by one-way ANOVA followed by Bonferroni's post test. P<0.05 was considered statistically significant.
RESULTS
GET-1 Transgenic Mouse Brain Contained Higher Endothelin-1 mRNA and Peptide Levels
Five independent GET-1 mouse lines were established among which lines 3804 and 3808 showed brain-specific expression (Figure 1B) while line 3804 carried higher transgene level. In all subsequent experiments, GET-1 mice referred to homozygous 3804 transgenic mice. Endothelin-1 mRNA in 2-month-old GET-1 brains (n = 8) was ~18-fold of that in NTg littermates (n = 7) (P<0.0005, Mann-Whitney test; quantitative real-time rt-PCR). Meanwhile, total ET-1 peptide level in heterozygous GET-1 brains were consistently increased to ~2-fold until 20 weeks of age (Figure 1C), indicating that transgene was continually expressed throughout these periods. While endothelial ET-1 mRNA and very low level of astrocytic ET-1 mRNA were present in NTg brain as we previously reported (data not shown) (Tsang et al, 2001), increased ET-1 mRNA was detected in GET-1 astrocyte-like cells (Figure 1D inset, thin arrows). These cells were confirmed to be astrocytes (Figure 1E inset, thin arrows) by GFAP ICC on adjacent sections, indicating that increased ET-1 mRNA was present only in astrocytes of GET-1 brains.
GET-1 Mice Appeared Normal Under Physiological Condition
GET-1 mice appeared normal and healthy and displayed no obvious gross anatomical or behavioral abnormalities. Homozygous and heterozygous GET-1 transgenic mice displayed normal fertility rate with very low lethality (Table 1). The average body weight of NTg and GET-1 mice between postnatal 11 to 12 weeks did not differ (28.2±0.6 g, n = 24 and 27.8±0.4 g, n = 30, respectively, for male mice; 22.0±0.4 g, n = 15 and 21.4±0.3 g, n = 28, respectively, for female mice), suggesting no growth disadvantage for GET-1 mice.
GET-1 cerebrovasculature was also examined for abnormal vascular development. Inspection of ink-filled cerebrovasculature revealed no difference (data not shown). All major branches were present and there were no missing vessels. Distribution of middle cerebral artery territory appeared to be identical for NTg and GET-1 mice.
Besides the lack of structural differences in their brains, GET-1 mice have mABP similar to that of age-matched NTg mice (Table 2). Absolute CBF measurement in various brain regions also revealed no difference between GET-1 and NTg mice. The values did not vary between hemispheres or between groups (Table 2). In addition, GET-1 brain water content was not different (Table 2).
GET-1 brain sections were also stained with H&E or anti-GFAP antibodies to assess for abnormal cytoarchitecture. Microscopic examination revealed no major histological differences (Figure 2A-2D). Counting of GFAP-stained cells in hippocampal areas indicated that there was no significant change in their densities (114.4±5.7 cells/mm2 (left) and 101.6±1.7 cells/mm2 (right) in NTg mice; 109.4±2.7 cells/mm2 (left) and 117±9.4 cells/mm2 (right) in GET-1 mice). These cells also appeared normal with no indication of hypertrophy, suggesting that astrocytic ET-1 overexpression caused no gliosis (Figures 2E and 2F).
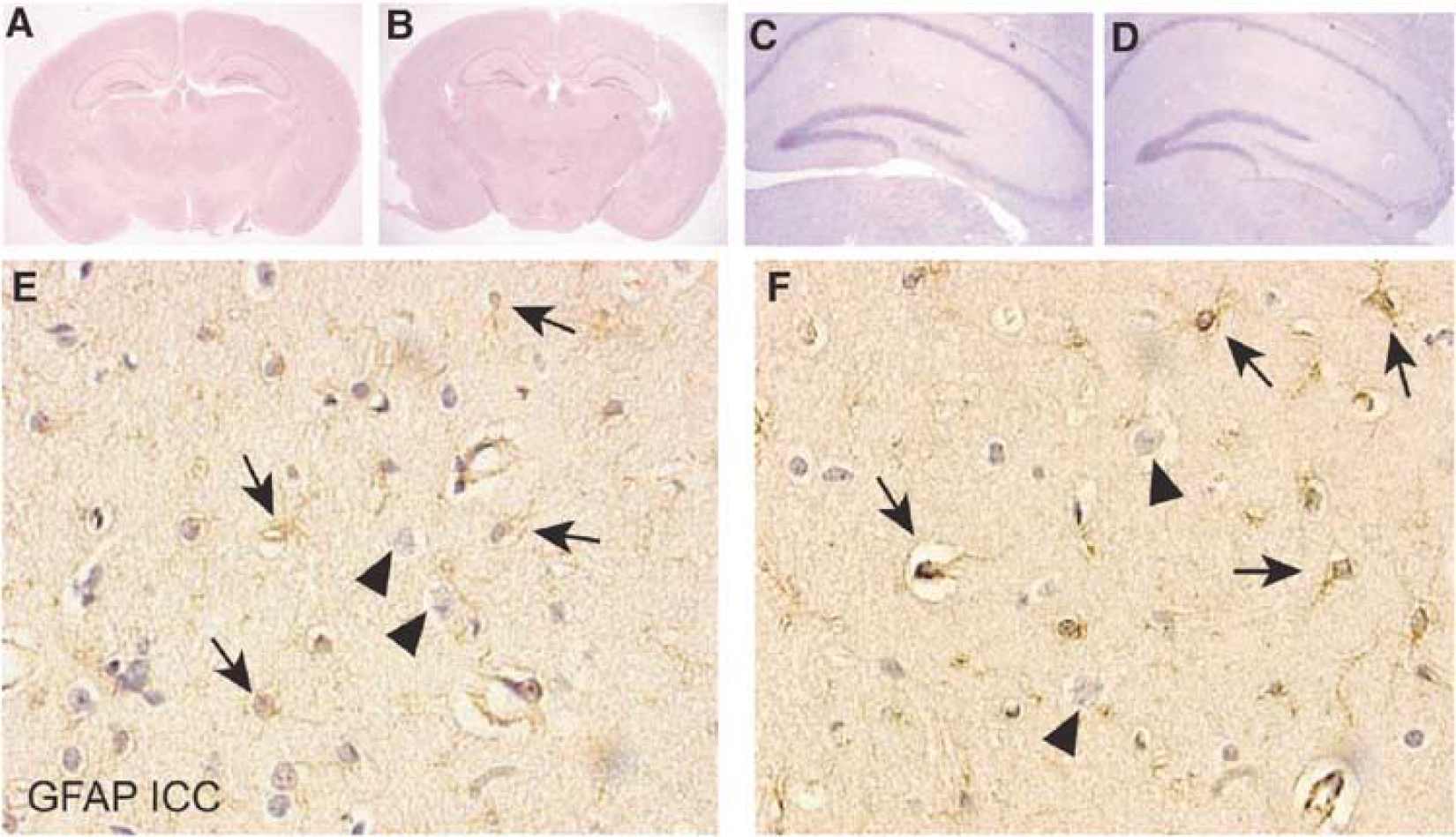
Cerebrovasculature and morphology of GET-1 mouse brains. (
Reproductive data of GET-1 mice
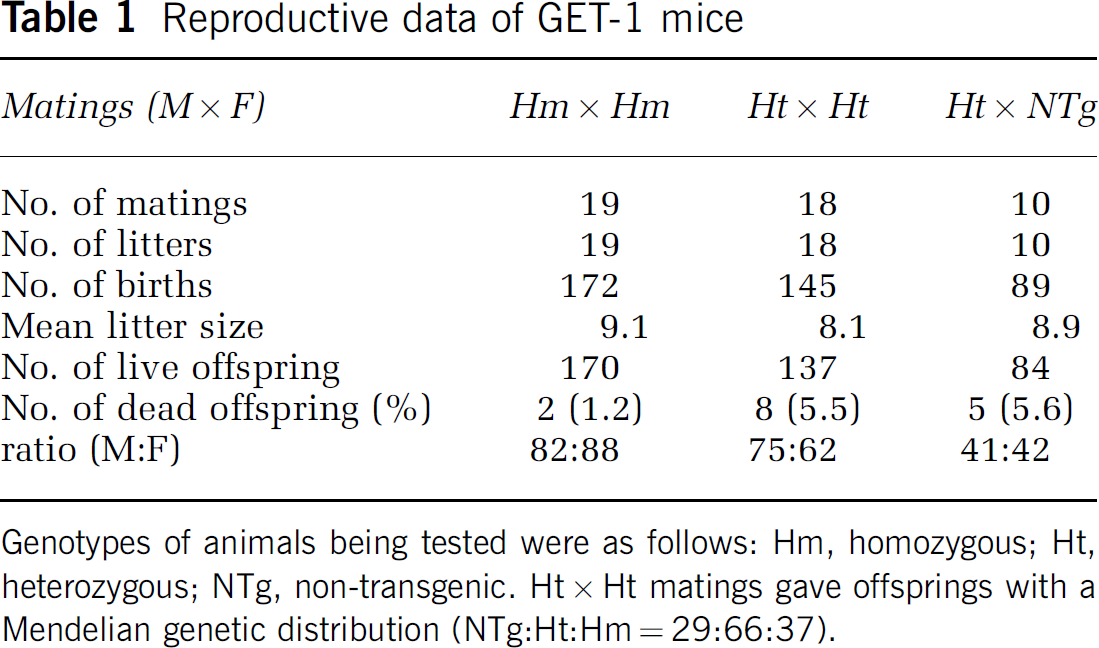
Genotypes of animals being tested were as follows: Hm, homozygous; Ht, heterozygous; NTg, non-transgenic. Ht ϗ Ht matings gave offsprings with a Mendelian genetic distribution (NTg:Ht:Hm = 29:66:37).
Physiologic data for NTg and GET-1 mice
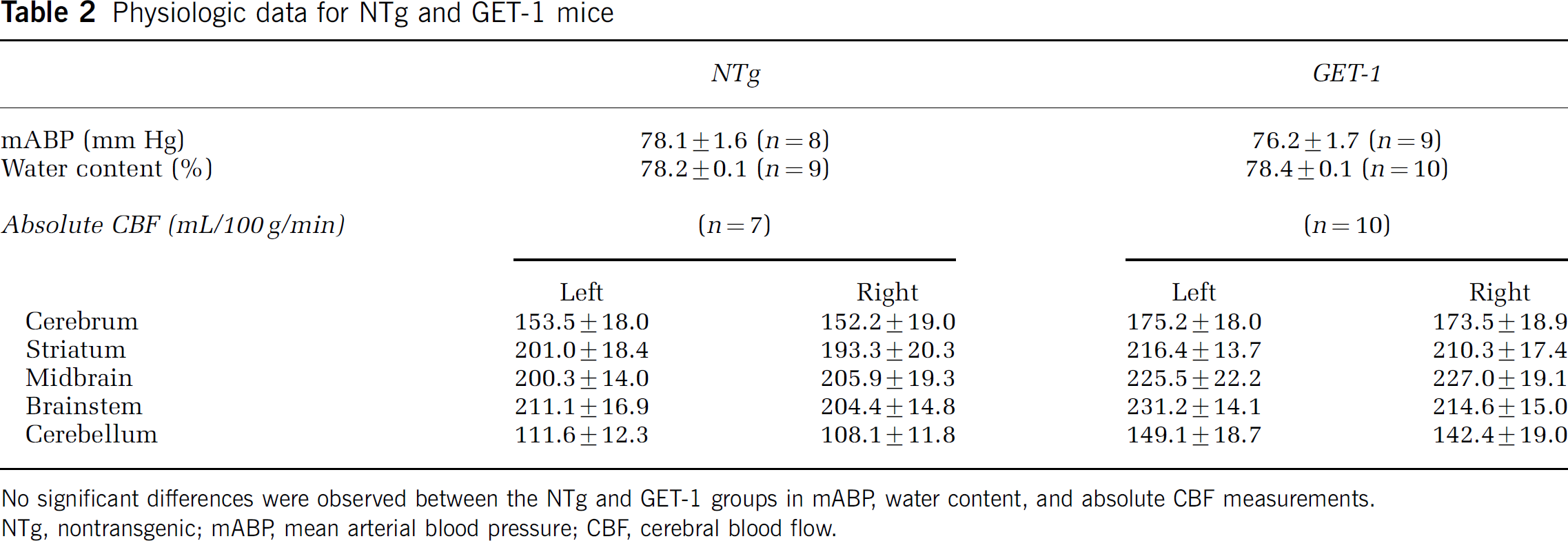
No significant differences were observed between the NTg and GET-1 groups in mABP, water content, and absolute CBF measurements.
NTg, nontransgenic; mABP mean arterial blood pressure; CBF, cerebral blood flow.
GET-1 Mice Displayed More Severe Neurologic Scores, Larger Infarct Size, Increased Brain Swelling and Increased Water Content after Middle Cerebral Artery Occlusion-Induced Cerebral Ischemia
To determine the effect of astrocytic ET-1 in ischemic stroke, mice were subjected to transient MCAO. Here, there was no significant difference in the mortality rate. Three out of 17 NTg mice died before neurologic scoring while 10 out of 12 GET-1 mice survived the transient MCAO (Table 3). During the experiment, rCBF and core body temperature did not differ (data not shown), indicating that the physiologic state before and after ischemia in these mice was similar. However, more severe neurologic impairments were observed in GET-1 mice 22 h after reperfusion (2.4±0.2, *P<0.002 by Mann-Whitney test, Table 4) whereas most NTg mice exhibited minor neurologic deficits (1.1±0.2, Table 4).
Mortality rate of NTg and GET-1 mice in various experiments after ischemia/reperfusion
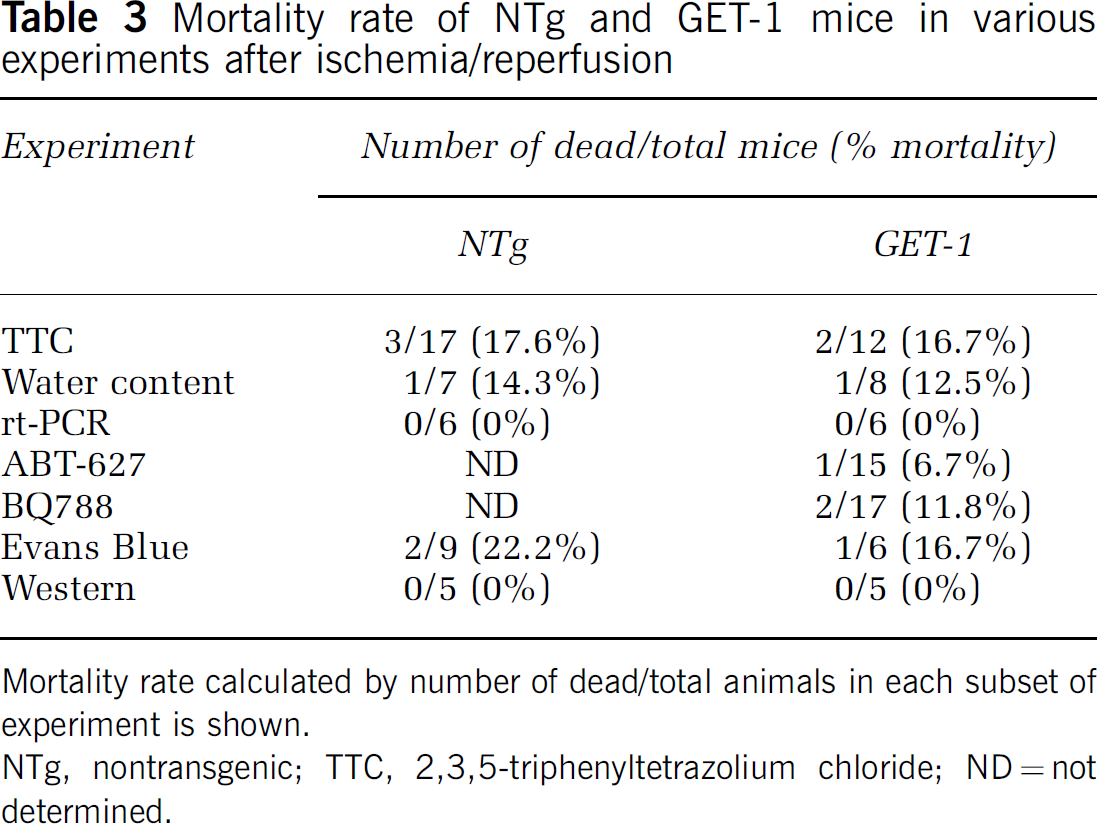
Mortality rate calculated by number of dead/total animals in each subset of experiment is shown.
NTg, nontransgenic; TTC, 2,3,5-triphenyltetrazolium chloride; ND = not determined.
More severe neurologic deficits in GET-1 mice after transient MCAO
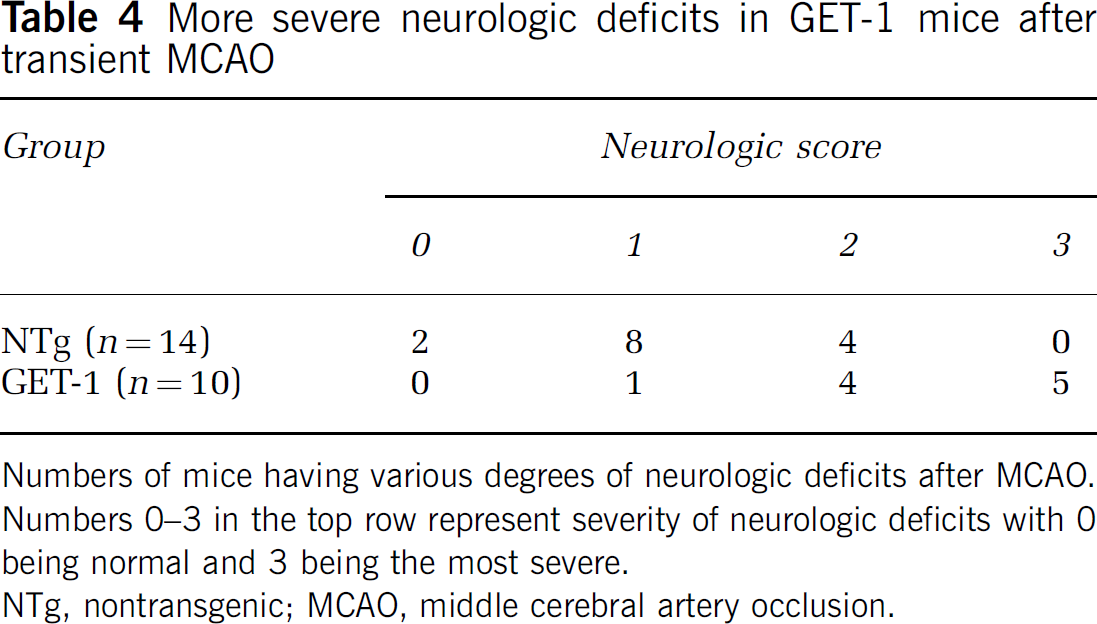
Numbers of mice having various degrees of neurologic deficits after MCAO.
Numbers 0–3 in the top row represent severity of neurologic deficits with 0 being normal and 3 being the most severe.
NTg, nontransgenic; MCAO, middle cerebral artery occlusion.
Moreover, TTC-staining analyses showed that infarct area % in brain slices #3 and #4 was significantly larger in GET-1 mice (Figures 3A and 3B). Similarly, infarct volume % in GET-1 mice was also significantly larger (Figure 3C) after correction for brain swelling while sham-operated mice did not display any infarct (data not shown). Moreover, significantly larger ipsilateral hemispheric swelling was observed in GET-1 mice (Figure 3D). In light of this, brain water content was also determined in a separate group of animals in another transient MCAO experiment. Here, one out of seven NTg mice died before neurologic scoring while all GET-1 mice survived the transient MCAO (Table 3). While water content in sham-operated brains and contralateral hemispheres of NTg and GET-1 mice were similar, increased water content was observed in both NTg and GET-1 ipsilateral hemispheres after transient MCAO (Figure 3E). More importantly, ipsilateral hemispheres of GET-1 mouse showed significantly higher water content, in agreement with the finding that GET-1 mice displayed more ipsilateral hemispheric swelling.
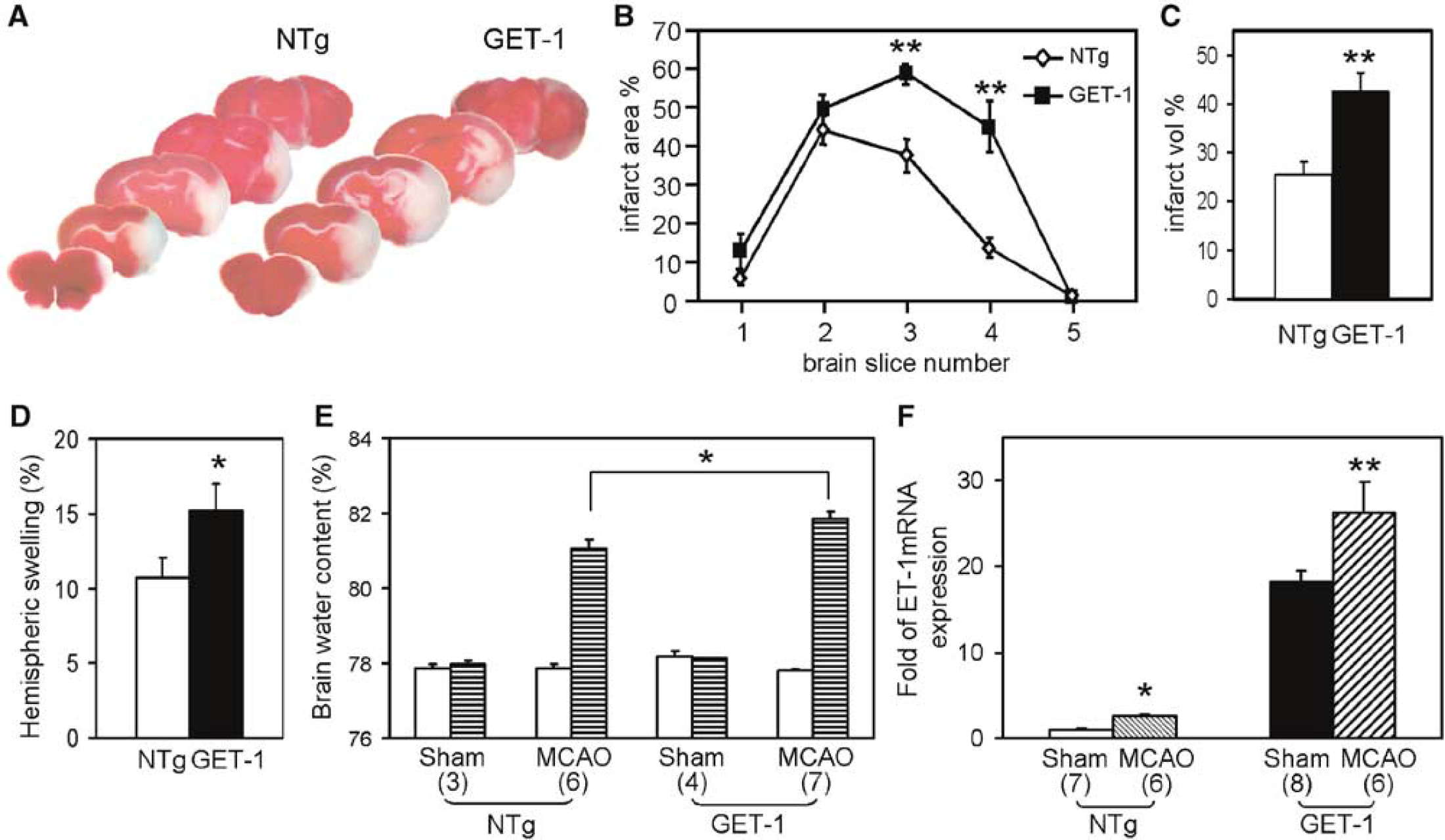
Greater infarct size and increased water content in GET-1 brains after transient middle cerebral artery occlusion (MCAO). (
Endothelin-1 mRNA Level Remained Higher in GET-1 Brains After Transient Middle Cerebral Artery Occlusion
Because ET-1 synthesis is regulated at the transcription level (Kuwaki et al, 1997) and ET-1 expression in normal mouse astrocytes was induced after H/I (Tsang et al, 2001), total (endogenous and transgenic) ET-1 mRNA levels after transient MCAO were measured by quantitative real-time PCR (Figure 3F). The mortality rate in both NTg and GET-1 animals in this subset of experiment was 0% after transient MCAO (Table 3). In NTg ipsilateral hemisphere, ET-1 mRNA level was increased to ~3-fold of that in NTg sham-operated group. More importantly, ET-1 mRNA levels in GET-1 ipsilateral brains further increased to ~25-fold, which was still higher than that in sham-operated GET-1 brains (~16-fold), indicating that ET-1 transgene contributed to significantly higher ET-1 mRNA level after ischemic brain injury.
Severity of Infarct, Hemispheric Swelling, and Ipsilateral Hemispheric Water Content Correlated Positively with Neurological Scores after Transient Middle Cerebral Artery Occlusion
Linear regression analysis showed a positive correlation between infarct volume % and hemispheric swelling (R2 = 0.64). Neurologic scores in individual mouse also correlated positively with infarct volume % (R2 = 0.65), hemispheric swelling (R2 = 0.67), and ipsilateral hemispheric water content (R2 = 0.65). Most importantly, the individual data plots showed the differences in infarct size, hemispheric swelling, ipsilateral hemispheric water content, and neurologic scores between NTg and GET-1 mice (data not shown).
ABT-627 But Not BQ788 Improved Neurologic Deficits and Reduced Infarct Size After Transient Middle Cerebral Artery Occlusion in GET-1 Mice
To determine if ETA receptor is involved, ABT-627 was administered 5 mins after the onset of ischemia because ET-1 level was significantly elevated in rat brain tissue after focal cerebral ischemia (Barone et al, 1994; Bian et al, 1994) while ET-1 expression is induced in astrocytes soon after H/I (Ho et al, 2001). ABT-627 is the active (+)-enantiomer of the competitive ETA-selective antagonist A-127722 that attenuated ischemic lesion size and cerebral edema after MCAO (Tatlisumak et al, 1998). ABT-627 inhibits binding to ETA and ETB receptors (K i = 0.034 nmol/L and 63 nmol/L, respectively) and potently blocks ETA receptor-mediated vasoconstriction (Jarvis et al, 2000). Because systemic administration of ABT-627 at 10 mg/kg can exert a significant antinociceptive effect (Jarvis et al, 2000) while oral administration at 10 mg/kg was protective against ischemic acute renal failure in rats (Kuro et al, 2000), ABT-627 treatment at 10 mg/kg was chosen in this experiment to study the involvement of ETA receptor in cerebral ischemia/reperfusion in GET-1 mice. ABT-627-treated GET-1 in mice showed a pronounced reduction in neurologic scores (Figure 4A) with generally smaller infarct size, reducing infarct volume by ~35% (Figure 4B). Despite these improvements, ABT-627 treatment had no significant effects on hemispheric swelling although there was a trend of decrease in hemispheric swelling (Figure 4C).
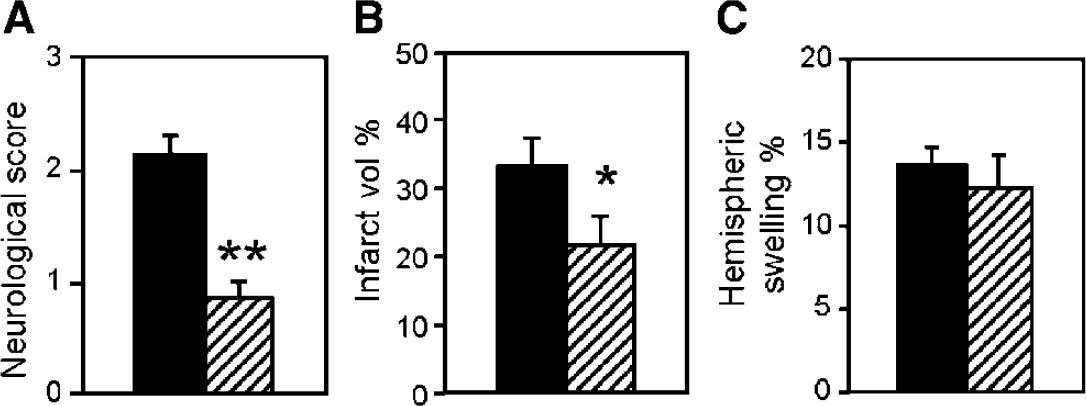
Improved neurologic deficits and reduced infarct size in GET-1 mice treated with ABT-627. (
On the other hand, the involvement of ETB receptor was also determined using BQ788, an ETB-specific antagonist. Pharmacologic characterization of this selective ETB antagonist showed that 1 mg/kg was an effective dose to block the effects elicited by ET-1 administration (Ishikawa et al, 1994). Here, BQ788 administration (1 mg/kg) did not induce any changes in the neurologic score, infarct size, or hemispheric swelling of GET-1 mice (data not shown).
Blood-Brain Barrier Disruption was Exacerbated in GET-1 Mice After Transient Middle Cerebral Artery Occlusion with Decreased Occludin Levels
Another experiment was performed to determine BBB integrity in NTg and GET-1 mice after MCAO. Here, two out of nine NTg mice died before neurologic scoring while one out of six GET-1 mice survived the transient MCAO (Table 3). At 22 h after MCAO and reperfusion, there was insignificant, low background level of EB extravasation in contralateral hemispheres of all experimental mice (Figure 5A). The amount of EB leakage was increased in NTg ipsilateral hemispheres, consistent with previous result that EB extravasation was increased after focal ischemia (Kondo et al, 1997). Most importantly, EB leakage was significantly elevated in GET-1 ipsilateral hemispheres (Figure 5A), indicating increased BBB permeability and therefore more BBB breakdown in GET-1 brains after MCAO.
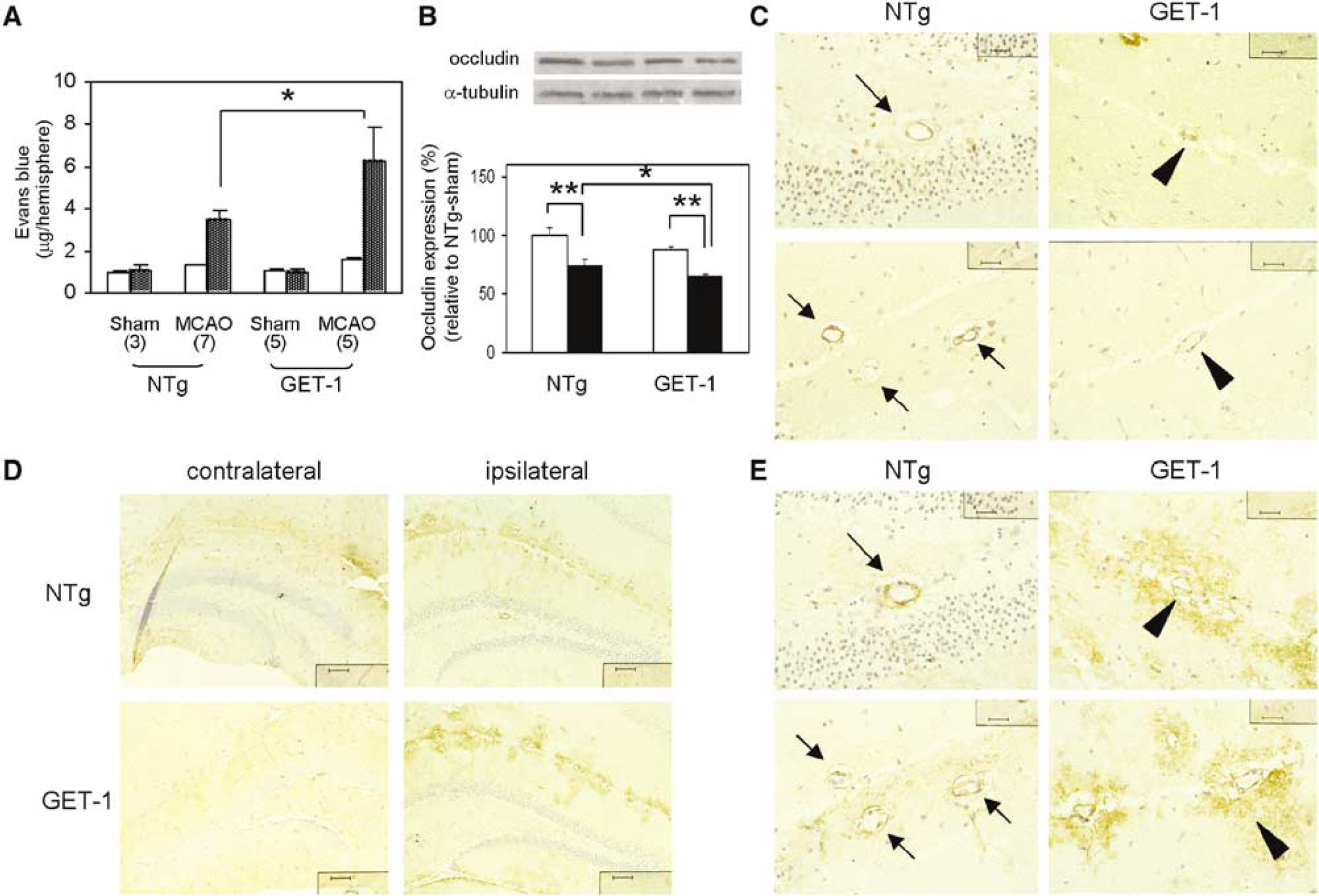
Increased blood-brain barrier (BBB) breakdown in GET-1 mice after transient middle cerebral artery occlusion (MCAO). (
The level of occludin, a protein associated with functional expression of tight junction in BBB, was also determined by Western blots. Occludin levels were similar in both sham-operated NTg and GET-1 brains (Figure 5B). In NTg ischemic hemisphere, occludin level was decreased while an even larger decrease was observed in ischemic hemisphere of GET-1 mice (Figure 5B), again suggesting increased BBB breakdown in GET-1 mice that was consistent with the EB extravasation results.
In line with the Western data, ICC results also showed downregulation of occludin in ipsilateral cerebral vessels in both NTg and GET-1 brains whereas the extent of decrease was more pronounced in GET-1 brains. In ipsilateral NTg hemisphere, occludin staining could still be seen quite prominently lining cerebral endothelial cells (Figure 5C, arrows). However, occludin staining was much weaker and more diffuse in GET-1 cerebral vessels (Figure 5C, arrow heads).
Changes in Expressions of Aquaporin 4 and Aquaporin 9 After Middle Cerebral Artery Occlusion
Because GET-1 brains showed increased hemispheric swelling and water content after transient MCAO, expression of AQP4, a water channel protein responsible for modulating water transport in the brain (Badaut et al, 2002), was also determined by Western blots. Similar to previous findings, AQP4 is present as different isoforms (32, 34, 36, and 38 kD) in mouse brains when resolved by urea-containing SDS-PAGE and probed with a C-terminal antibody (Neely et al, 1999). Therefore, intensities of all four bands were quantitated and added together to yield total AQP4 levels. Here, both sham-operated NTg and GET-1 ipsilateral hemispheres gave similar AQP4 levels (Figure 6B), suggesting GET-1 brains were not different from NTg brains under normal conditions, in agreement with data obtained from water content and BBB studies. Upon transient MCAO, total AQP4 level had no significant change in NTg brains despite the presence of brain edema (Figure 6B). More surprisingly, total AQP4 level instead dropped in spite of the increased cerebral edema in GET-1 brains (Figure 6B).
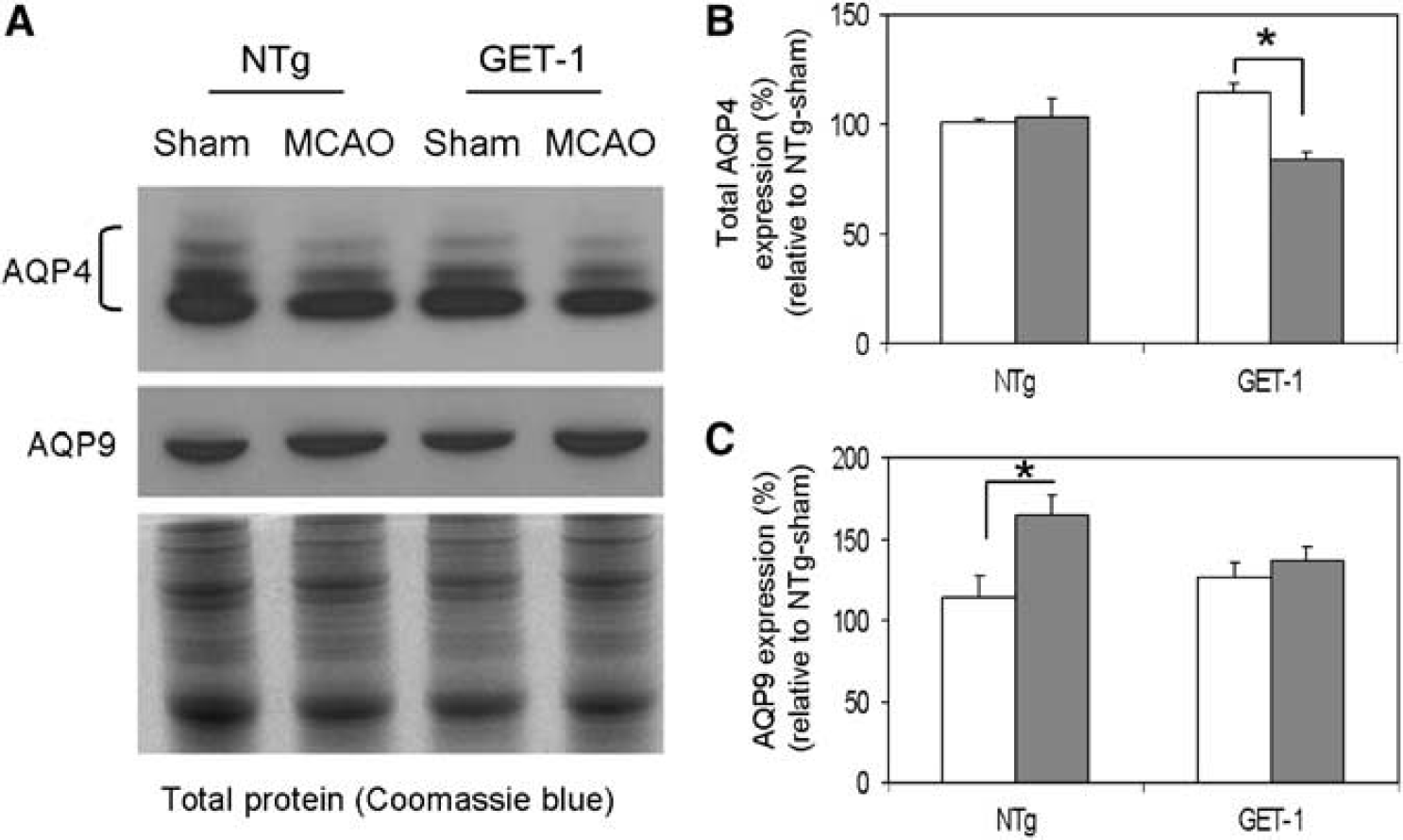
Western blots data. (
The expression of AQP9, another member of the aquaporin family whose expression is found in astrocytic processes in mouse brains (Badaut et al, 2001), was also examined. Western blot analyses showed that there was no significant difference in AQP9 expression between sham-operated NTg and GET-1 brains (Figure 6C). Upon transient MCAO, AQP9 content in ipsilateral cerebral hemisphere was highly increased in NTg mice (Figure 6C). Yet, there was not any significant change in AQP9 content in GET-1 ipsilateral cerebral hemisphere after transient MCAO (Figure 6C).
Increased Aquaporin 4 Staining in GET-1 Astrocytic End-Feet After Middle Cerebral Artery Occlusion
To further examine the expression of AQP4 in the brain after transient MCAO despite the unexpected Western data, AQP4 cellular localization was determined by ICC. Aquaporin 4 staining in contralateral hippocampus of both NTg and GET-1 brains after transient MCAO was localized in astrocytic end-feet in contact with cerebral vessels thereby displaying their outlines (Figure 5D, contralateral), as previously reported (Badaut et al, 2002). However, AQP4 immunolabeling was nearly abolished in the ischemic core and peri-infarct border in both NTg and GET-1 brains (data not shown), similar to previous reports (Amiry-Moghaddam et al, 2003; Badaut et al, 2001). Near the ipsilateral hippocampal areas, AQP4 staining surrounding cerebrovessels became less polarized and more patchy when compared with that in the contralateral side (Figure 5D). The outlines of cerebral vessels were not distinct. More importantly, intensity of AQP4 staining in these areas was much increased in GET-1 brains (Figure 5D). At higher magnification, little AQP4 immunostaining was associated with peri-vascular processes facing the endothelial cells, suggesting loss of AQP4 pool, which is similar to previous observations (Amiry-Moghaddam et al, 2003). However, increased AQP4 staining was very pronounced and diffuse with loss of polarization in GET-1 swollen astrocytic processes (Figure 5E, arrowhead), suggesting upregulation of AQP4 in astrocytic end-feet after ischemic brain injury. In addition, it is clear that occludin signals could be found inside cerebral vessels lining endothelial cells whereas AQP4 signals were localized in astrocytic processes surrounding the vessels in the brain parenchyma (compare Figures 5C and 5E).
DISCUSSION
After H/I brain injury, ET-1 level is increased in astrocytes as well as vascular endothelial cells (Tsang et al, 2001). Previously, the role of endothelial ET-1 and exogenous ET-1 application in cerebral ischemic damage and their effect on BBB integrity during ischemia were reported (Volpe and Cosentina, 2000). However, the contribution of astrocytic ET-1 to ischemic brain injury is not clearly understood. So far, there is no report of suitable models to address the contribution of ET-1 produced by endothelial cells or astrocytes to cerebral ischemia. Here, we report for the first time the effect of ET-1 produced by astrocytes on BBB integrity and brain injury during cerebral ischemia/reperfusion. Despite increased cerebral ET-1 levels, GET-1 mice showed no obvious evidence of abnormal brain development under normal condition. Their cerebrovasculature was indistinguishable from that of NTg mice. Interestingly, overexpression of this potent vasoconstrictor did not affect absolute CBF under normal condition although intracerebral injection of ET-1 has been used to induce focal cerebral ischemia (Hunter et al, 1995). Indeed, it has been shown that ET-1 does not contribute significantly to cerebrovascular tone under normal condition (Patel et al, 1996). Previous studies also showed that endothelin antagonists had no significant effects on cerebral arterial or pial arteriolar caliber and did not alter CBF in vivo (Foley et al, 1994; Patel et al, 1995). Therefore, under normal condition it is possible that the constitutively overexpressed ET-1 peptide levels in GET-1 mice did not bring about any changes in CBF. Furthermore, no increase in mABP was observed in GET-1 mice under normal condition. This can be explained by our EB experiments that under normal condition, astrocytic ET-1 does not affect BBB integrity and therefore it is likely that astrocytic ET-1 does not cross the BBB to affect systemic blood pressure. However, it is also possible that ET-1 transgenic mice have increased nitric oxide in their brains because ET-1 may stimulate nitric oxide synthesis (Kobari et al, 1994) as a compensatory mechanism for ET-1 overexpression under normal condition.
Although GET-1 brains appeared normal at both macroscopic and microscopic levels under normal condition, GET-1 mice were more susceptible to ischemic brain damage induced by transient MCAO. In addition, GET-1 mice showed increased EB extravasation and decreased occludin levels indicating increased BBB permeability and therefore more BBB breakdown after transient MCAO. Recently, it has been shown that ET-1 released by astrocytes can increase BBB permeability (Abbott, 2002). The further increase in ET-1 mRNA level after ischemic brain injury in GET-1 brain may contribute to the significantly increased BBB permeability, which agrees well with the observations that GET-1 mice displayed more brain swelling, increased water content and therefore increased edema after transient MCAO. Because the degree of edema seems to correspond with the severity of ischemia; that is, the more severe the ischemia, the greater the breakdown of BBB and edema formation (Ayata and Ropper, 2002), this may explain why GET-1 mice have larger infarct size and more severe neurologic deficits after transient MCAO. Our results suggest that an increase in BBB breakdown in GET-1 mice after ischemic brain injury results in cerebral edema and brain swelling, which may in turn lead to larger infarct and more severe neurologic deficits. More importantly, our results are the first to provide clear in vivo evidence that astrocytic ET-1 has a significant pathophysiologic role in cerebral ischemia/reperfusion.
Results of our antagonist studies showed that pathways elicited by ET-1 via ETA but not ETB are involved in cerebral ischemia/reperfusion injury, in agreement with other studies (Dawson et al, 1999; Matsuo et al, 2001). ABT-627 administration can normalize the vascular effects of ET-1 because ET-1 is known to induce vasoconstriction of cerebral vessels resulting in decreased cerebral blood flow and neuronal damage (Macrae et al, 1993; Willette et al, 1990). It was able to greatly improve neurologic impairment and partially reduce infract size in GET-1 mice to a level comparable to those of the NTg mice. However, it did not significantly reduce brain swelling contributed by astrocytic ET-1 in GET-1 mice although there was a slight trend of decrease in brain swelling after ABT-627 treatment. This finding is in agreement with a previous report where MCAO-induced ischemic lesion but not edema was reduced by ETA antagonists (Dawson et al, 1999) although there is a contradicting finding that an ETA antagonist can reduce brain edema after MCAO (Matsuo et al, 2001). It is possible that BBB breakdown and edema-induced ischemic injury because of astrocytic ET-1 may involve other ET-1-mediated secondary mechanisms (Bentzer et al, 2002). One possibility is that increased ET-1 level in the brain after MCAO can alter gap junctional permeability among astrocytes (Blomstrand et al, 1999) which in turn affect BBB maintenance and astrocytic edema resulting in brain edema. Another explanation for the lack of effect on brain swelling would be that other compensatory agents such as nitric oxide may act on the cerebral microvasculature or other cells in the CNS.
We also looked into the possibility of AQP4 involvement in astrocytic ET-1-induced edema and BBB breakdown after transient MCAO. In rodents, AQP4 is the principal astrocyte water channel (Solenov et al, 2004). Astrocytic expression of AQP4, which plays a key role in cerebral water homeostasis (Badaut et al, 2002), is upregulated in experimental models of brain injury and swelling (Papadopoulos et al, 2002). Indeed, AQP4 expression correlates with BBB disruption and astrocytic hypertrophy (Vizuete et al, 1999) while increased AQP4 transcripts parallels brain edema after MCAO (Taniguchi et al, 2000). Previous studies also showed that deletion of AQP4 resulted in improved neurologic outcome and partially protected AQP4-deficient mice from brain swelling in response to ischemic stroke (Manley et al, 2000) while astrocytes isolated from these mice showed reduced osmotic water permeability (Solenov et al, 2004), providing direct evidence for a role of AQP4 in brain edema formation. Our studies showing increased AQP4 staining in astrocytic end-feet associated with worse neurologic deficits, increased hemispheric swelling, and increased water content in GET-1 brains after transient MCAO agreed well with previous observations. Similarly, our data that increased AQP4 immunostaining in astrocytic end-feet in GET-1 brains with increased BBB breakdown also agreed with the suggestion that BBB disruption appeared to be the most efficient inductor of AQP4 in hypertrophic astrocytes (Vizuete et al, 1999). Yet, AQP4-mediated transcellular water movement is also crucial for fluid clearance in vasogenic edema (Papadopoulos et al, 2004). Therefore, increased AQP4 staining around cerebrovessels in GET-1 brains indicating high AQP4 content also suggested a compensatoy response to edema formation (Papadopoulos et al, 2004), highlighting the need for increased edema fluid clearance from the brain parenchyma in GET-1 mice.
However, at the protein level, total AQP4 level remained unchanged in NTg brains while it actually dropped in GET-1 brains with increased AQP4 staining after transient MCAO. A mismatch between AQP4 immunostaining and Western blotting data has also been observed in dystrophin null mice in which reduction in AQP labeling is not associated with any changes in brain AQP4 abundance (Vajda et al, 2002). This difference was suggested to be related to translocation of AQP4 distribution (Vajda et al, 2002). Whether similar or even opposite events happen in GET-1 mice after transient MCAO is unclear. Actually, not much molecular-level information is available regarding potential regulators of AQP4 expression. It has been suggested that polarized expression of AQP4 in astrocytes may be regulated by factors released by neurons and/or endothelial cells (Papadopoulos et al, 2002). Whether increased astrocytic ET-1 can influence AQP4 expression which in turn results in more water transport activity and therefore edema formation after ischemic brain injury remains to be clarified.
Besides AQP4, AQP9 has also been implicated in the regulation of postischemic edema (Badaut et al, 2001). AQP 9 is an aquaglyceroporin that is permeated by water and glycerol (Agre and Kozono, 2003) and is recently shown to be expressed in mouse brain (Badaut et al, 2001). Moreover, AQP9 immunostaining is increased in reactive astrocytes after ischemia/reperfusion (Badaut et al, 2001). This is potentially beneficial in lactate acidosis during ischemia because AQP9 enables astrocytes to take up excess lactate and therefore favors lactate clearing from the extracellular space although this will in turn lead to water influx into the astrocytes (Badaut et al, 2001). Similar to previous reports, our Western data also showed that AQP9 level is increased in NTg ipsilateral cerebral hemisphere after transient MCAO. Nonetheless, no such change was observed in GET-1 brains that showed higher water content after transient MCAO. This may suggest that GET-1 brains with overexpression of ET-1 in astrocytes failed to increase its AQP9 protein expression to cope with lactate acidosis induced by ischemia. Because no aquaporin has been identified in neurons (Agre and Kozono, 2003), this suggest that lactate buffering capacity in GET-1 astrocytes may be dampened and these astrocytes may not be able to handle ischemia-induced lactate accumulation in the brain parenchyma, which can lead to more edema after ischemic stroke.
Although the involvement of ET-1 in water balance in the kidney has been shown (Wong et al, 2003; Sonntag et al, 2004), our study is the first report to show a role of ET-1 in brain water balance and aquaporin expression. Here, we showed that transgenic mice overexpressing ET-1 in the astrocytes exhibited no major abnormal phenotypes under normal condition. However, they were more susceptible to focal cerebral ischemic injury, displaying further BBB breakdown, increased brain edema, and water content with increased AQP4 expression leading to larger infarct size and more severe neurologic deficits. Results from ET antagonist, AQP4, and AQP9 studies suggest that a combination of the use of ETA antagonist as well as pharmacological manipulations of AQP4 and AQP9 would potentially be one of the optimal therapeutic strategy in stroke. Our model combined genetic and pharmacological approaches to further dissect the cell-type specific, that is, astrocytic, ET-1 contribution to BBB dysfunction, brain water accumulation, and cerebral ischemia/reperfusion injury.
Footnotes
Acknowledgements
The authors thank Marcella Ma for performing microinjection experiments, Brian Lam for his assistance in absolute CBF measurement and Michael Koon for helping in mABP measurements. The authors also thank Dr Ruth Wu-Wong (Abbott Laboratory) for providing ABT-627, Dr Mark Knepper (NIH) for providing AQP4 antibody and Professor KF So (Department of Anatomy, The University of Hong Kong) for the use of Microscope-assisted Counting Set-up and the NeuroLucida software.