
Abstract
The present study was designed to elucidate the effects of the chemokine monocyte chemoattractant protein (MCP-1) on blood–brain barrier (BBB) permeability. Experiments were conducted under in vitro conditions (coculture of brain endothelial cells and astrocytes) to study the cellular effects of MCP-1 and under in vivo conditions (intracerebral and intracerebroventricular administration of MCP-1) to study the potential contribution of MCP-1 to BBB disruption in vivo. Our results showed that MCP-1 induces a significant increase in the BBB permeability surface area product for fluorescein isothiocyanate (FITC)-albumin under in vivo conditions, particularly during prolonged (3 or 7 days) exposure (0.096±0.008 versus 0.031±0.005 μL/g min in controls at 3 days, P<0.001). Monocyte chemoattractant protein-1 also enhanced (17-fold compared with control) the permeability of the in vitro BBB (coculture) model. At the cellular level, MCP-1 causes alteration of tight junction (TJ) proteins in endothelial cells (redistribution of TJ proteins determined by Western blotting and loss of immunostaining for occludin, claudin-5, ZO-1, ZO-2). Monocyte chemoattractant protein-1-induced alterations in BBB permeability are mostly realized through the CCR2 receptor. Absence of CCR2 diminishes any effect of MCP-1 on BBB permeability in vitro and in vivo. The permeability surface area product for FITC-albumin after 3 days exposure to MCP-1 was 0.096±0.006 and 0.032±0.007 μL/g min, in CCR2+/+ and CCR2−/− mice, respectively (P<0.001). Monocytes/macrophages also participate in MCP-1-induced alterations in BBB permeability in vivo. Monocytes/macrophages depletion (by clodronate liposomes) reduced the effect of MCP-1 on BBB permeability in vivo ∼2 fold. Our results suggest that, besides its main function of recruiting leukocytes at sites of inflammation, MCP-1 also plays a role in ‘opening’ the BBB.
Introduction
Blood–brain barrier (BBB) disruption, with subsequent increased vascular permeability and leukocyte migration, is associated with numerous diseases and conditions that affect central nervous system (CNS) such as multiple sclerosis, HIV encephalomyelitis, Alzheimer disease, trauma, and stroke (Halliday et al, 2000; Becker, 2001; Langford and Masliah, 2001). A variety of the factors can alter BBB permeability. For example, histamine and substance P may induce rapid and transient opening of the BBB, while thrombin and vascular endothelial growth factor (VEGF) are implicated in more prolonged opening (van Hinsbergh and van Nieuw Amerongen, 2002). Depletion of energy metabolism, mobilization of intracellular Ca2+, generation of reactive oxygen species, and activation of matrix metalloproteinases are also associated with alterations in BBB permeability (Rosenberg et al, 1998; Ye et al, 1999; Lum and Roebuck, 2001). However, increased microvascular permeability during inflammatory responses is closely correlated with the expression of proinflammatory mediators (Stanimirovic and Satoh, 2000; Petty and Lo, 2002). While BBB disruption may lead to the entry of leukocytes into brain, some recent studies indicate that migration of leukocytes may contribute to the persistence of increased vascular permeability (Merrill and Murphy, 1997; Couraud, 1998). How leukocyte migration may regulate vascular permeability is still under investigation, but some data indicate that the signaling molecules involved in regulating migration, such as cytokines, adhesion molecules, and chemokines, could have a prominent role (Merrill and Murphy, 1997; Stanimirovic and Satoh, 2000; Dietrich, 2002).
Chemokines are proinflammatory mediators mainly involved in the selective driving of leukocytes into brain parenchyma. High perivascular chemokine expression is found in many pathological settings accompanied by inflammation, providing a chemoattractant gradient for leukocyte influx (Murphy, 1994; Rollins, 1997). However, chemokines may also be involved in endothelial cell activation (upregulation of adhesion molecules) and regulation of endothelial permeability (Matsumoto et al, 1997; Weber, 2003). One of the most commonly expressed chemokines in the CNS during inflammation is monocyte chemoattractant protein-1 (MCP-1, CCL2). MCP-1 is a member of the CC subfamily of chemokines. There is strong evidence that MCP-1 is involved in the recruitment of monocytes/macrophages and activated lymphocytes into the brain during neuropathological states (Menicken et al, 1999). Monocyte chemoattractant protein-1 is mostly expressed in perivascular space and brain parenchyma during CNS inflammation but it is also present in CSF in several CNS inflammatory states (stroke, meningitis, multiple sclerosis) (Mastroianni et al, 1998; Losy and Zaremba, 2001; Sindern et al, 2001; Chen et al, 2003; Sorensen et al, 2004). Besides its role as a chemoattractant, some recent studies have shown that MCP-1 may also regulate brain endothelial permeability in vitro by altering tight junction (TJ) proteins, and modulate the expression of endothelial adhesion molecules and leukocyte integrins as well as cytokine production during CNS inflammation (Tekstra et al, 1999; Stamatovic et al, 2003).
The present study further assesses the ability of MCP-1 to regulate BBB permeability. To fully understand its effects two model (in vitro and in vivo) systems were used. The in vitro studies used co-cultures of astrocytes and endothelial cells in a Transwell chamber system (in vitro model of BBB). In the in vivo (mouse) studies, murine recombinant MCP-1 was administered by intracerebral (IC) or intracerebroventricular (ICV) injection. Intracerebral injections were used to simulate situations in which MCP-1 is expressed in perivascular space and brain parenchyma. Intracerebroventricular administration was used to study situations characterized by MCP-1 presence in cerebrospinal fluid (CSF).
Materials and methods
All procedures were performed in strict accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Michigan.
Brain Endothelial Cell Culture
Mouse brain microvascular endothelial cells (mBMEC) were prepared using a previously described protocol (Stamatovic et al, 2003). Briefly, 4 to 6-week-old wild-type (WT) on CD-1 or 50:50 C57BL/6 × 129Sv background and CCR2−/− mice on 50:50 C57BL/6 × 129Sv background (generous gift from Dr Moore Department of Internal Medicine, and Dr Kunkel Department of Pathology, University of Michigan, Ann Arbor) were euthanized by decapitation under isoflurane anesthesia. The brains were removed and the cerebral cortices devoid of white matter and leptomeniges were minced in Hanks balanced salt solution (HBSS, Invitrogen Corp., Carlsbad, CA, USA) and homogenized gently in Dounce type homogenizer. The homogenates were suspended in 18% Dextran solution (Dextran 60–90,000 USB, Cleveland, OH, USA) and centrifuged to remove the myelin. The supernatants were carefully aspirated, and the pellets were resuspended in HBSS and layered over a preformed Percoll gradient (Percoll, Pharmacia, Peapack, NJ, USA) and centrifuged at 1661g for 11 mins. After centrifugation, the top layer containing the microvessels was collected, washed once with HBSS, and then digested in HBSS solution containing 1 mg/mL collagenase/dispase (Roche, Indianapolis, IN, USA), 10 U/μl DNAase I (Sigma Chemical, St Louis, MO, USA) and 1 μg/ml Nα-p-tosyl-L-lysine chloromethyl ketone (TLCK, Sigma Chemical, St Louis, MO, USA) for 40 mins at 37°C. Mouse brain microvascular endothelial cells were cultured in Dulbecco's modified Eagles medium (DMEM, Invitrogen Corp, Carlsbad, CA, USA) supplemented with 10% inactivated fetal calf serum, 2.5 μg/mL heparin (Sigma Chemical, St Louis, MO, USA), 20 mmol/L HEPES, 2 mmol/L glutamine, 1 × antibiotic/antimycotic (all purchased from Invitrogen Corp, Carlsbad, CA, USA), endothelial cell growth supplement (BD Bioscience, San Jose, CA, USA) and grown in six-well plates coated with collagen type IV (BD Bioscience, San Jose, CA, USA). Application of this protocol typically produces primary endothelial cell cultures that are approximately 99% pure (as determined by immunocytochemistry, anti-PECAM-1 antibody BD Bioscience, San Jose, CA, USA).
Astrocyte Cell Culture
Cerebral astrocyte cultures were prepared from 1-day-old mice according to the method of Hertz, with minor modifications (Hertz et al, 1982; Andjelkovic et al, 1999a). The mice were either CD-1 strain or CCR2−/− and CCR2+, both on a 50:50 C57BL/6 × 129Sv background. After dissection of cerebral cortices and trypsin digestion (45 mins at 37°C in HBSS, 1 × trypsin (Invitrogen Corp., CA, USA) and 10 U/μl DNAse I (Sigma Chemical, St Louis, MO, USA). Cells were washed in HBSS, resuspended in complete astrocyte media (DMEM, 10% inactivated FBS, 1 × glutamine, 1 × antibiotic/antimycotic (A/A), (Invitrogen Corp, Carlsbad, CA, USA)) and seeded in 2.5 × 107 per 75 cm2 tissue culture flask. Monolayers were grown under an atmosphere of 5% CO2/95% air at 37°C. Two weeks after the initial plating, flasks of cells were shaken at 0.2g for 2 h at 4°C. After this time, the supernatant, containing mainly microglia, was removed. Cultures were then shaken for an additional 18 h at 37°C. The resulting supernatant, which contained predominantly neurons was again removed. After this time, cultures were approximately 99% positive for GFAP by immunocytochemistry. First or second passage was used in the experiments.
In Vitro Model of the Blood–Brain Barrier. CoCulture of Astrocytes and Mouse Brain Microvascular Endothelial Cells
For coculturing, the cells were plated on polyester membranes with 0.4 μm pores and an insert diameter of 12 mm (Transwell, Costar Corning Inc., Corning, NY, USA). Mouse astrocytes (1 × 105) passage 1–2 were seeded on the upper surface of the membrane. After 6 h, when cells were attached, inserts were turned over and placed in a 12-well culture plate. Mouse brain microvascular endothelial cells (densities of 2.5 × 105/cm2) were then seeded on the inner, cell-free membrane side previously treated with collagen IV (BD Bioscience, San Jose, CA, USA). Cocultures were maintained at 37°C under normoxic conditions of 95% air/5% CO2. Three to four days after initial coculturing with astrocytes, the monolayer of mBMEC had transendothelial electrical resistances (TEERs) of ∼400 Ω cm2. Cocultured cells were treated with MCP-1 added into the lower and/or upper compartment of the Transwell dual chamber system at a concentration of 100 nmol/L. In a previous study, we found that this concentration had a prolonged effect on brain endothelial cell barrier function but did not have any toxic effect on brain endothelial cells as assayed by cell death (Stamatovic et al, 2003). In the current study, we also found that this concentration of MCP-1 also had no toxic effect on astrocytes (evaluated by Live/Dead Viability/Cytotoxicity Assay Kit, Molecular Probes Inc, Eugene, OR, USA).
Intracerebroventricular and Intracerebral Injection of Recombinant Murine Monocyte Chemoattractant Protein-1
In vivo experiments used either CD-1 mice or CCR2+/+ and CCR2−/− mice both on a 50:50 C57BL/6 × 129Sv background. The mice, 8 to 10 weeks old, were anesthetized with 4% chloral hydrate (400 mg/kg, intraperitoneal injection) and placed in a stereotactic frame (David Kopf Instruments, Tujunga, CA, USA). A burr hole was drilled in paracranium and infusion was made via a 26-gauge cannula attached to a 10-μL Hamilton syringe (volume controlled by MicroSyringe pump controller, World Precision Instruments Sarasota, FL, USA). The stereotaxic coordinates for IC injection (right brain parenchyma) were: −2.5 mm posterior to bregma, 1.8 mm lateral from midline, and 2.5 mm in depth from the skull surface. For ICV injection (right ventricle), the coordinates were: −0.4 mm posterior to bregma, 1 mm lateral from midline, and 3.5 mm in depth from the skull surface. Recombinant MCP-1 (PreproTech, Rocky Hill, NJ, USA) dissolved in 5 μL saline solution for a total dose of 5 to 25 μg or the same volume of saline was delivered over 1 mins. After injection, the cannula was held in place for an additional 2 mins before retraction. The burr hole was then sealed with bone wax and the animal allowed to recover. The effect of MCP-1 on the BBB permeability was tested 6, 12, 24, and 48 h after MCP-1 injection.
For continuous MCP-1 administration, Alzet osmotic minipumps designed to deliver 1 μL/h for 3 days or 0.5 μL/h for 7 days were used (Durect Corporation, Cupertino, CA, USA). The osmotic minipumps delivered either murine MCP-1 (5 μg/μL) or PBS via cannulas localized at the same stereotaxic coordinates as used for IC and ICV injections.
Brain Water Content
Brain tissue water content was measured in whole brain by the wet/dry method. Briefly, mice were killed at the end of the experiment by decapitation under deep isoflurane anesthesia. The brain was quickly removed, weighed wet, oven-dried at 100°C for 48 h, and reweighed. The brain water content (%) was then calculated as ((wet weight−dry weight)/wet weight) × 100%.
Fluorescent Angiography: Quantification of Fluorescein Isothiocyanate-Albumin Leakage
Fluorescent angiography was performed, according to the Cavaglia's method with minor modifications (Cavaglia et al, 2001). Wild-type (CCR2+/+) and CCR2−/− mice on 50:50 C57BL/6 × 129Sv background or CD-1 mice, between 6 and 8 weeks of age were anesthetized with intraperitoneal injection of 40 mg/kg pentobarbital. A catheter was placed in a femoral vein for injection of 10 mL/kg of FITC-albumin, (MW 69 kDa; Sigma, St Louis, MO, USA). The FITC-albumin was dissolved in phosphate buffered saline (PBS) in concentration of 10 mg/mL and infused slowly over 5 mins. To avoid systematic blood pressure elevation, the same volume of blood was withdrawn from a catheterized femoral artery. At 20 mins after injection, the animal was decapitated and the brain removed. Brains were fixed for 18 h in 4% paraformaldehyde, cryoprotected with sequential immersions in 10% and 20% sucrose solutions, and then cut into 50-μm-thick coronal sections with a freezing microtome. Sections were mounted onto microscope glass slides, coverslipped with Vectashield mounting medium (Vector Lab., Burlingame, CA, USA) and examined on a Zeiss confocal microscope (LSM 510 Zeiss, Germany). Microscope data were acquired with a × 2.5 objective with constant laser power (45% of laser power), pinhole, zoom, focus, gain and duration of image capturing. Image analysis was performed as described by Bultmann et al (1994). NIH 1.61 software was used for image analysis. A total of 20 brain slices from each animal were analyzed (10 in rostral and 10 in caudal direction from place of injection). Corresponding slices from control brain served as the reference image. After shading correction, the reference image was projected in overlay mode on test image recorded from the respective slice and then the reference image was subtracted from test image. Subtracted images were subjected to median filtering procedure with kernel size of 3 × 3 to reduce background noise. Pixels with intensity ⩽5 were set zero. For every image processed in this way, the mean intensity was calculated using the formula
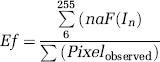
The result termed as ‘degree of extravasation’ [E (f)] is the measure of FITC-albumin extravasation per brain slice at a given time point of an experiment; aF represents the absolute frequency and I the intensity of pixel (6…. 255).
In Vivo Permeability Assay
Blood–brain barrier integrity in mice was also assessed by measurement of the blood–brain transfer coefficient for FITC-albumin (K i ) using a method modified from a previously described procedure (Ohno et al, 1980). Briefly, FITC-albumin was injected as bolus into a femoral vein 20 mins before the end of the experiment. Serial arterial blood samples were then taken from a femoral artery every 5 mins from 0 to 20 min to determine the plasma profile of FITC-albumin. At the end of experiment, the mice were killed by decapitation. Brain were rapidly removed and dissected into two parts (right and left hemispheres). The hemispheres were weighed and homogenized in 50 mmol/L TRIS buffer solution (pH 7.4). The homogenates were centrifuged 955g for 30 mins, and the supernatant collected. Methanol was added to the collected supernatant (1:1) and mixture was centrifuged again under the same conditions. The fluorescence intensity of the supernatant as well as the fluorescence intensity of the plasma samples was measured with a fluorescent reader (Bio-Tek Instruments, Inc., Winooski, VT, USA; emission 485 nm and excitation 540 nm) and the concentration was calculated by using a standard curve. The Ki for FITC–albumin was determined using the equation of Ohno et al (1980):
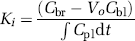
where Cbr is the concentration of FITC-albumin in brain tissue at the time of decapitation (ng/g), Cbl is the concentration of FITC-albumin (ng/mL) in the last blood sample, Vo is the regional blood volume (mL/g), T is the duration of the experiment (min) Cpl is the arterial concentration of FITC-albumin over time t, dt denotes integration over time; Vo was determined in a second set of animals where mice were decapitated 1 min after intravenous injection of FITC-albumin. Assuming no extravasation during this short circulation time, Vo=Cbr/Cbl. Owing to the fact that the K i values measured in this study are <1% of cerebral blood flow, these values are essentially identical to permeability surface area (PS) products (Fenstermacher et al, 1981).
In Vitro Permeability Assay
In vitro permeability assay was performed on mouse brain endothelial cells cocultured with astrocytes. The permeability experiments were initiated by the addition of FITC-albumin to the apical (donor) chamber containing 0.4 mL of DMEM (Invitrogen Corp., Carlsbad, CA, USA). The basal (receiving) chamber contained 1.2 mL of DMEM, and 0.2 ml of this fluid was sampled and replaced with fresh DMEM at 1 h intervals from 0 to 6 h. The permeability (P; cm/min) of the monolayer during any time interval (T) will be calculated as
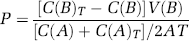
where C(B) and C(B)T are the concentrations (ng/mL) of FITC-albumin in the basal (receiving) chamber at the start and at the end of the time interval, respectively, and V(B) is the volume of the basal chamber (in mL). C(A) and C(A) T are, respectively, the concentrations of FITC-albumin in the apical (donor) chamber at the start and at the end of the time interval and (C(A)+C(A) T )/2 is the average concentration over the time interval. T is the duration of the time interval (in mins), while A is the area of the filter (in cm2). All samples were read on the fluorescent reader (Bio-Tek Instruments, Inc., Winooski, VT, USA; emission 485 nm and excitation 540 nm). The concentration of FITC-albumin in samples was calculated from a standard curve derived using known concentrations of tracer.
Transendothelial Electrical Resistance
Electrical resistances of established coculture of astrocytes and mBMEC were measured with an Endohom-12 electrical resistance apparatus (World Precision Instruments Inc., Sarasota, FL, USA). Cocultured cells were treated with MCP-1 added into the lower and/or upper compartment of the Transwell dual chamber system in a concentration of 100 nmol/L and TEER was measured at intervals over 6 h. The resistance of a blank filter was subtracted to calculate final TEER values (Ω cm2). All experiments were performed in triplicate. The results were expressed as means±s.e. of five independent experiments.
Monocytes/Macrophages Depletion
Mononuclear phagocytes were depleted in vivo by injection of clodronate liposomes (CLODR-LIP, a gift from GmbH, Mannheim, Germany) into the lateral tail vein (0.1 mL/10 g body weight). This dose has been previously shown to eliminate splenic and hepatic macrophages within 24 h and 90% of blood monocytes within 18 h (van Rooijen N and van Nieuwmegen R, 1984; Sunderkotter et al, 2004). At 18 h before treatment with MCP-1, CLODR-LIP was slowly injected intravenously under pentobarbital anesthesia (intraperitoneal injection; 40 mg/kg). As a control, liposomes combined with PBS instead of the clodronate solution was used in another set of animals.
Immunohistochemistry
Cultured cells were fixed in 4% paraformaldehyde for 20 mins at 20°C while brain samples were fixed in 4% paraformaldehyde for 18 h and then cryoprotected with sequential immersions in 10% and 20% sucrose solutions. After fixation, samples were preincubated in blocking solution (5% bovine serum albumin (BSA), 0.05% Tween and PBS), and then incubated overnight with primary antibody (rabbit anti-occludin, mouse anti-ZO-1, anti-claudin-5, Zymed, South San Francisco, CA, USA), anti-ZO-2 or anti-CD45 (BD Bioscience, San Jose, CA, USA) at 4°C. The reaction was visualized by fluorescein-conjugated anti-mouse (Sigma Chemical, St Louis, MO, USA; dilution 1:200) or anti-rabbit antibody (Vector Lab., Burlingame, CA, USA; dilution 1:100). For double label immunostaining, samples were further preincubated in blocking solution and then incubated with rat anti-mouse CD31 antibody (BD Bioscience, San Jose, CA, USA). Reaction was visualized by Texas red conjugated anti-rat antibody (Vector Lab., Burlingame, CA, USA). All samples were viewed on a confocal microscope (LSM 510 Zeiss, Germany).
Western Blotting
Samples were lysed in radioimmunoprecipitation assay (RIPA) buffer (10 mmol/L TRIS, 140 mmol/L NaCl, 1% Triton, 1% Na deoxycholate, 0.1% SDS, 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF) and 1 μg/mL aprotinin, 1 mmol/L NaVO4), homogenized and centrifuged. Insoluble material was removed and the total protein concentration in the resulting supernatant was calculated using a Pierce protein assay kit (Pierce, Rockford, IL, USA). Equivalent amounts of each protein sample were electrophoretically separated on a 7.5% or a 12% SDS-polyacrylamide gel and transferred to Trans-Blot nitrocellulose membrane (BioRad, Hercules, CA, USA). Rabbit polyclonal anti-occludin, mouse monoclonal anti-ZO-1, and anti-claudin-5 (Zymed, South San Francisco, CA, USA), and mouse monoclonal anti-ZO-2 (BD Biosciences, San Jose, CA, USA) antibodies were used according to the manufacturer's recommendation. The results were visualized with a chemiluminescent HRP substrate kit (Pierce, Rockford, IL, USA).
Triton-X Extraction of Membrane-Associated Proteins
Membrane-associated proteins were extracted from mBMEC according to a protocol modified from Fey et al (1984). Briefly, confluent monolayers of mBMEC (from Transwell insert) were overlaid with extraction buffer for 20 mins at 4°C on a gently rocking platform. The extraction buffer contained 0.5% Triton X-100, 10 mmol/L Tris-HCl pH 7.4, 100 mmol/L NaCl, 300 mmol/L sucrose, plus proteinase inhibitor mixture containing phenylmethylsulfonyl fluoride, iodoacetamide, benzamidine (each 1 μmol/L), aprotinin, leupeptin, and pepstatin A (each 20 μg/mL; Roche, Indianapolis, IN, USA). The soluble supernatant was collected and this fraction was defined as the Triton-X soluble fraction. The residue of cells with well preserved nuclei and cytoskeleton fibers adherent to the culture vessels was gently washed twice with Tris buffer saline (TBS) with the protease inhibitors and then lysed with the RIPA buffer (10 mmol/L TRIS, 140 mmol/L NaCl, 1% Triton, 1% Na deoxycholate, 0.1% SDS, 0.5 mmol/L PMSF and 1 μg/mL aprotinin (Sigma, St Louis, MO, USA). The extract was collected and this fraction was defined as the Triton X-100 insoluble fraction.
Statistics
Statistical analyses were performed on the commercial available software, Stat-View (SAS Institute Cary, NC, USA). One-way ANOVA was used to compare means between experimental groups. A Dunnett test was used to determine significance between groups.
Results
Effect of Monocyte Chemoattractant Protein-1 on Blood–Brain Barrier Permeability
In preliminary experiments, CD-1 mice received either an IC or an ICV injection of murine recombinant MCP-1 (5 to 25 μg) and the effects on leukocyte infiltration and FITC-albumin extravasation were monitored after 24 h. Lower concentrations (5 to 20 μg) induced FITC-albumin leakage but failed or modestly induced leukocyte infiltration. Only when 25 μg MCP-1 was injected were there marked increases in BBB permeability and leukocyte infiltration (data not shown). Because of these data, 25 μg MCP-1 was chosen for further experiments. Monocyte chemoattractant protein-1 was administrated IC or ICV and the effect on FITC-albumin extravasation was monitored 6, 12, 24, and 48 h later. Figures 1A and 1B summarizes FITC-albumin extravasation after IC administration of MCP-1. Extravasation was significantly enhanced in MCP-1-treated animals compared with controls by 6 h after injection. Peak MCP-1-induced extravasation was observed at 12 h after both IC (Figure 1A) and ICV (E(f)=1.9±0.3; P<0.001 versus control) administration.
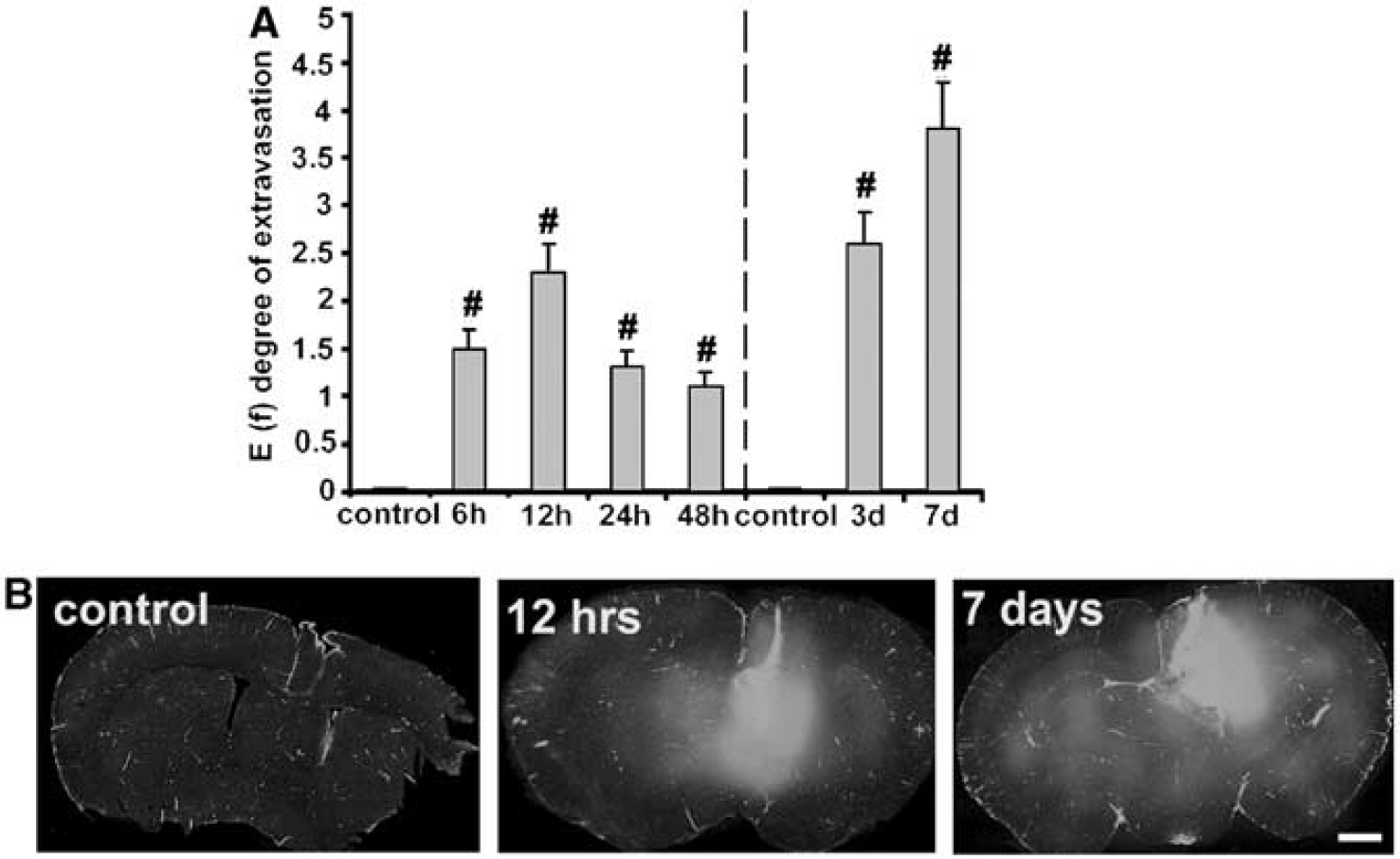
Intracerebral administration of monocyte chemoattractant protein (MCP-1) induces fluorescein isothiocyanate (FITC)-albumin leakage in the brain. FITC-albumin leakage into mouse brain was assayed at 6,12, 24 and 48 h after an acute dose of recombinant murine MCP-1 (25 μg) or after long-term infusion for 3 (5 μg/h) or 7 days (2.5 μg/h). Mice treated with saline were used as control. (
Long-term administration of MCP-1 (5 μg/h for 3 days or 2.5 μg/h for 7 days) also caused pronounced extravasation of FITC-albumin, irrespective of whether the MCP-1 was given IC (Figure 1A) or ICV (data not shown). Quantitatively, the effect of long-term administration was similar to or slightly greater than the peak effect found after acute administration (Figure 1). The degree of extravasation with IC and ICV MCP-1 infusions was similar (at day 7, E(f) values were 3.8±0.5 and 3.5±0.6, respectively).
The effect of MCP-1 on BBB permeability was also assessed by measurement of a PS product for FITC-albumin. A single dose of MCP-1- (25 μg) injected IC induced an increase in the PS product in the ipsilateral hemisphere at 6 and 12 h with the effect waning 24 and 48 h after injection (Figure 2). Similar data was obtained after ICV injection of MCP-1 (peak effect at 12 h, MCP-1 treated 0.09±0.02 versus 0.035±0.01 μL/g/min in controls). The contralateral hemisphere did not show significant alterations in BBB permeability compared with control. There were profound effects on the FITC-albumin PS product when animals were exposed to MCP-1 long-term for 3 (5 μg/h) or 7 days (2.5 μg/h). Increase in BBB permeability was present in both ipsilateral (injected) and contralateral hemispheres after IC injection (Figure 2). Similar increases were also found after ICV infusion of MCP-1 where the FITC-albumin PS products after 3 days 0.11±0.01 and 0.07±0.01 μL/g min in the ipsi- and contralateral hemispheres compared with 0.035±0.01 and 0.033±0.005 μg/g min in controls.
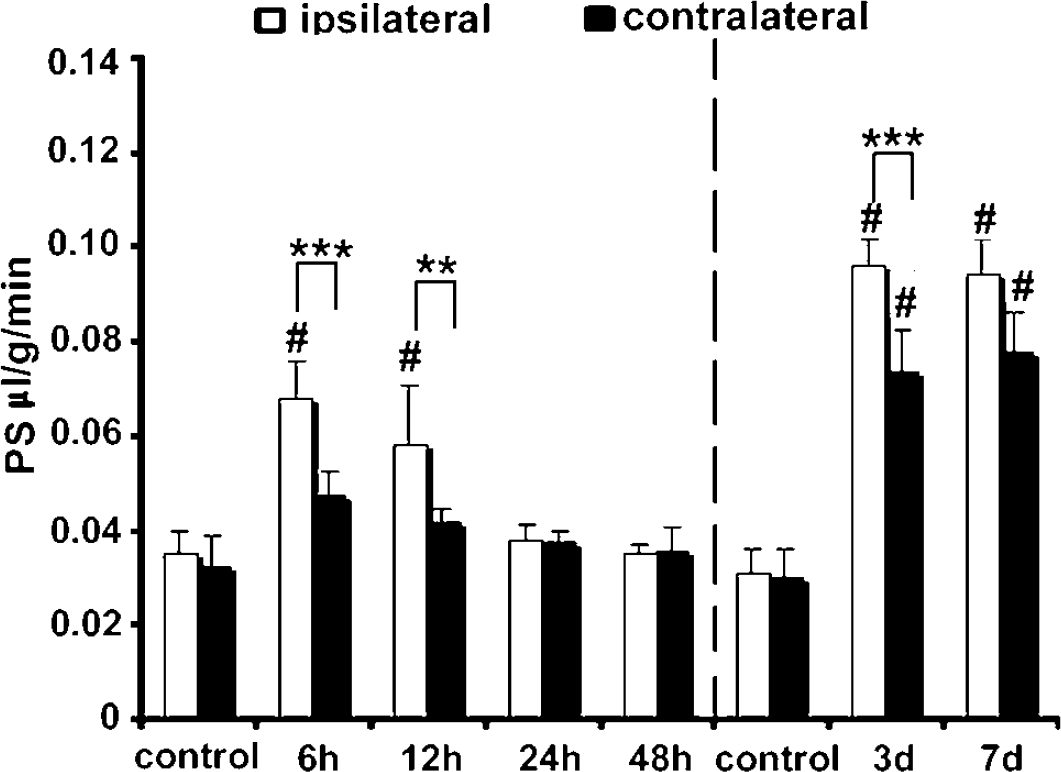
Intracerebral administration of monocyte chemoattractant protein (MCP-1) increases blood–brain barrier (BBB) permeability (in vivo study). Permeability surface area (PS) products for fluorescein isothiocyanate (FITC)-albumin in MCP-1 and saline-treated control mice 6, 12, 24, and 48 h after an acute MCP-1 dose (25 μg) or after long-term infusion for 3 (5 μg/h) or 7 days (2.5 μg/h). Monocyte chemoattractant protein-1 was given by intracerebral (IC) administration and the PS products were measured in ipsilateral and contralateral hemispheres. Values are mean±s.d. # indicates significant difference between ipsilateral hemisphere MCP-1-treated mice and ipsilateral hemisphere of control animals at the P<0.001 level; ** and *** indicate significant difference between ipsilateral and contralateral hemisphere in the same animals at the P<0.01 and P<0.001 level, respectively.
Marked BBB disruption can result in vasogenic brain edema. Acute MCP-1 (25 μg) administration did not affect brain water content when administered IC or ICV. However, long-term administration for 3 or 7 days caused significant (P<0.01) increases in percentage of brain water content. Thus, after IC infusion of MCP-1 water content was 82.9%±1.8% and 83.2%±1.9% at 3 and 7 days versus 78.9%±2.5% and 78.6%±2.5% in controls at those time points. Intracerebroventricular infusions of MCP-1 caused similar increases in water content (∼5%; P<0.01 versus controls at both time points).
The effects of MCP-1 on BBB permeability in vivo were mimicked in studies on an in vitro BBB model (cocultures of astrocytes and brain endothelial cells derived from CD-1 mice). Under regular experimental condition, the TEER value was approximately 400 Ω cm2. The TEER values started to decrease at 1 h after the addition of 100nmol/L MCP-1 (from 409±33 at the beginning of experiment, to 354±20 Ω cm2). The effect becomes more profound as time passes (243±23 Ω cm2 after 6 h of treatment with MCP-1, P<0.001; Figure 3A). This progressive decrease in TEER was matched by an increase in permeability to FITC-albumin (Figure 3B), which increased from 0.2±0.05 at the first hour of MCP-1 treatment to 8.9±0.8 × 10−3 cm/min after 6 h of MCP-1 treatment.
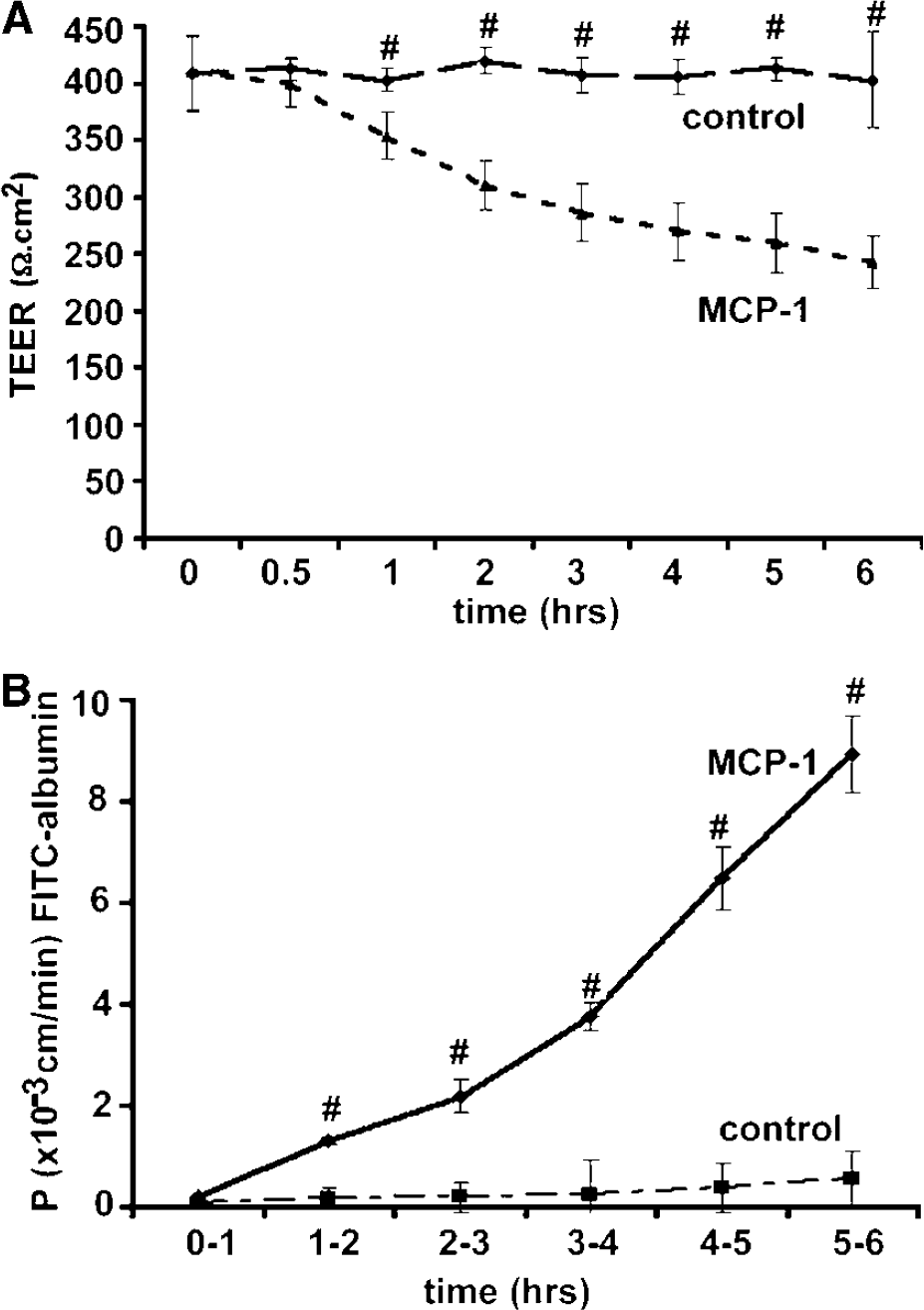
Monocyte chemoattractant protein-1 increases blood–brain barrier (BBB) permeability (in vitro study). Cocultures of astrocytes and mouse brain microvascular endothelial cells (mBMEC) were exposed to MCP-1 (100 nmol/L) for 6 h. MCP-1 was added to the lower (abluminal side), upper (luminal side), or both compartments. Untreated cocultures served as controls.
Morphological and Biochemical Changes in the Tight Junctions on Monocyte Chemoattractant Protein-1 Administration
Western blotting and immunohistochemistry were used to examine whether MCP-1 induces increases in BBB permeability in vivo (CD-1 mice) by altering TJ complex proteins. For Western blotting, samples were taken from the ipsilateral (area around the MCP-1 injection site) and the contralateral hemisphere. As shown in Figure 4A, IC MCP-1 administration caused a marked decrease in the expression of occludin, claudin-5, ZO-1, and ZO-2. This result was confirmed by immunohistochemistry where staining for these junction proteins was decreased or completely lost in the ipsilateral hemisphere (where the BBB was disrupted) compared with the contralateral hemisphere (where BBB integrity was undamaged). An example of this in an animal that received MCP-1 for 3 days (5 μg/h) is shown in Figure 4B.
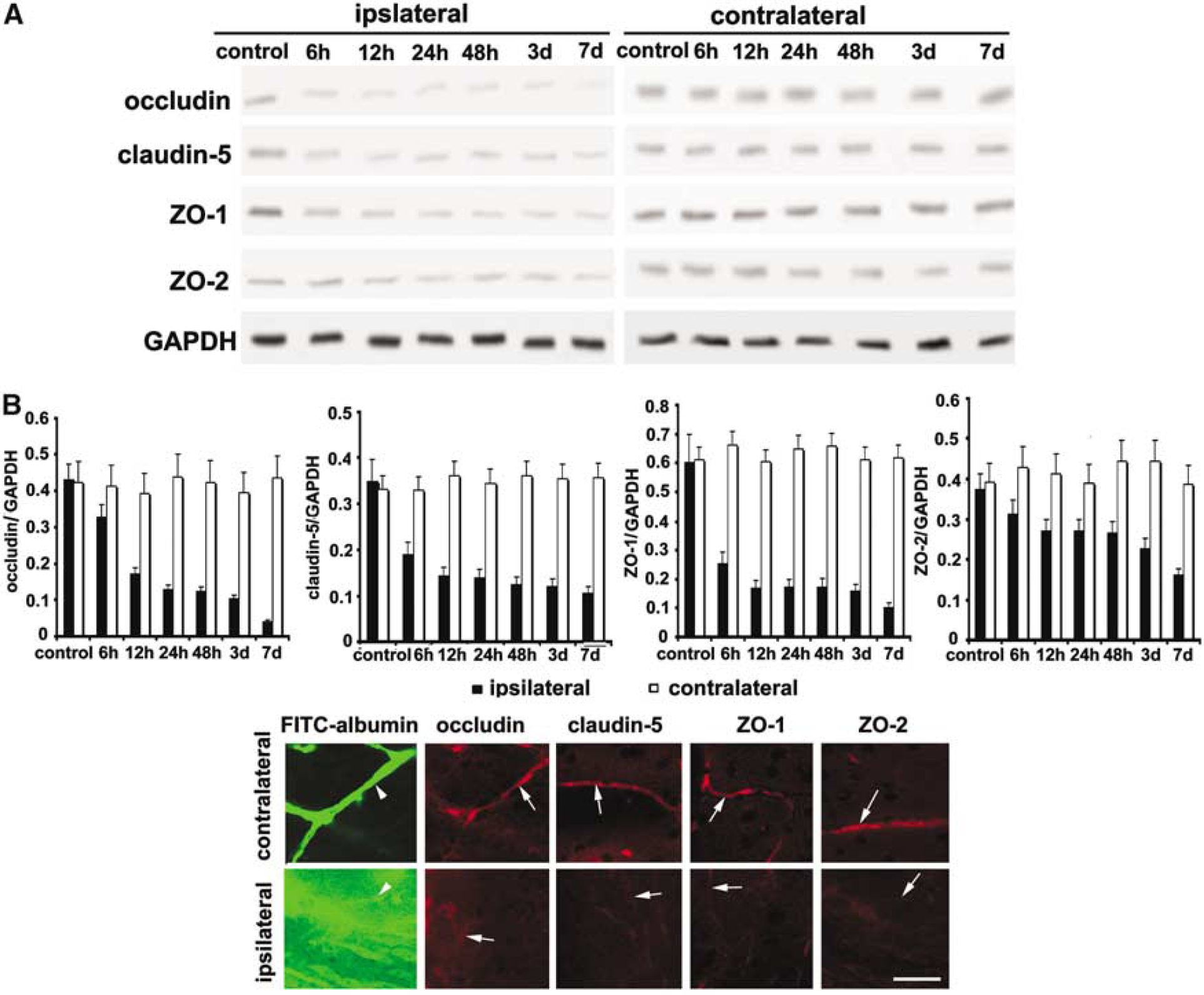
Intracerebral (IC) administration of monocyte chemoattractant protein-1 (MCP-1) alters tight junction (TJ) complex proteins (in vivo study). (
The effect of MCP-1 on TJ proteins was also examined in the in vitro BBB model. Cells were exposed to MCP-1 (100 nmol/L) for 6 h (the time of the peak BBB permeability effect). Monocyte chemoattractant protein-1 induced loss (occludin), fragmentation (claudin-5, ZO-1, ZO-2), or comb-like (ZO-1) staining of TJ proteins and promoted intercellular gap formation (Figure 5A). Biochemical (fractional) analysis of TJ proteins in the brain endothelial cells cocultured with astrocytes showed that MCP-1 induced a shift of occludin, claudin-5, ZO-1, and ZO-2 from Triton X-100 soluble into Triton X-100 insoluble fraction (Figure 5B). This redistribution in TJ proteins may result from an internalization of TJ protein into a cytoplasmic compartment.
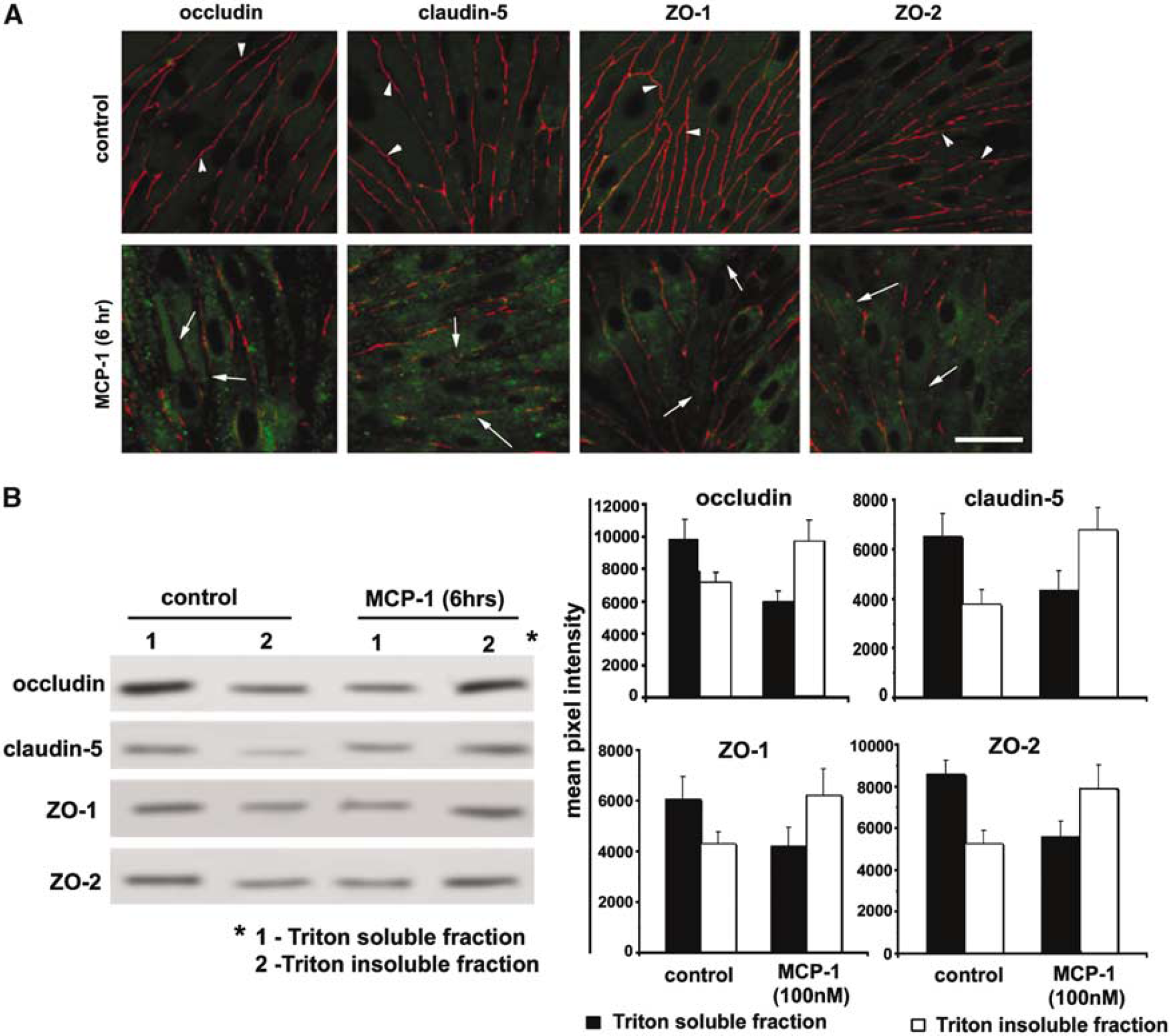
Morphological and biochemical alterations in tight junction (TJ) proteins induced by monocyte chemoattractant protein-1 (MCP-1) (in vitro study). (
Receptor-dependent effects of Monocyte Chemoattractant Protein-1 on Blood–Brain Barrier Permeability
The role of chemotactic cytokine receptor 2 (CCR2) in MCP-1-induced BBB disruption was first investigated in CCR2 knockout mice. In acute experiments, MCP-1 (25 μg) was administered IC, and BBB permeability was measured after 12 h. In long-term experiments, MCP-1 was given for 3 days (5 μg/h) by the same route and then BBB permeability was measured. Blood–brain barrier disruption was assessed by extravasation of FITC-albumin, either by determining the E(f) (Figure 6A) or the PS product (Figure 6B). Compared with WT mice (CCR2+/+), the extravasation of FITC-albumin after acute or long-term administration of MCP-1 was much less in CCR2−/− mice (Figures 6A and 6B). Indeed, the FITC-albumin extravasation in CCR2−/− mice treated with MCP-1 for 3 days was not significantly different from saline-treated controls (Figures 6A and 6B). Similar data were obtained after ICV administration of MCP-1. Thus, 12 h and 3 days after MCP-1 administration, the E(f) for FITC-albumin was 9- and 5-fold greater in CCR2+/+ mice compared with CCR2−/− mice. Similarly, the FITC-albumin PS product was ∼3-fold greater in CCR2+/+ mice compared with CCR2−/− mice at both time points.
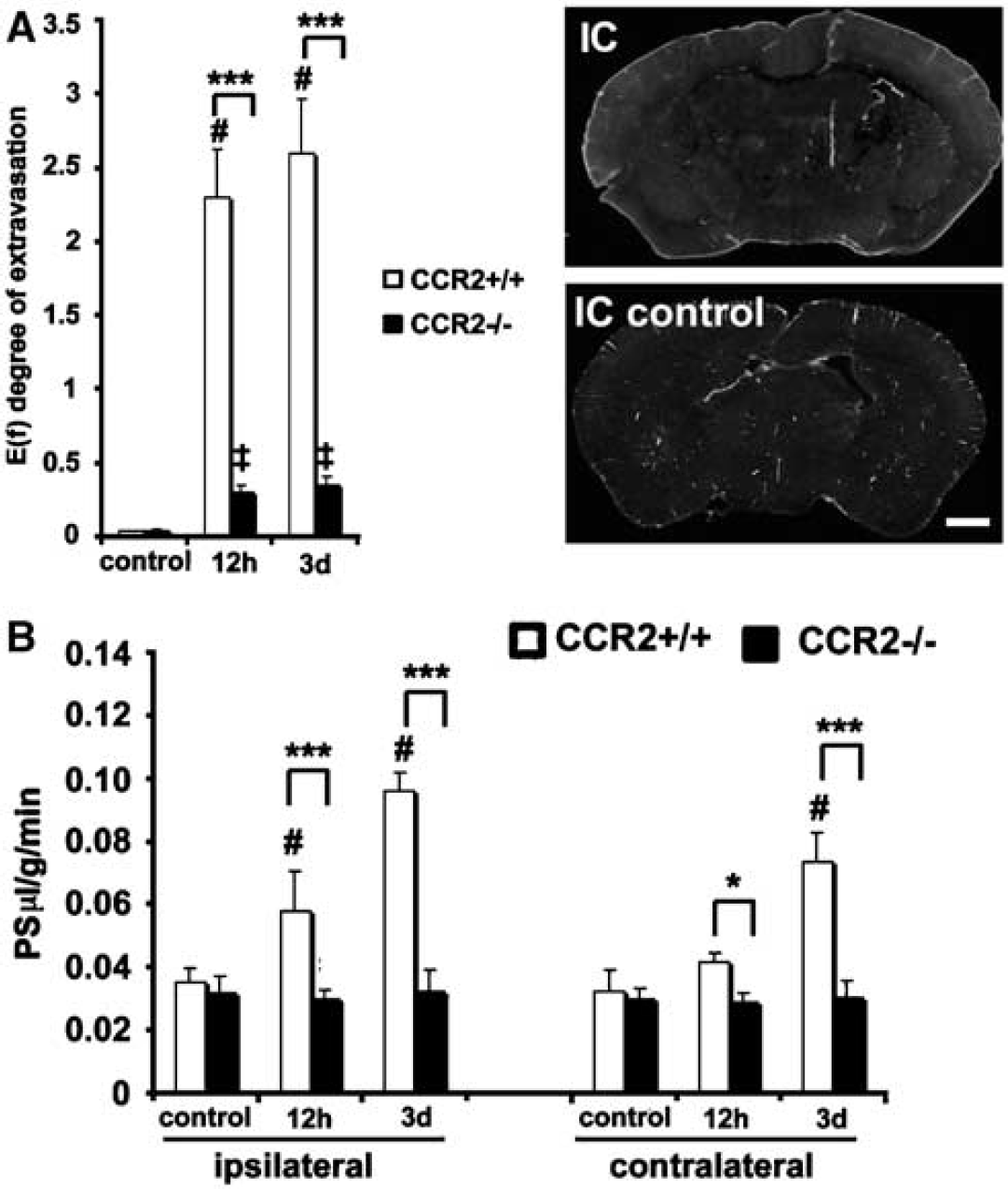
Effect of monocyte chemoattractant protein-1 (MCP-1) on blood–brain barrier (BBB) permeability in the absence of chemotactic cytokine receptor 2 (CCR2). CCR2+/+ or CCR2−/− mice received a single injection of MCP-1 (25 μg) or a long-term infusion (5 μg/h). Monocyte chemoattractant protein-1 was administered by intracerebral administration. Controls received only PBS. Fluorescein isothiocyanate (FITC)-albumin extravasation was examined 12 h after the acute injection and 3 days after the onset of the long-term infusion. In
The role of CCR2 on MCP-1-induced BBB disruption was also tested in the in vitro BBB model. The model was established using astrocytes and mBMEC from either WT mice (CCR2+/+ on 50:50 C57BL/6 × 129Sv background) and mBMEC or CCR2 knockout mice (CCR2−/−, same background). In cocultures established with CCR2+/+ astrocytes and brain endothelial cells, MCP-1 caused a marked time-dependent increase in FITC-albumin permeability (Figure 7), similar to that found with cells derived from CD-1 mice (Figure 3). Cocultures of CCR2+/+ endothelial cells and CCR2−/− astrocytes also showed similar changes. In contrast, when cocultures used CCR2 −/− endothelial cells with CCR2+/+ astrocytes or CCR2−/− astrocytes, MCP-1 did not elicit these permeability changes (Figure 7).
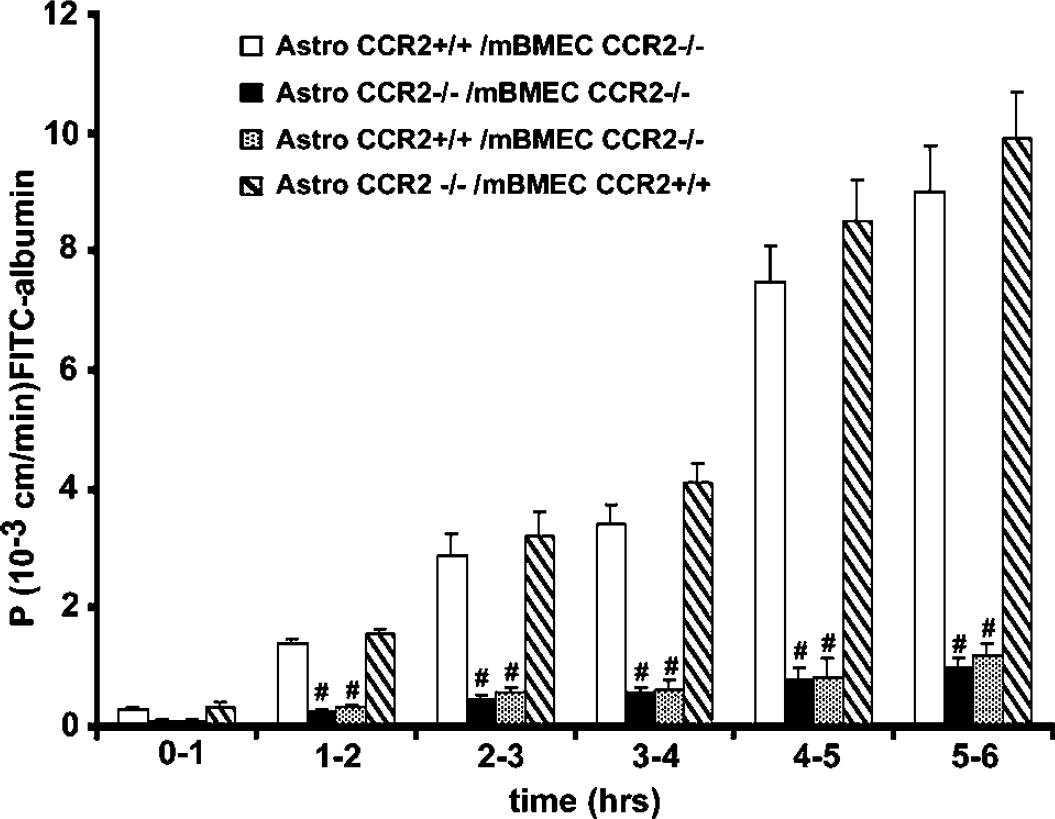
Effect of monocyte chemoattractant protein-1 (MCP-1) on the permeability of an in vitro blood–brain barrier (BBB) model in the presence/absence of chemotactic cytokine receptor 2 (CCR2). Mouse brain microvascular endothelial cells and astrocytes prepared from either CCR2 −/− or +/+ mice were cocultured (in vitro BBB model) and then exposed to MCP-1 (100 nmol/L L) for 6 h. The permeability (P) of the model to fluorescein isothiocyanate (FITC)-albumin was assessed at hourly intervals. Monocyte chemoattractant protein-1 enhanced the permeability of the barrier if CCR2+/+ endothelial cells were used irrespective of the source of the astrocytes (CCR2+/+ or −/−). MCP-1 had no effect on the permeability of the barrier if CCR2−/− endothelial cells were used, again, irrespective of the source of the astrocytes. Values are means±s.d., # indicates a significant difference at the P<0.001 level between each of the groups with CCR2+/+ endothelial cells and those with CCR2−/− endothelial cells.
Monocyte/macrophages and Monocyte Chemoattractant Protein-1 Induced Blood–Brain Barrier Disruption
Although MCP-1 has direct effects on BBB permeability through endothelial CCR2, it is possible that effects of MCP-1 on other cell types may contribute to enhanced BBB disruption in vivo. We first examined whether there was a correlation between leukocyte infiltration (leukocytes were labeled with CD45) and BBB ‘leakage’ (FITC-albumin extravasation). Blood–brain barrier leakage was scored on a scale from 0 to 5 (0—no leakage, 1—modest leakage, 2—mild leakage, 3—leakage, 4—completely open). The presence of CD45+ positive cells was scored on a scale of 0 to 4 (0—no cells, 1—<10 cells, 2–10 to 100 cells, 3–100 to 200 cells, 4—>200 cells). There was a highly significant correlation (r=0.966) between MCP-1-induced leukocyte infiltration and BBB opening. In particular, there was a high correlation between the highest number of infiltrating leukocytes and the greatest degree of BBB opening. To examine whether recruitment of monocytes by MCP-1 might contribute to BBB disruption, CD-1 mice were depleted of monocytes using clodronate liposomes (CLODR-LIP) before treatment with MCP-1. Liposomally incorporated PBS (PBS-LIP) was used as a control, MCP-1 was administered intracerebrally as an acute injection (25 μg), and BBB permeability assessed at 12 h, or as a long-term infusion (5 μg/h) and the permeability assessed at 3 days. In mice treated with PBS-LIP, acute and long-term MCP-1 treatment induced BBB disruption (Figure 8). In mice depleted of monocytes/macrophages with CLODR-LIP, MCP-1 still induced some BBB disruption but this was ameliorated compared with PBS-LIP animals (Figure 8). Monocyte/macrophage depletion also reduced the BBB disruption induced by ICV administration of MCP-1 (data not shown).
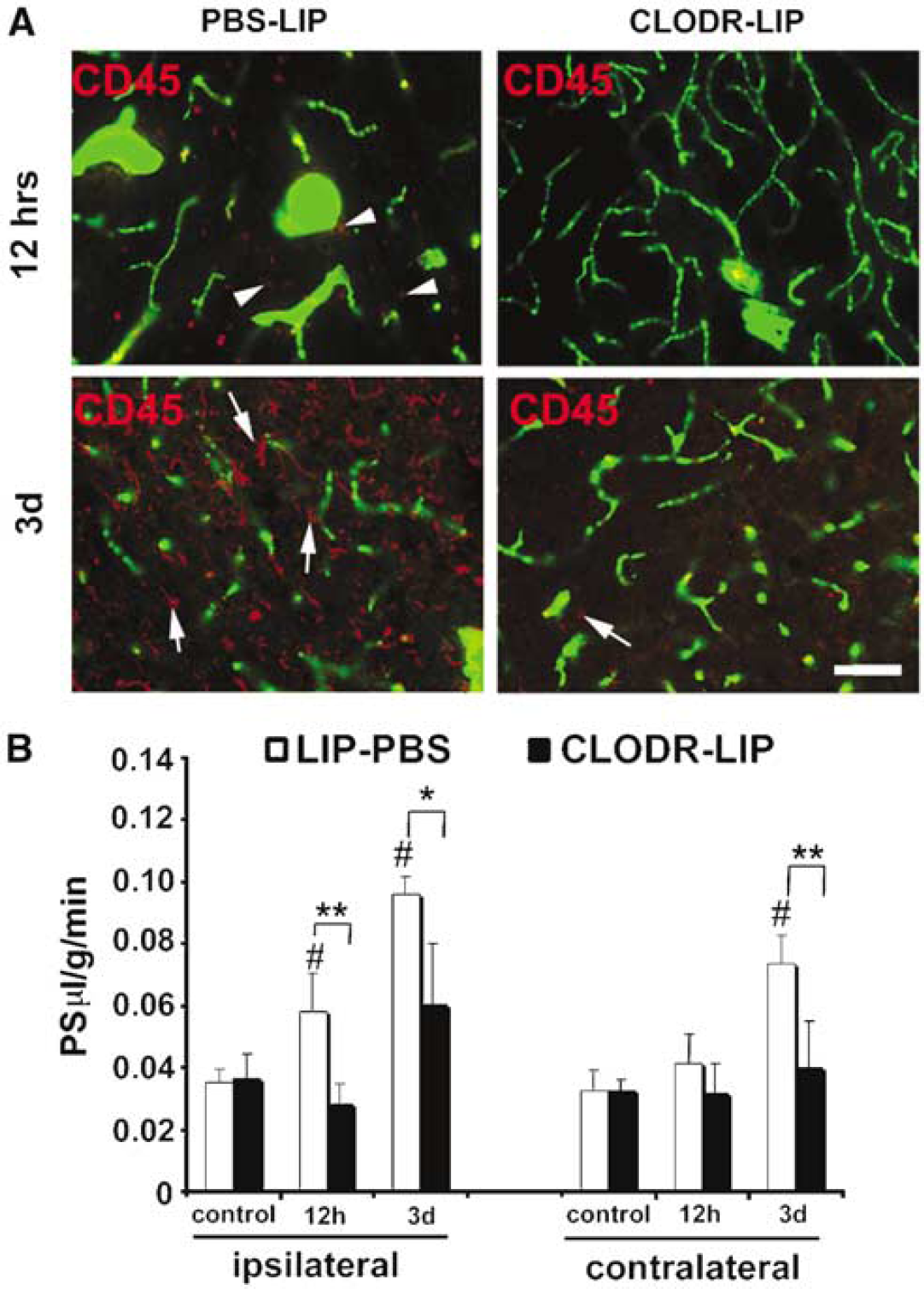
Effects of intracerebral administration of monocyte chemoattractant protein-1 (MCP-1) on blood–brain barrier (BBB) permeability in normal and monocyte-depleted mice. (
Discussion
Chemokines are overexpressed in inflammation-related disorders associated with a disturbed BBB. This has led to the suggestion that chemokines may affect BBB permeability. However, only a few studies have provided evidence that chemokines participate in altered endothelial permeability (Biffl et al, 1995; Bell et al, 1996; Matsumoto et al, 1997). The data in the current study strongly support this hypothesis, and indicate for the first time that MCP-1 can induce alterations in BBB permeability under in vitro and in vivo conditions. In particular, our study clearly shows that (a) MCP-1 induces significant increases in BBB permeability and brain water content; (b) MCP-1-induced BBB disruption is associated with alterations in the TJ complex; (c) MCP-1 does not show any effect on BBB permeability in the mice lacking the MCP-1 receptor, CCR2; and (d) MCP-1 induces recruitment of monocytes, which in turn contribute to alterations in BBB permeability. The implications of these findings are discussed below.
There is a CNS inflammatory response in a diverse range of insults, including trauma, cerebral infarction, multiple sclerosis, viral infection, and neurodegeneration (Miller and Meucci, 1999; Feuerstein et al, 2000). The classic hallmarks of brain inflammation are breakdown of the BBB, edema formation, tissue infiltration by peripheral blood cells, activation of immunocompetent cells, as well as intrathecal release of numerous immune mediators such as interleukins and chemotactic factors (Martiney et al, 1998). While it is a commonly accepted concept that increased BBB permeability leads to unrestricted passage of molecules into brain and leukocyte infiltration, some recent studies indicate that migration of leukocytes may contribute to the persistence of increased vascular permeability (Merrill and Murphy 1997; Couraud, 1998; Petty and Lo, 2002). How leukocyte migration may regulate vascular permeability is still under investigation, but some data imply that molecules involved in regulating such migration, including cytokines, adhesion molecules, and chemokines, could have a prominent role (Stanimirovic and Satoh, 2000; Dietrich, 2002).
Proinflammatory cytokines such as IL-1α, IL-1β, TNF-α, IL-6, GM-CSF are recognized as major contributors to BBB breakdown, causing increased BBB permeability under in vitro and in vivo conditions (Yang et al, 1999; Blamire et al, 2000; Paul et al, 2003). However, several studies examining the effect of IL-1β on BBB permeability have found a strong correlation between IL-1β-induced expression of chemokines, such as CINC1 and MIP-2, and increased BBB permeability, and that the neutralizing antibodies to MIP2 and CINC1 can prevent BBB breakdown (Anthony et al, 1998; Campbell et al, 2002). In 1997, Matsumoto et al (1997) also demonstrated that administration of neutralizing antibody to the chemokine IL-8 significantly attenuates BBB breakdown and brain edema during an ischemic/reperfusion brain injury. Taken together, these findings suggest that chemokines could play a key role in mediating BBB breakdown. Our results provide further support for this hypothesis.
The chemokine, MCP-1 is considered as one of the most potent chemotactic factors for monocytes during CNS inflammation. The expression of MCP-1 is closely correlated with monocyte infiltration during clinical manifestations of multiple sclerosis, stroke, or CNS trauma. Several studies have also suggested a possible effect of MCP-1 on BBB permeability but direct evidence, until now, does not exist (Bell et al, 1996; Stamatovic et al, 2003). High concentrations of MCP-1 are present in the perivascular space of the BBB to direct infiltration of monocytes but brain endothelial cells also possess CCR2 receptors, suggesting that MCP-1 may regulate BBB permeability to facilitate monocyte transmigration during inflammation. Our previous in vitro study on endothelial monocultures showed that MCP-1 can alter brain endothelial permeability (Stamatovic et al, 2003) but whether it would have a similar effect in vivo was still open to question. To better understand the role of MCP-1 in regulating BBB permeability during a CNS inflammation, we treated the mice with either MCP-1 or saline by IC or ICV injection. Both IC and ICV routes of administration were included because MCP-1 can be present in brain parenchyma or CSF in CNS inflammatory states. The dose of MCP-1 administered was based on our preliminary data. Our selected concentration of MCP-1 was enough to induce monocyte infiltration and increase in BBB permeability in a time-dependent manner. Compared with some other studies where chemokine effects on the BBB have been investigated, our administered MCP-1 dose is significantly higher, but with this dose we achieve the effect of MCP-1 during inflammation including a robust infiltration of leukocytes. Smaller concentrations of chemokines (including also MCP-1) had no or modest effect on leukocyte movement (Biffl et al, 1995; Bell et al, 1996 and our preliminary data presented above).
In this study, two methods were used to assess the effect of MCP-1 on BBB permeability to FITC-albumin in vivo. The first examined extravasation on histological sections, while the second examined such extravasation using tissue homogenates. Qualitatively, the two techniques produced similar results (e.g., a similar temporal profile of disruption). Quantitatively, there were differences in the fold change in permeability measured by the two techniques. This may reflect the fact that whole hemispheres were homogenized in the latter technique, resulting in the inclusion of tissue unaffected by the MCP-1 injected.
These effects of MCP-1 on BBB disruption in vivo were also replicated in an in vitro BBB model (endothelial cell/astrocyte cocultures). We have previously shown that MCP-1 enhances the permeability of endothelial monocultures (Stamatovic et al, 2003). However, such cultures can be criticized for not displaying full BBB characteristics (e.g., low TEERs; problems with forming tight junctions or absence of expression some transport proteins). The inclusion of astrocytes results in a preparation with more BBB characteristics, including a higher TEER (∼400 versus 130 Ω cm2 in the previous study). The inclusion of astrocytes also enabled examination of whether MCP-1 may have effects on BBB permeability via actions on astrocytes as well as endothelial cells. The importance of examining potential multiple sites of action are emphasized by our findings on the role of monocytes in BBB disruption (see below).
In monocultures of mBMEC, MCP-1-induced enhanced permeability was associated with changes in TJ complexes proteins. In particular, there was a morphological change in the distribution in the TJ proteins occludin, ZO-1, ZO-2, and claudin-5 with MCP-1 treatment and a shift in the distribution of these proteins from Triton-X100 soluble to Triton-X100 insoluble fraction (Stamatovic et al, 2003). This latter shift is thought to reflect an internalization of TJ proteins into a cytoplasmic compartment, and a resultant increase in permeability, on exposure to MCP-1. In the current study, similar changes were found in the coculture in vitro BBB model. Occludin, ZO-1, ZO-2, and claudin-5 all shifted towards the Triton-X 100 insoluble fraction with MCP-1 treatment and there was a disruption in the normal cellular distribution pattern of these proteins. Importantly, we also found evidence for alterations in these junction proteins with MCP-1 in vivo. Immunohistochemistry and Western blotting both indicated a loss of TJ proteins in mice injected either with IC or ICV with MCP-1. Thus, it appears that the effects of MCP-1 on BBB permeability are caused by alterations in TJ complexes. Alterations in these complexes are implicated in BBB breakdown in many CNS pathological states including malaria, viral infection, multiple sclerosis, experimental allergic encephalitis, ischemia/reperfusion injury, and trauma (Brown et al, 1999; Mark and Davis 2002; Ng et al, 2003; Kirk et al, 2003). Furthermore, direct intracerebral injections of some proinflammatory cytokines (e.g., IL-1β, TNF-α, and IL-6), which are known to modify BBB, also cause structural changes in endothelial TJs (Bolton et al, 1998).
The main receptor for MCP-1 is CCR2 and we have previously found that CCR2 is involved in changes in MCP-1-induced alteration in the permeability of endothelial monocultures (Stamatovic et al, 2003). The current study demonstrates that CCR2 is involved in MCP-1-induced BBB disruption in vivo. Thus, MCP-1-induced extravasation of FITC-albumin was absent in CCR2 knockout mice. Similarly, in the in vitro BBB model, the marked enhancement of FITC-albumin permeability with MCP-1 treatment found in cocultures of endothelial cells and astrocytes derived from CCR2+/+ mice was absent in cocultures derived from CCR2−/− mice. Importantly, MCP-1 induced barrier disruption in cocultures where astrocytes were CCR2−/− but endothelial cells were CCR2+/+, but it did not induce disruption if endothelial cells were CCR2−/− and astrocytes were CCR2+/+. Thus, although astrocytes express CCR2, barrier disruption in this model was dependent on endothelial CCR2.
Our results suggest that MCP-1/CCR2 axis has a very important role in regulating inflammation and it could be a very promising target for many CNS inflammatory diseases including stroke and trauma where inflammation participates in secondary brain injury. The effects of CCR2 deletion (or inhibition) on ischemia-induced brain injury (and BBB disruption) have yet to be examined. However, a few studies, primarily on experimental allergic encephalomyelitis, have indicated that absence or inhibition of CCR2 may limit brain damage (Izikson et al, 2000; Huang et al, 2001).
In addition to direct effects via endothelial CCR2, MCP-1 appears to affect BBB permeability by other indirect mechanisms in vivo. Depletion of circulating monocytes and activated macrophages by CLODR-LIP attenuated the effect of MCP-1 on BBB permeability in vivo. Thus, MCP-1 might exert its effect on the BBB partially by recruiting monocytes into brain parenchyma, which in turn increases BBB permeability. Chemotactic cytokine receptor 2 is present on monocytes as well as endothelial cells (Fantuzzi et al, 1999; Andjelkovic et al, 1999b). The effects of CCR2 deletion on MCP-1-induced BBB breakdown in vivo may, therefore, reflect an inhibition of both the endothelial and monocyte actions of MCP-1.
Taken together, the present in vitro and in vivo results provide strong evidence that, beside their main function of recruiting leukocytes at sites of inflammation, chemokines play a role in modulating BBB permeability. To the best of our knowledge, this is the first detailed study of MCP-1 influence on the BBB showing that MCP-1 alters BBB permeability in vivo by direct and indirect effects (endothelial cell receptor and monocyte mediated). Together with our previous study on MCP-1 (Stamatovic et al, 2003), this helps to provide a clearer picture of how MCP-1 induces BBB breakdown. The observation that MCP-1 is abundantly expressed in the CNS during pathological states associated with BBB disruption leads us to hypothesize that MCP-1 could be a major factor regulating BBB integrity. Monocyte chemoattractant protein-1 and its receptor CCR2 are potentially an important new target for the prevention of BBB disruption. Future studies should address methods of preventing MCP-1 production and inhibiting of CCR2 and examining whether this will result in reducing inflammation-mediated brain injury such as occurs in trauma or stroke.
Footnotes
Acknowledgements
The authors thank Danica Petrovic-Djergovic (Department of Neurosurgery) and David Oldfield (Department of Pediatrics, University of Michigan Medical School) for their technical assistance.