
Abstract
Oestrogen is a complex hormone whose role as a neuroprotectant in experimental stroke has been published in numerous studies. However, although some clinical studies report a reduction in stroke incidence by oestrogen replacement therapy in postmenopausal women, others have found increased mortality and morbidity after stroke. Diathermy occlusion of the proximal middle cerebral artery (MCAO), one of the most reproducible rodent stroke models, has consequently been employed to investigate physiologic and supraphysiologic doses of 17β-oestradiol on ischaemic brain damage. Lister Hooded rats were ovariectomised (OVX) and a 21-day release pellet (placebo, 0.025 or 0.25 mg 17β-oestradiol) implanted in the neck. At 2 weeks after OVX, animals underwent MCAO and were perfusion fixed 24 hours later. Neuronal perikaryal damage was assessed from haematoxylin and eosin-stained sections and in adjacent sections, axonal pathology was assessed with amyloid precursor protein and Neurofilament 200 (NF-200) immunohistochemistry. 17β-Oestradiol induced a dose-dependent increase in neuronal perikaryal damage, 0.025 and 0.25 mg 17β-oestradiol increased damage by 20% (P<0.05) and 27.5% (P<0.01) compared with placebo. 17β-Oestradiol did not influence axonal pathology compared with placebo. Our results suggest that 17β-oestradiol treatment can exacerbate brain damage in experimental stroke. Thus, further investigation into the role of oestrogen and the mechanisms which mediate its effects in stroke is required.
Introduction
Oestrogen, the female sex hormone, is known to influence cerebral ischaemic damage. In previous experimental stroke studies the hormone has been shown to act as a neuroprotective agent (Hurn and Macrae, 2000). The main model used to investigate 17β-oestradiol has been the intraluminal thread (ILT) model in rats: pretreatment with 17β-oestradiol has been shown to be neuroprotective with transient ILT ischaemia (Simpkins et al, 1997, Rusa et al, 1999) and permanent ILT ischaemia (Dubal et al, 1998).
Clinical studies designed to investigate the influence of oestrogen replacement therapy (ERT) on stroke have, however, not all showed the beneficial influence of oestrogen found in experimental studies. A few observational studies have reported protective effects of ERT. Finucane et al (1993) found that postmenopausal hormone-treated women showed reduced risk of occurrence of stroke and also reduced death rates. Paganini-Hill and Perez Barreto (2001) reported that women on long-term ERT treatment and women who had a recent history of using ERT showed significantly less risk of suffering a stroke. However, other studies have reported the opposite effect. The Framingham Heart Study (Wilson et al, 1985), an observational study, found that postmenopausal women on ERT had an increased risk of stroke, while a placebo-controlled randomised trial found that oestradiol did not reduce stroke mortality rates and, in fact, increased the risk of fatal stroke (Viscoli et al, 2001). A further observational study suggested that low-dose oestrogen treatment decreased the risk of coronary events and stroke, but that a higher dose may increase the risk of stroke (Grodstein et al, 2000). In addition, a comparative study from the Women's Health Initiative has found that the detrimental effects of hormone replacement therapy (HRT) (including increased risk of stroke) outweigh any beneficial effects (Rossouw et al, 2002).
Previous studies reporting the beneficial influence of ERT on cognition (Smith et al, 2001) stimulated our interest in the effect of 17β-oestradiol on subcortical projections after stroke. Oestrogen has the ability to enhance regeneration of nerve fibres in the periphery after crush injury (Islamov et al, 2002) and protect mature hippocampal neurons against fibrillar A1β-induced neurotoxicity by a mechanism involving changes in the composition and stability of the cytoskeleton (Shah et al, 2003). Because oestrogen receptors are thought to be located on axons, as observed using immunoelectron microscopy in the rat hippocampus (Milner et al, 2001), and because oestrogen's antioxidant activity could limit oxidative stress, particularly lipid peroxidation, in myelinated fibre tracts, we have investigated whether 17β-oestradiol reduces axonal pathology after middle cerebral artery occlusion (MCAO). The study of ischaemic white matter damage has been largely ignored despite 50% of the human brain being composed of white matter. This has been primarily because of the lack of methodology relating to white matter damage after experimental stroke. However, recently, methods have been developed to allow quantification of white matter damage using amyloid precursor protein (APP) immunohistochemistry (Imai et al, 2002), previously validated as a marker of axonal injury (Kawarabayashi et al, 1991).
Consequently, proximal occlusion of the MCA by electrocoagulation, one of the most reproducible and established rat stroke models (Tamura et al, 1981), has been used to study the effect of 17β-oestradiol on both neuronal perikaryal (grey matter) damage and on axonal (white matter) damage.
Methods
All experiments were performed under a project licence from the Home Office and were subject to the Animals (Scientific Procedure) Act, 1986. Lister Hooded rats were used because the present study is part of a larger programme of work on oestrogen that includes some behavioural testing (e.g. Morris water maze), where the visual acuity of pigmented strains is a requirement.
Ovariectomy and 17β-Oestradiol Pretreatment
Female Lister Hooded rats, 12-week-old, were anaesthetised with 5% halothane and then maintained on a facemask at 2% in oxygen–nitrous oxide (30:70). All animals were injected subcutaneously with 0.02 ml 50 mg/mL carprofen (Rimadyl) for postoperative analgesia and then underwent bilateral ovariectomy via a dorsolateral approach. A 21-day release pellet (Innovative Research of America) containing placebo (n=7), 0.025 mg 17β-oestradiol (n=8) or 0.25 mg 17β-oestradiol (n=8) was implanted subcutaneously at the nape of the neck.
Middle Cerebral Artery Occlusion
At 2 weeks after ovariectomy, the animals underwent permanent left MCAO using an intracranial approach and diathermy occlusion (Tamura et al, 1981). Anaesthesia was initially induced with 5% halothane and the animals were then mechanically ventilated with 1% to 2% halothane in oxygen–nitrous oxide (30:70). Rectal temperature was maintained at 37°C±0.5°C throughout. A cannula was inserted into the femoral artery to monitor physiologic parameters. A proximal occlusion was performed via a subtemporal approach. The temporal muscle was retracted and the skull exposed. A craniectomy was performed and the dura was removed to expose the MCA. The MCA was electrocoagulated from the origin of the lenticulostriate arteries to a point just distal to the inferior cerebral vein before being transected to ensure complete occlusion. The wound was then sutured, the anaesthesia withdrawn and the animal allowed to recover. At 24 hours after occlusion, terminal blood samples were taken in duplicate to measure 17β-oestradiol plasma levels using a radioimmunoassay (Diagnostic Products Corporation, Wales, and UK). Samples were frozen at −20°C until use. The 24-hour end point was based on the fact that the infarct has reached its maximum size by 24 hours in this model (Dawson, 1993; Gill, 1992).
Tissue Processing and Histology
The animals were transcardially perfused with heparinised saline followed by 4% paraformaldehyde in phosphate buffer. The brains were postfixed for 24 hours in the skull and then for 24 hours out of the skull, before being processed for paraffin embedding. Sections (6 μm) collected at 8 predetermined coronal levels throughout the MCA territory were stained with haematoxylin and eosin (H&E) to assess neuronal perikaryal damage. Infarct volumes were derived by integration of areas of ischaemic damage transcribed onto line diagrams at the 8 predetermined coronal levels (end points for integration 12.5 mm anterior to and 0.05 mm posterior to the interaural line) (Osborne et al, 1987). They were then quantified using an MCID image analysis system (Imaging Research, St Catherine's, Ontario).
Immunohistochemistry
Axonal pathology was assessed on adjacent sections by immunohistochemistry using both anti-APP and antineurofilament 200 (NF-200) antibodies. Briefly, sections from the 8 predetermined coronal levels were selected for each antibody and stained using monoclonal APP (1:400 Chemicon) or monoclonal NF-200 (1:10,000 Sigma). Sections were cleared, dehydrated, and then microwaved to increase epitope exposure. Endogenous peroxide activity was quenched using 3% H2O2 in methanol for 30 mins. The sections were washed in PBS before being blocked for 1 hour using 10% normal horse serum and then incubated overnight with the primary antibody at 4°C. After 2 × 5-min washes in fresh PBS, sections were incubated for 1 hour with secondary antibody (rat absorbed anti-mouse IgG raised in horse). Vectastain ABC kit was applied for 1 hour before visualisation of staining using DAB peroxidase substrate kit. Amyloid precursor protein-stained sections were counterstained with haematoxylin.
The APP staining was analysed using a 65-point scoring system for the presence (1) or absence (0) of APP accumulation in axonal swellings in 65 different axon-rich areas (Imai et al, 2002). NF-200 immunostaining was used to transcribe changes in axonal staining patterns (axonal swellings, granulated irregular staining in axons) or loss of staining in the ipsilateral hemisphere compared with the staining pattern in the contralateral hemisphere. The areas of abnormal NF-200 staining were mapped onto line diagrams at the eight predetermined stereotaxic coronal planes. The volume of axonal pathology was then calculated using an MCID image analyser to measure areas of axonal pathology at each of the stereotaxic coordinates, with the volume of damage derived using integration points (as previously stated for H&E assessment of infarct volume).
Statistical Analysis
Data are presented as mean±s.d. for plasma oestradiol, physiologic variables, volume of neuronal perikaryal damage and NF-200 staining for axonal pathology, and as median with confidence intervals for APP score.
Statistics used were ANOVA, followed by Dunnett's test for multiple comparisons for neuronal perikaryal damage and for NF-200 axonal pathology. Amyloid precursor protein score was analysed by the Mann–Whitney nonparametric test, while physiologic variables were analysed with the Student's unpaired t-test.
Results
Physiologic Variables and Plasma 17β-Oestradiol Levels
Table 1 illustrates the physiologic parameters measured during the study. It was found that animals receiving 17β-oestradiol treatment did not gain body weight to the same extent as the placebo-treated animals. This effect was found to be dose related but there was no correlation found between weight change and subsequent volume of neuronal perikaryal damage (r2 values: 0.01 for placebo, 0.11 for 0.025 mg 17β-oestradiol and 0.22 for 0.25 mg 17β-oestradiol). A small reduction in plasma glucose was apparent in the high-dose oestradiol group during surgery. End terminal plasma glucose levels from samples collected 24 hours later showed no difference between the groups.
Plasma 17β-oestradiol levels and physiologic variables for all groups
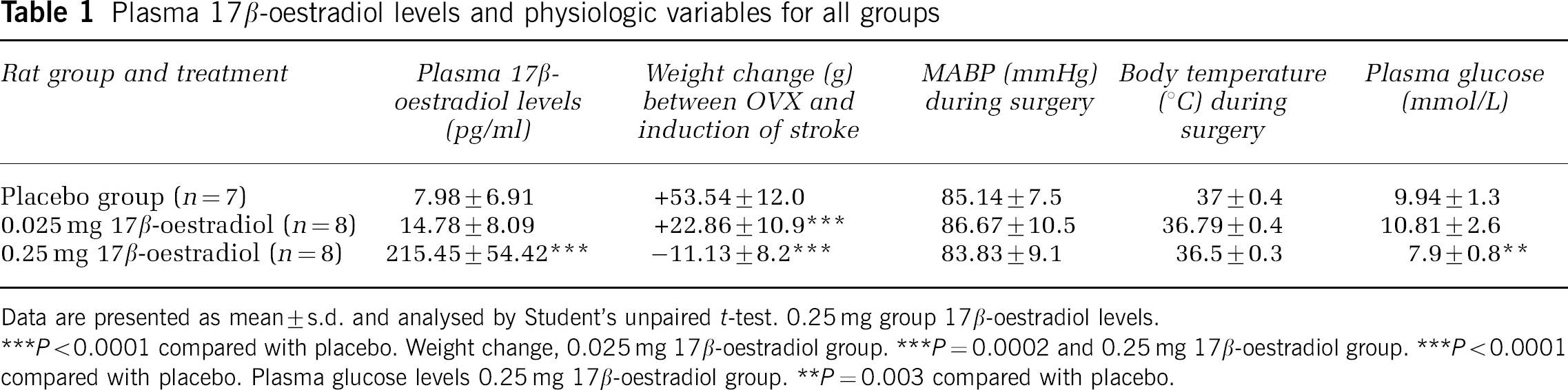
Data are presented as mean±s.d. and analysed by Student's unpaired t-test. 0.25 mg group 17β-oestradiol levels.
P<0.0001 compared with placebo. Weight change, 0.025 mg 17β-oestradiol group. ***P=0.0002 and 0.25 mg 17β-oestradiol group. ***P<0.0001 compared with placebo. Plasma glucose levels 0.25 mg 17β-oestradiol group.
P=0.003 compared with placebo.
Neuronal Perikaryal Damage
17β-Oestradiol induced a significant, dose-related increase in neuronal perikaryal damage in both the low-dose (0.025 mg pellet) and high-dose (0.25 mg pellet) groups compared with the placebo group (Figure 1). The increase in damage in the low-dose group was 20% (P<0.05) and in the high-dose group was 27.5% (P<0.01) compared with placebo. Neuronal perikaryal damage in this model was present in both cortex and caudate nucleus. The 17β-oestradiol-induced increase in neuronal perikaryal damage was mainly within the cortex (Figures 1A and 1C), with a statistically significant increase detected with the high dose of oestradiol (P<0.01). When the data are examined at different coronal levels throughout MCA territory, the oestradiol-treated groups revealed increased areas of damage in the core MCA territory, with less difference between groups at the rostral and caudal poles as shown in Figure 1B.

(). *P<0.05, **P<0.01 versus placebo for that region (ANOVA followed by Dunnett's test for multiple comparisons). (
Composite images illustrating the extent of damage after diathermy occlusion of the proximal MCA and the expansion in neuronal perikaryal damage in the oestradiol-treated groups, are shown in Figure 1C.
Axonal Pathology
Figure 2A shows the immunostaining patterns found with APP and NF-200. Accumulation of APP in damaged axons, specifically in axonal swellings and bulbs, was counted as axonal pathology. NF-200 staining of damaged axons resulted in either disruption of smooth, ordered neurofilaments in axons, thus appearing more granulated and irregular, or loss of NF-200 staining compared with the contralateral hemisphere.

(), using irregular NF-200 immunohistochemistry as a marker of axonal pathology. (
17β-Oestradiol did not influence axonal pathology at either dose, as measured by APP immunostaining using the 65-point scoring system. The median APP scores were 19 (12.6 to 21.1, 95% CI) for placebo, 19 (16.7 to 21.8, 95% CI) for the 0.025 mg 17β-oestradiol group and 17 (15.2 to 18.8, 95% CI) for the 0.25 mg 17β-oestradiol group (Figure 2B).
Similarly, NF-200 immunostaining showed no effect of 17β-oestradiol on the volume of tissue displaying axonal damage. The volume of axonal damage was 239±39.3 mm3 for placebo, 260±16.7 mm3 for 0.025 mg 17β-oestradiol, and 255±16.3 mm3 for 0.25 mg 17β-oestradiol (not significant versus placebo, Figure 2C).
There was no significant correlation between infarct volume (neuronal perikaryal damage) and axonal pathology as scored by APP in any of the groups. Using NF-200, significant correlations between infarct volume and axonal pathology (volume of NF-200 irregular staining) were found for the placebo group (r2=0.6097, P=0.0382) and the low-dose oestrogen group (r2=0.6504, P=0.0156), but not the high-dose oestrogen group (r2=0.09600, P=0.4552), albeit the correlations were strongly influenced by one point in both placebo and low-dose groups (Figure 2D). The relationship between infarct volume and volume of NF-200 irregular staining was influenced by both low- and high-dose oestrogen such that with oestrogen treatment, the extent of axonal damage did not match the increased infarct volumes.
Discussion
The major finding in the present study is that 17β-oestradiol exacerbates ischaemic damage in a dose-related manner after permanent MCAO. There was an increase in neuronal perikaryal damage with both physiologic (14.8±8.1 pg/mL) and supraphysiologic (215.5±54.4 pg/mL) plasma levels of 17β-oestradiol. These results are contrary to our hypothesis and were surprising in light of previous reports in the literature of oestrogen-mediated neuroprotection (Hurn and Macrae, 2000). More recent studies have revealed deleterious effects of oestrogen in models of cerebral ischaemia. For examples, 17β-oestradiol increased brain damage after global ischaemia in mice (Harukuni et al, 2001) and in diabetic rats (Santizo et al, 2002), and after distal MCAO in normotensive rats (Carswell et al, 2004). However, the majority of studies have shown that 17β-oestradiol replacement ameliorates ischaemic damage (Simpkins et al, 1997; Yang et al, 2000; Dubal et al, 1998; Rusa et al, 1999; for a review, see Hurn and Macrae, 2000). Differences in experimental protocols of these studies and the present study probably explain the different results. There are two main differences in experimental protocol, namely, the use of the ILT model (neuroprotection studies) versus the use of the diathermy model (present study), and the use of transient MCAO (most of the neuroprotection studies) versus permanent MCAO (present study). Cerebral blood flow in the reperfusion phase after transient MCAO has been shown to be improved by oestrogen (McCullough et al, 2001). In addition, oxidative stress, which is increased during the reperfusion phase after prolonged severe ischaemia, is reduced by oestrogen (Behl et al, 1997). Thus, the lack of reperfusion in the present study may explain the lack of a beneficial influence and thereby allow the unmasking of a detrimental influence of 17β-oestradiol. To our knowledge, there are only two studies which have used permanent MCAO to show oestrogen-induced neuroprotection in normotensive, female rats, both of which use the permanent ILT model (Dubal et al, 1998; Yang et al, 2000). The ILT and diathermy models of permanent MCAO have distinctly different characteristics and one explanation for the different results could be the completeness of the MCAO using the diathermy model. The electrocoagulation and subsequent section of the MCA precludes any possible residual flow through the MCA. In the ILT model, even when the thread is left permanently in place at the origin of the MCA (permanent ILT), the MCA itself is still patent and there is still a possibility of a small residual blood flow through the MCA, which could be improved by oestrogen. The second distinct difference between the two models relates to this point in that the endothelium and smooth muscle of the MCA are intact in the ILT model compared with being totally destroyed after electrocoagulation. Oestrogen receptors are present on endothelial cells (Stirone et al, 2003) and oestrogen-induced vasodilatation may be mediated via endothelial nitric oxide synthase (Pelligrino et al, 2000). Thus, the lack of benefit with oestrogen in the present study might be related to the completeness of occlusion and a lack of reperfusion with the diathermy model, thereby allowing detrimental effects of oestrogen to be unmasked.
Other considerations for the different results include treatment regimen and strain used. Regarding dose, supraphysiologic doses of oestrogen have failed to induce neuroprotection (Rusa et al, 1999) or improve perfusion (Pelligrino et al, 1998) and have been reported to exacerbated ischaemic damage (Wallace et al, 2003) in some published studies. Regarding the delivery system and treatment duration, neuroprotection has been observed using similar subcutaneous oestrogen implants to the present study (Hurn and Macrae, 2000), but only one of these studies has the implant in place for a similar duration before MCAO as in the present study, and that study uses a transient MCAO model (Rusa et al, 1999). The strain used in the present study is not likely to explain the different results since exacerbation of ischaemic damage after MCAO has been reproduced in Wistar Kyoto (Carswell et al, 2004) and in Sprague–Dawley (Wallace et al, 2003) rats.
The mechanisms behind the detrimental effects of 17β-oestradiol are currently unclear. They may involve an action on neuronal cell bodies because our two methodological approaches found no significant effects of 17β-oestradiol on axonal pathology. The volume of irregular NF-200 staining (axonal damage) correlated significantly with the volume of neuronal perikaryal damage. Oestrogen influenced the relationship between volumes of neuronal perikaryal damage and axonal damage (irregular NF-200 staining) such that with oestrogen treatment the extent of axonal damage did not match the increased infarct volumes. Further investigation is required to elucidate the biologic significance of this. A plausible mechanism to explain exacerbation of neuronal perikaryal damage, but not axonal damage, is the enhancement of excitotoxicity. This could be mediated by an upregulation or potentiation of NMDA receptors, which are found on neuronal cell bodies, but not on axons (Jones and Baughman, 1991). 17β-Oestradiol has been shown to increase NMDA receptor mRNA levels in the cerebral cortex (Brann et al, 1993), increase NMDA-binding sites in CA1 pyramidal cells (Woolley et al, 1997) and increase NMDA-dependent Ca2+ transients (Pozzo-Miller et al, 1999).
Though the present results are surprising, they provide some insight into the published results from recent randomised clinical trials in postmenopausal women that have shown that oestrogen alone increased the risk of second stroke (Viscoli et al, 2001) and that conjugated equine oestrogen plus medroxyprogesterone increased overall health risks including an increase in stroke rate (Rossouw et al, 2002). Thus, oestrogen clearly has the potential to both ameliorate or exacerbate brain injury and the overall effect of the hormone is possibly determined by a complex interplay of opposing mechanisms that will be influenced by the completeness and duration of blood vessel occlusion, the nature of the brain injury, and the duration and dose of oestrogen treatment. With millions of women worldwide taking ERT, HRT or the contraceptive pill, it is essential to identify the mechanisms responsible for the detrimental effects of oestrogen uncovered in this study, so that new oestrogen-related therapies can be developed that will selectively mediate the beneficial effects but not the detrimental effects of oestrogen in the injured brain.
Footnotes
Acknowledgements
We would like to thank L Gallagher for her excellent technical assistance.