
Abstract
The inhibitory activity of myelin-associated glycoprotein (MAG) on neurons is thought to contribute to the lack of regenerative capacity of the CNS after injury. The interaction of MAG and its neuronal receptors mediates bidirectional signaling between neurons and oligodendrocytes. The novel finding that an anti-MAG monoclonal antibody not only possesses the ability to neutralise the inhibitory effect of MAG on neurons but also directly protects oligodendrocytes from glutamate-mediated oxidative stress-induced cell death is reported here. Furthermore, administration of anti-MAG antibody (centrally and systemically) starting 1 hour after middle cerebral artery occlusion in the rat significantly reduced lesion volume at 7 days. This neuroprotection was associated with a robust improvement in motor function compared with animals receiving control IgG1. Together, these data highlight the potential for the use of anti-MAG antibodies as therapeutic agents for the treatment of stroke.
Keywords
Introduction
It is well established that CNS neurons have a limited capacity for regrowth and repair after injury. However, albeit limited, spontaneous functional recovery is often observed in a number of stroke patients and also in rodent models of cerebral ischemia (Corbett and Nurse, 1998; Hunter et al, 1998, 2000). Inhibitory molecules contained in myelin-including myelin-associated glycoprotein (MAG), Nogo, and oligodendrocyte myelin glycoprotein (OMgp)—are thought to contribute to the inability of the brain to recover from injury by preventing neurite outgrowth (Woolf and Bloechlinger, 2002). It is therefore possible to envisage that the endogenous capacity to repair may be enhanced by the neutralisation of this inhibitory environment. Indeed, neutralisation of MAG has been shown to promote regeneration in peripheral (Torigoe and Lundborg, 1998; Mears et al, 2003) and central nerves (Wong et al, 2003) after injury. In addition, anti-Nogo antibodies have been shown to promote regeneration in a number of CNS lesion paradigms (Schnell and Schwab, 1993; Thallmair et al, 1998; Brosamle et al, 2000) and to improve functional recovery after focal cerebral ischemia in the rat (Papadopoulos et al, 2002; Wiessner et al, 2003). Together, these data support the hypothesis that neutralising components of the inhibitory environment may offer a new therapeutic strategy for the treatment of acute neuronal injury and other neurodegenerative diseases.
Although the signalling pathways for MAG, OMgp, and Nogo-66 may converge in neurons (Wang et al, 2002), there are also differences in the neuronal receptors used by these molecules, suggesting different functional activity (Vinson et al, 2001, 2003; Niederost et al, 2002; Vyas et al, 2002). Interestingly, MAG is likely to play a role in oligodendrocyte function that may be distinct from that of Nogo or OMgp. In oligodendrocytes, the cytoplasmic region of MAG has been shown to interact with several molecules involved in cytoskeletal organisation and signal transduction (Umemori et al, 1994; Jaramillo et al, 1994; Kursula et al, 2000, Kursula et al, 2001). Antibody engagement of oligodendrocyte cell surface MAG leads to activation of Fyn kinase, a molecule key to oligodendrocyte differentiation and survival and to myelination (Umemori et al, 1994). Furthermore, MAG knockout animals exhibit defects in myelin that resemble degenerative changes observed in brains of multiple sclerosis and encephalomyelitis patients (Lassman et al, 1997). Therefore, in addition to a role in preventing neuronal regeneration, MAG is also involved in maintaining the structure and function of oligodendrocytes and myelin.
In the present studies using an anti-MAG antibody (Poltorak et al, 1987; Mears et al, 2003; Wong et al, 2003), we report that in addition to neutralisation of the inhibitory effect of MAG on neurons, the antibody also directly protected oligodendrocytes from oxidative stress-induced cell death in vitro. Moreover, both central and systemic administration of the anti-MAG antibody after focal cerebral ischemia in the rat resulted in a rapid improvement in motor function and was associated with a substantial reduction in lesion area. These studies highlight the potential for the use of anti-MAG antibodies as therapeutic agents for the treatment of stroke.
MATERIALS AND METHODS
Materials
Anti-MAG monoclonal antibody, control mouse IgG1, and anti-myelin basic protein (MBP) and Tau 1 were purchased from Chemicon (Hampshire, UK). Anti-glycogen-associated protein 43 (GAP43) was purchased from Sigma (Dorset, UK). Anti-egr-1 antibody was purchased from Santa Cruz Technologies (CA, USA). A Western blot of rat tissue lysates, prepared under nonreducing conditions, was purchased from Chemicon (Hampshire, UK).
In Vitro Assays
Western blotting was performed using standard techniques. The membrane was probed with anti-MAG antibody at 1 μg/mL followed by anti-mouse horse radish peroxidase (HRP) and detection using enhanced chemiluminescence (ECL, Amersham, Bucks, UK).
Each in vitro assay was performed on at least three independent occasions. For cell adhesion assays, MAG-Fc was coated onto a 96-well dish at 10 μg/mL. After washing, triplicate wells were incubated with different concentrations of anti-MAG antibody. Wells were then washed and 400,000 calcein-labelled cerebellar granule neurons (prepared as described, Vinson et al, 2001) were added per well. Plates were centrifuged at 18 g for 1 min and then incubated at room temperature for 30 min. Wells were washed and the plate was read using Ascent software. Results are presented as mean and s.d. of a representative experiment.
Neurite outgrowth experiments were performed as described (Vyas et al, 2002). Membranes prepared from Chinese hamster ovary (CHO) cells expressing human MAG (Vinson et al, 2001) or wild-type CHO cells were coated onto a 96-well plate at 0.12mg/mL for 4 hours at room temperature. Wells were then incubated with anti-MAG antibody for 1 hour at room temperature and then washed once with phosphate-buffered saline (PBS). Cerebellar granule neurons (CGN) were plated (20,000 cells/well) and incubated overnight. Cells were fixed, stained, and quantified as described (Vinson et al, 2001).
Cultures of primary oligodendrocytes were established from the cortex of P0-P2 Sprague-Dawley rat pups as described (Vinson et al, 2001) and differentiated in culture for 5 days, at which point they extended out myelin-like membranes expressing myelin markers and cell surface MAG (Vinson et al, 2001). For RT-PCR, RNA was extracted from cells using TRIzol (Invitrogen, Paisley, UK). One microgram of total RNA from cells or rat brain was reverse transcribed using standard methods. Myelin-associated glycoprotein exists in two isoforms, long and short, due to alternative splicing of exon 12 and usage of different stop codons. The following polymerase chain reaction (PCR) primers, spanning exon 12, were used (forward, 5′-CCCG AATTCCGAATCTCTGG; reverse, 5′-CTCAGCCAGCTCCT CTGTCAGGG), giving products of 180 bp (MAG-long) and 225bp (MAG-short). Positive control templates were 250 pg of plasmid DNA containing either MAG-long or MAG-short fragments that had been TA cloned into pCR2.1TOPO vector (Invitrogen, Paisley, UK). Negative controls were RT reactions containing no RNA (not shown).
For survival assays, oligodendrocytes were seeded in 48-well plates (5 × 104 per well) in 500 μL Sato′s medium (400ng/mL T3, 400ng/mL T4, 2mmol/L glutamine, 50U/mL penicillin, and 50 μg/mL streptomycin, 5mL N2 supplement (Invitrogen, Paisley, UK)) with 0.5% fetal calf serum and were used at 5 days in vitro. Glutamate (2 mmol/L final concentration) and antibodies were diluted in cystine-free Dulbecco's minimal essential media, and cystine (0.2 mmol/L final concentration) in 1N HCl. All incubations were performed at 37°C for 16 hours. Glutamate/antibodies were all added simultaneously. Cell survival was quantified by colorimetric 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide assay (Skaper et al, 1990). Absorbance was measured at 570 and 630 nm with a SpectroMAX 250-ELISA plate reader. Granule neurons were prepared from 8-day-old rat cerebellum. Cells were plated in 48-well tissue culture plates coated with poly-d-lysine ± myelin. Eight days after cell plating, cultures were incubated for 30 min, at room temperature, with Mg2+-free Locke's solution (+ 100mmol/L glutamic acid ±10, 5, 1, 0.5, or 0.1 μg/mL anti-MAG or purified mouse IgG1). After 30 min, the medium was removed and basal Eagle's medium containing penicillin/streptomycin, 2 mmol/L glutamine, and 25 mmol/L KCl plus antibody was added to the cells. Cell survival was quantified 24 hours later by measuring lactate dehydrogenase (LDH) release into the culture medium (Roche, Lewes, UK) following the manufacturer's instructions.
Intracerebral Ventricular Cannulation
All experiments were conducted according to the requirements of the United Kingdom Animals (Scientific Procedures) Act (1986) and conformed to GlaxoSmithKline ethical standards.
Under halothane anaesthesia, intracerebral ventricular cannulae were positioned in the left lateral cerebral ventricle (coordinates: 1.6 mm from the midline, 0.8 mm caudal from the bregma, 4.1 mm from the skull surface, incisor bar –3.2 mm below zero). Seven days after surgery, correct cannula placement was verified by an intense drinking response to angiotensin II (100 ng, Simpson et al, 1978). Nine days later, animals underwent focal cerebral ischemia.
Focal Cerebral Ischemia
Transient (90-min) focal cerebral ischemia was induced in male Sprague-Dawley rats, each weighing between 300 and 350 g. Under halothane anaesthesia, middle cerebral artery occlusion (MCAO) was performed using the intraluminal thread technique as described previously (Longa et al, 1989). Animals were maintained normothermic throughout the surgical procedure. Only those animals displaying tight circling behaviour 1 hour after MCAO were included in the study. Animals were maintained for 1 week, at which time animals were killed by transcardial perfusion of 0.9% saline followed by 4% paraformaldehyde in 100 mmol/L phosphate buffer and processed for cresyl fast violet (CFV) staining, egr-1, MBP, or Tau 1 immunohistochemistry as described previously (Irving et al, 2001). The area of neuronal and white matter damage was measured using an Optimas 6.1 imaging package as described previously (Irving et al, 2001). The operator was masked to treatment groups. Data are expressed as mean area (mm2)±s.d.
Dosing Regime
Anti-MAG monoclonal antibody and mouse IgG1 isotype control antibody were dialysed against sterile 0.9%. sodium chloride overnight and concentrated (0.5 mg/mL). Intracerebral ventricle administration: animals received 2.5 μg of anti-MAG antibody or 2.5 μg mouse IgG1 intracerebral ventricular 1, 24, and 72 hours after MCAO (5 μL per dose). Intravenous administration: animals received 200 μg of anti-MAG antibody or 200 μg mouse IgG1 intravenously 1 and 24 hours after MCAO. The experimenter was masked to the identity of each dosing solution.
Behavioural Outcomes
Neurological score: 1 hour, 1, 2, and 7 days after MCAO, animals underwent neurological assessment as described previously (Hunter et al, 2000) with the addition of grip strength measurements (scores 2 for good right forelimb grip and 1 for weak grip). The total score is 27 for a normal animal. For the intravenous study, this neurological score was modified further with grip strength being scored from 0 to 3, motility 0 to 4, general condition 0 to 4, and circling 0 to 5. The total score is 33 for a normal animal. Data are presented as median values or box plots (median and 25:75 quartiles). Statistical analysis was Kruskal-Wallis ANOVA.
Beam walking was used as a measure of hindlimb and forelimb coordination by means of distance travelled across an elevated 100-cm cylindrical beam as previously described (Virley et al, 2000). Before MCAO, rats were trained to cross the beam from start to finish and baseline measurements were taken the day before MCAO. For testing, each rat was given 2 trials 24 hours and 7 days after MCAO; the data represent a mean±s.d. of the two trials. Statistical analysis was ANOVA followed by Student's t-test.
Immunohistochemistry
Immunohistochemistry was conducted using standard procedures as described previously (Irving et al, 2001). Incubation with the primary antibody was conducted (MBP 1:750, Tau 1 1:500, GAP43 1:750, egr-1 1:200) overnight at 4°C. Using analySIS imaging system software, the number of egr-1 positive nuclei were counted per 25 μm2 area. Twelve areas were measured from the contralateral and ipsilateral: striatum, hindlimb cortex, and cingulate cortex in two separate sections per animal (n = 6/group). The operator was masked to treatment groups. The data were presented as mean±s.d. number of nuclei/25 μm2. Statistical analysis consisted of a one-way ANOVA with repeated measures (Statistica v 6.0, Stat Soft Inc.).
RESULTS
Anti-MAG Monoclonal Antibody Blocks MAG-Mediated Inhibition of Neurite Outgrowth and Protects Oligodendrocytes From Cell Death
The affinity of anti-MAG antibody for rodent MAG was found to be 0.7 nmol/L using Biacore technology (data not shown). In Western blots across a panel of rat tissues, a single band was seen only in the brain, indicating a high degree of specificity for MAG (Figure 1A). In assays examining MAG-neuron interaction, almost maximal blockade of neuronal adhesion to MAG occurred at 0.3 μg/mL (Figure 1B). At 0.5 μg/mL the antibody blocked MAG-mediated inhibition of neurite outgrowth (Figure 1C). These data confirmed and extended previous findings showing that the antibody was of high affinity and specificity and that it neutralised the effect of MAG on neurons at low concentrations (Yang et al, 1996; Vinson et al, 2001).
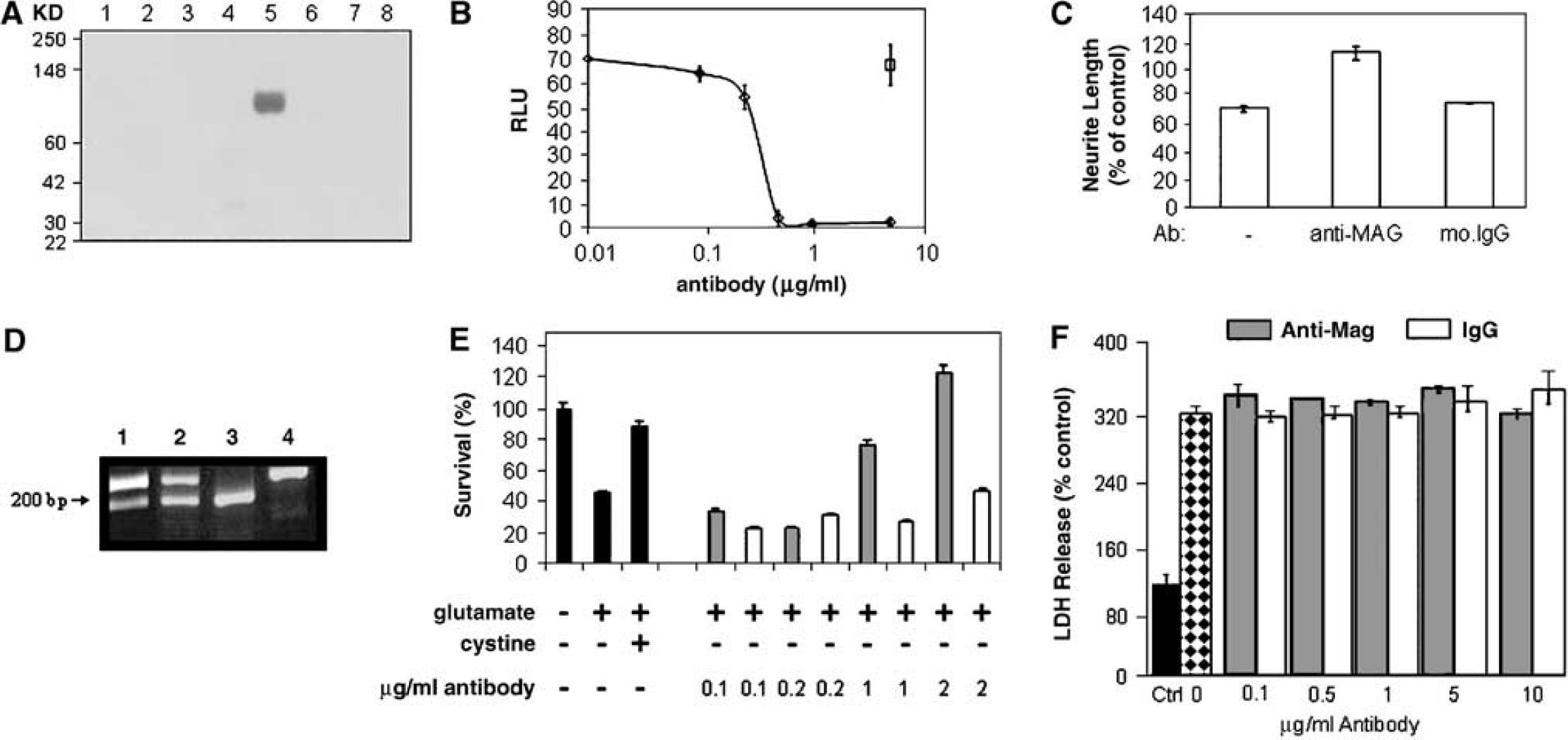
Anti-MAG neutralises the effect of myelin-associated glycoprotein (MAG) on neurons and promotes oligodendrocyte survival. (
The anti-MAG antibody was then used to examine its ability to protect oligodendrocytes from glutamate-mediated oxidative cell death. At 6 days in vitro, the oligodendrocytes used in these studies, like adult rat brain, expressed both long and short isoforms of MAG at the RNA level (Figure 1D), and stained positive for MAG at the cell surface (Vinson et al, 2001). These cultured oligodendrocytes displayed other features of the mature phenotype, including expression of galactocerebroside, CNPase, and Nogo-A (unpublished observations). Treatment with 2 mmol/L glutamate in cystine-free medium for 16 hours reduced cell viability by 60% (Figure 1E). Death was not prevented by the N-methyl-d-aspartate (NMDA) receptor antagonist MK801 or by the kainate/α-amino-3-hydroxy-5-methylisoazole-4-propionic acid (AMPA) receptor antagonist 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX; data not shown), but was completely reversed by the addition of cystine (Figure 1E). Anti-MAG antibody, but not control IgG1 reduced the degree of cell death (Figure 1E). In contrast, anti-MAG antibody treatment (0.1 to 10 μg/mL) failed to protect cerebellar granule neurons in culture against glutamate toxicity when cultured in the presence or absence of a myelin substrate (Figure 1F).
Central Administration of Anti-MAG Antibody Provides Functional Recovery and Neuroprotection Following Stroke
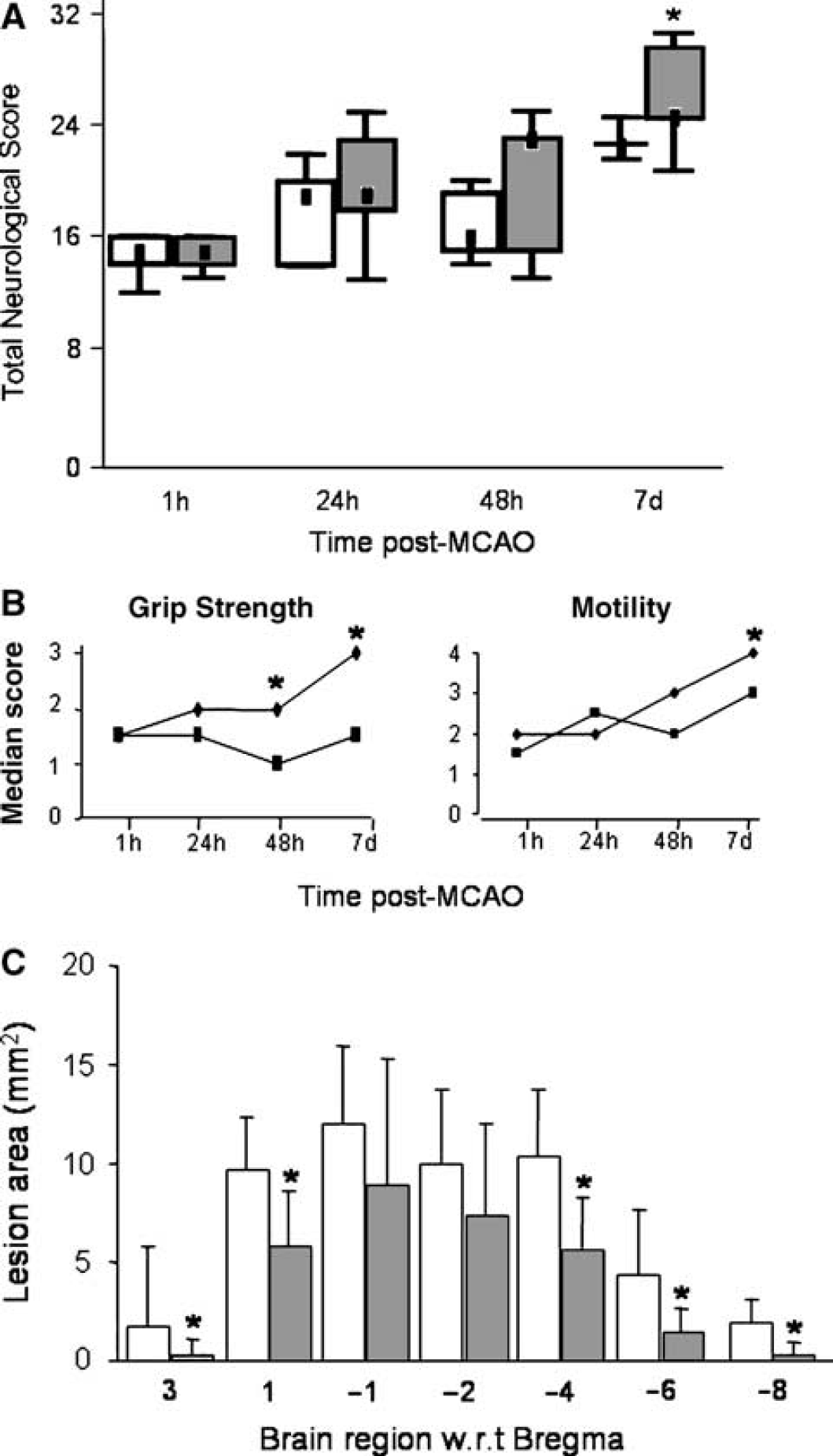
To investigate the effect of the anti-MAG antibody in a transient MCAO model, antibody was administered directly into the left lateral ventricle to avoid the complication of blood-brain barrier passage. At 1 hour after MCAO, animals in both treatment groups showed marked impairment in neurological score (Figure 2A). However at 24 hours, 48 hours, and 7 days after MCAO, animals treated with anti-MAG antibody showed significantly improved total neurological score compared with those treated with control IgG1 (Figure 2A). This improvement was mainly attributed to improved performance in the following tests: paw placement, circling, motility, horizontal bar (Figure 2B) grip strength (24 hours, P = 0.049; 48 hours, P = 0.0495; 7 days, P = 0.243), inclined plane (24 hours, P = 0.006; 48 hours, P = 0.006; 7 days, P = 0.169), and visual forepaw reaching.
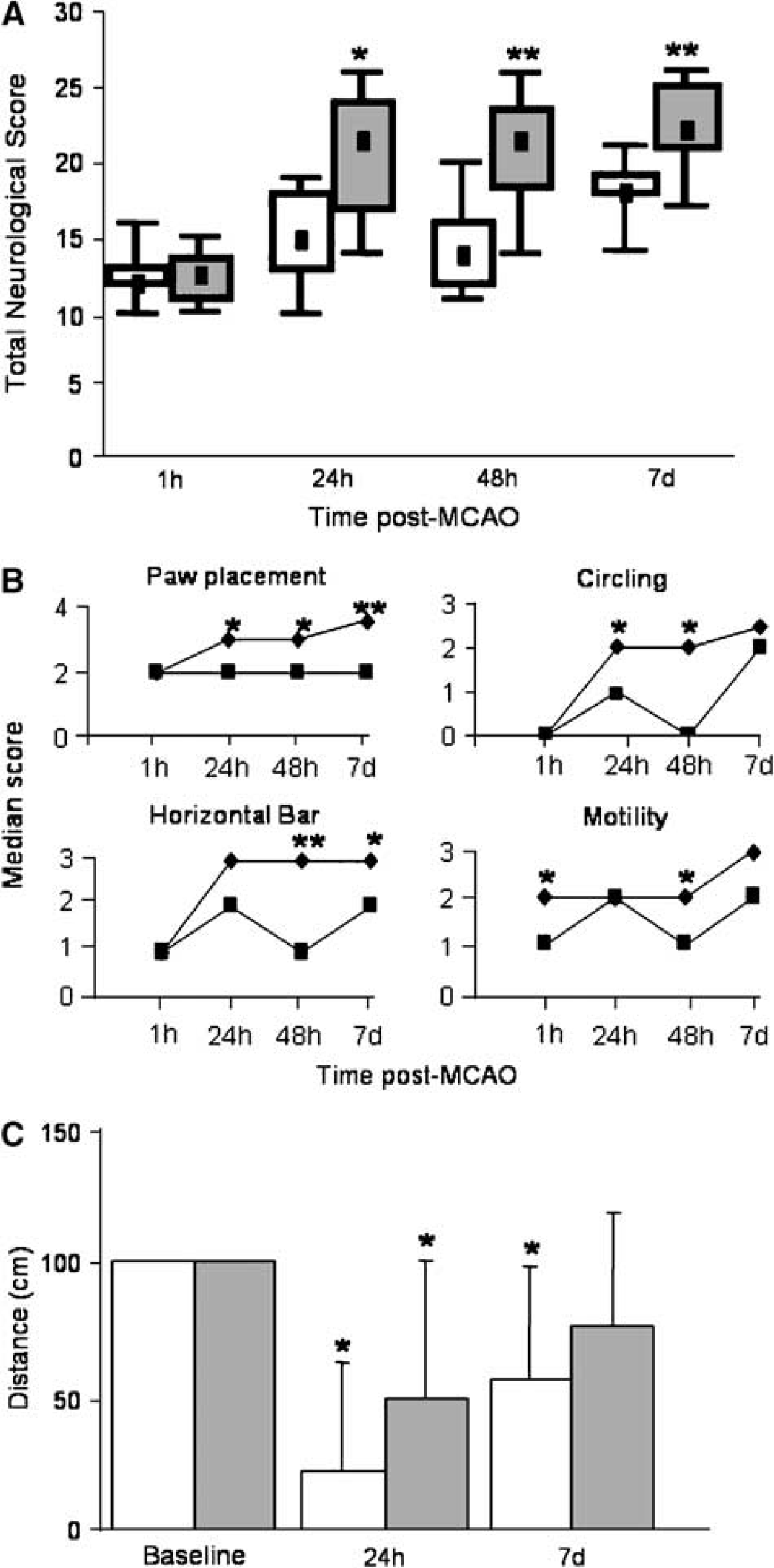
Central administration of anti-MAG antibody (2.5 μg) improves motor function after middle cerebral artery occlusion (MCAO). (
Before surgery all animals were trained to cross a cylindrical beam (100 cm). At 24 hours after surgery, anti-MAG-treated animals travelled twice the distance of their IgG1-treated counterparts (the mean distance travelled being 50 and 22 cm, respectively). At 7 days after MCAO, the performance of animals treated with IgG1 remained significantly impaired compared with baseline (P = 0.01). In contrast, the performance of animals treated with anti-MAG antibody improved and was not significantly different from baseline (P = 0.07). These data show that anti-MAG antibody treatment accelerated recovery of this beam walking task compared with IgG1-treated controls (Figure 2C).
Improvement in functional recovery was accompanied by a marked reduction in lesion area measured 7 days after transient MCAO. Animals treated with anti-MAG antibody showed reduced lesion areas (approximately 40% to 50%) as detected by CFV staining (Figure 3A) compared with IgG1-treated controls. In addition, a similar reduction in the area of myelin degradation (as delineated by disrupted MBP immunostaining) was observed in anti-MAG-treated animals (Figures 3B and 3C). Tau 1 immunostaining in oligodendrocytes is thought to be indicative of cellular integrity after stroke (Irving et al, 2001). Tau 1 is increased in oligodendrocytes up to 24 hours after MCAO in the ischaemic hemisphere (Irving et al, 1997; Valeriani et al, 2000), but is lost as the white matter degenerates at 1 week following MCAO (Irving et al, 2001). The number of Tau 1 positive, histologically normal, oligodendrocytes present within and around the ischaemic lesion was greater following anti-MAG compared with IgG1-treated animals (Figure 3D). The ability of the anti-MAG antibody to reduce Tau 1 loss in oligodendrocytes within the lesion area suggests that treatment may have maintained a degree of oligodendrocyte integrity.
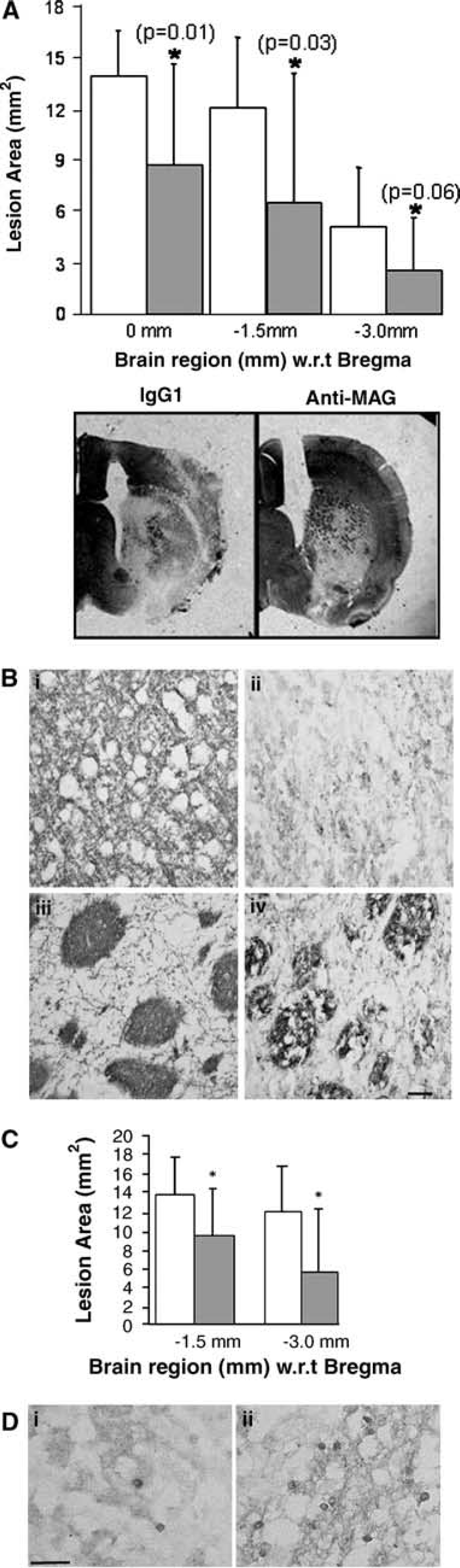
Central administration of anti-MAG antibody (2.5 μg) reduced lesion area. (
GAP43 is routinely used to investigate axonal sprouting following ischemia (Stroemer et al, 1998); however, in the present study no increased GAP43 staining was detected in IgG1 or anti-MAG-treated animals. egr-1 staining has previously been shown to correlate with recovery of function after trauma (Vargo and Marshall, 1996) and neuronal plasticity (Wang et al, 1994; Wallace et al, 1995). egr-1 immunoreactivity was increased in the contralateral cingulate cortex and ipsilateral hindlimb cortex in anti-MAG-treated animals compared with IgG1-treated animals 7 days after MCAO (Figures 4A-4C).
Therapeutic Potential for Anti-MAG Antibodies: Intravenous Administration of Antibody Provides Functional Recovery and Neuroprotection
At 48 hours after MCAO, animals treated with anti-MAG antibody showed significant improvement in paw placement (P = 0.048) and grip strength (P = 0.033). At 7 days after the onset of cerebral ischemia, animals treated with anti-MAG antibody continued to improve (paw placement, P = 0.041; grip strength, P = 0.048; motility, P = 0.05) and showed significant improvement in total neurological score (Figures 5A and 5B).
Despite the effect of the anti-MAG antibody on motor function being less robust when administered systemically, a marked reduction in lesion area was detected. Anti-MAG antibody treatment significantly reduced lesion area by 25% to 70% (as determined by CFV) compared with isotype controls when examined 7 days after MCAO (Figure 5C). At low levels, peripherally administered antibodies are able to enter the CNS (Poduslo and Curran, 2001) and have been shown to bind CNS antigens (Bard et al, 2000). Furthermore, the level of penetration may be greater when the blood-brain barrier is compromised, however it is worth noting that the systemic dose of antibody administered was likely to result in brain concentrations significantly lower than that achieved by direct administration. Nonetheless, the effect of the antibody on lesion size was similar in magnitude to those treated with anti-MAG antibody by intracerebral ventricle administration and other reported putative neuroprotective agents (Ma et al, 1998; Mackay et al, 1996; Minematsu et al, 1993).
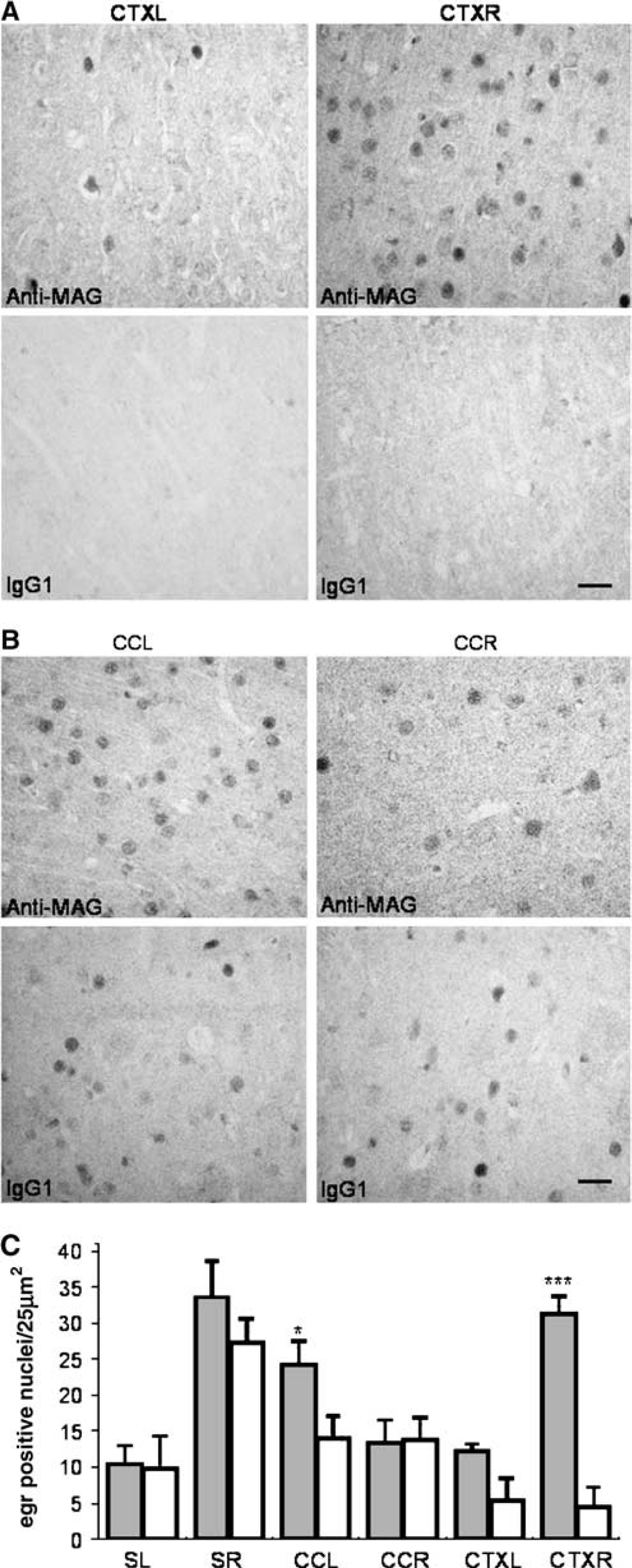
Anti-MAG antibody increased the number of egr-1 positive nuclei in the CCL and CTXR. Photomicrographs showing egr-1 immunostaining in the (
Systemic administration of anti-MAG antibody (200 μg intravenous) improved function and reduced lesion area. (
DISCUSSION
The present study demonstrates that an anti-MAG monoclonal antibody not only neutralises the inhibitory effect of MAG on neurite outgrowth but also protects oligodendrocytes from glutamate-mediated oxidative toxicity in vitro. Furthermore, when administered (centrally or systemically) after MCAO in the rat, this anti-MAG antibody provides a robust and rapid improvement in functional recovery, which is associated with a marked reduction in lesion area and myelin degradation across many different brain levels 7 days after transient MCAO.
The degree of neuroprotection offered by the anti-MAG antibody was both marked and intriguing. Because of the role that MAG plays in oligodendrocyte integrity and myelination, we investigated the potential that the anti-MAG antibody may act to protect oligodendrocytes/myelin from ischaemic injury or indeed directly protect neurons from excitotoxic stress. While oligodendrocytes have been reported to express AMPA and kainate receptors (Steinhausen and Gallo, 1996) and AMPA/kainate receptor activation can induce oligodendrocyte cell death in vitro (Sánchez-Gómez and Matute, 1999; Deng et al, 2003), the response of oligodendrocytes to focal ischemia appears to be predominantly free radical mediated (Irving et al, 1997). Oxidative glutamate toxicity towards cells lacking functional NMDA receptors can also be a component of the excitotoxicity-initiated cell death pathway (Schubert and Piasecki, 2001). Previous studies have shown that after MCAO, the number of Tau 1 positive oligodendrocytes increased as early as 40 min after the onset of ischemia indicative of stress in these cells (Irving et al, 1997). In this study, the number of Tau 1 positive cells was significantly decreased by the spin trap agent α-phenyl-tert-butylnitrone (PBN) but not by AMPA or NMDA receptor antagonists at doses known to reduce ischaemic injury (Irving et al, 1997). Oligodendrocytes are extremely sensitive to oxidative stress, and glutamate has been shown to induce death through glutathione depletion and thus oxidative stress rather than receptor-driven excitotoxicty (Oka et al, 1993; Back et al, 1998). Here we report for the first time that an anti-MAG antibody has the ability to protect oligodendrocytes from glutamate-mediated oxidative stress in vitro. Our observation that the anti-MAG antibody failed to directly protect neurons while protecting oligodendrocytes from glutamate toxicity in vitro suggests that protection of oligodendrocytes may contribute to the neuroprotection seen in vivo. This is supported by a reduction in the degree of myelin breakdown (as detected by decreased MBP immunostaining) in anti-MAG-treated animals compared with controls.
Oligodendrocytes and neurons are interdependent for function and survival and communicate via cell surface receptors and soluble factors (Fields and Stevens-Graham, 2002). Myelination is critical for neuronal maturation and function and demyelination leads to axonal atrophy (Ferguson et al, 1997; Lovas et al, 2000). Furthermore, oligodendrocytes secrete factors that promote neuronal survival such as brain-derived neurotrophic factor, neurotrophin-3, insulin-like growth factor-1, neuregulins, and glial cell-derived neurotrophic factor (Du and Dreyfus, 2002; Wilkins et al, 2003). It is tempting to speculate that protection of oligodendrocytes after stroke may result in the maintenance of myelin structure and promotion of neuronal survival and function. The recent findings of Lappe-Siefke et al (2003), indeed, support the idea that oligodendrocytes support axonal function and survival throughout adult life, independent of myelin.
Increased synaptogenesis and neuronal sprouting have previously been reported in the contralateral cingulate cortex and tissue immediately adjacent to the lesion and correlated with functional recovery after MCAO (Stroemer et al, 1995, Stroemer et al, 1998). In the present study however, no increase in GAP43 staining was detected in IgG1- or anti-MAG-treated animals 1 week after MCAO. It is possible that this reflects differences in the model and species used in the two studies, that is, normotensive animals were used in the current study versus spontaneously hypertensive rats in the Stroemer studies. Furthermore, the model used in the current study has subcortical and cortical damage compared with the discrete cortical lesion used by Stroemer et al (1995), (1998). Egr-1 is essential for synaptic plasticity in the hippocampus (Wallace et al, 1995; Jones et al, 2001) and has been shown previously to correlate with recovery of function following trauma (Vargo and Marshall, 1996). Increased egr-1 immunostaining was observed in the present study within the contralateral cingulate cortex and tissue immediately adjacent to the lesion, indicative of modulated plasticity in these regions. These data, coupled with the neuroprotection offered by the anti-MAG antibody and the previously reported ability of this antibody to stimulate regrowth following injury (Torigoe and Lundborg, 1998; Mears et al, 2003; Wong et al, 2003), highlight the fact that anti-MAG antibodies may offer both a neuroprotective and regenerative strategy for the treatment of stroke. In the present studies the anti-MAG antibody was administered 1 hour after the onset of ischemia. It is currently unclear whether the enhanced functional recovery observed in the present study was solely due to the resulting neuroprotection or a combination of protection and enhanced plasticity. Studies to further investigate the effect of anti-MAG on neurite outgrowth and plasticity in longer-term studies are underway.
Here we report the novel finding that an anti-MAG antibody can enhance oligodendrocyte survival following oxidative stress-induced injury in vitro. Administration of the anti-MAG antibody to rats following transient MCAO induced a rapid improvement in functional outcome and marked reduction in lesion area. Moreover, the ability of the antibody to protect the brain from ischaemic injury when administered intravenously demonstrates the potential for anti-MAG antibodies as therapeutic agents for the treatment of stroke.
Footnotes
Acknowledgements
The technical expertise provided by M Duxon, SJ Hadingham, CA Campbell, R Morrow, T Wattam, and G Northern is greatly appreciated. Thanks also go to Rabinder Prinjha for valuable discussions and a careful review of the manuscript.