
Abstract
Posttraumatic hyperthermia (PTH) is a noninfectious elevation in body temperature that negatively influences outcome after traumatic brain injury (TBI). We sought to (1) characterize a clinically relevant model and (2) investigate potential cellular mechanisms of PTH. In study I, body temperature patterns were analyzed for 1 week in male rats after severe lateral fluid percussion (FP) brain injury (n=75) or sham injury (n=17). After injury, 27% of surviving animals experienced PTH, while 69% experienced acute hypothermia with a slow return to baseline. A profound blunting or loss of circadian rhythmicity (CR) that persisted up to 5 days after injury was experienced by 75% of brain-injured animals. At 2 and 7 days after injury, patterns of cell loss and inflammation were assessed in selected brain thermoregulatory and circadian centers. Significant cell loss was not observed, but PTH was associated with inflammatory changes in the hypothalamic paraventricular nucleus (PVN) by one week after injury. In brain-injured animals with altered CR, reactive astrocytes were bilaterally localized in the suprachiasmatic nucleus (SCN) and the PVN. Occasional IL-1β+/ED-1+ macrophages/microglia were observed in the PVN and SCN exclusively in brain-injured animals developing PTH. In animals with PTH there was a significant positive correlation (r=0.788, P<0.01) between the degree of postinjury hyperthermia and the total number of cells positive for inflammatory markers within selected thermoregulatory and circadian nuclei. In study II, a separate group of animals underwent the same injury and temperature monitoring paradigm as in study I, but had additional physiologic data obtained, including vital signs, arterial blood gases, white blood cell counts, and C-reactive protein levels. All parameters remained within normal ranges after injury. These data suggest that PTH and the alteration in CR of temperature may be due, in part, to acute reactive astrocytosis and inflammation in hypothalamic centers responsible for both thermoregulation and CR.
Introduction
Traumatic brain injury (TBI) is associated with functional and lifelong deficits in up to 70,000 to 90,000 survivors each year who require continued treatment and care (Consensus Conference, 1999; Kersel et al, 2001). In both clinical and experimental studies, hyperthermia in the acute postinjury phase is one of the factors that has been associated with worsened outcome after TBI (Dietrich, 1992; Dietrich et al, 1996; Chatzipanteli et al, 2000; Jiang et al, 2002). Posttraumatic hyperthermia (PTH), also known as neurogenic fever, is a syndrome that persists for weeks to months after brain injury and has been reported in 4% to 37% of patients with moderate to severe TBI (Sazbon and Groswasser, 1990; Meythaler and Stinson III, 1994; Childers et al, 1994; Thompson et al, 2003). In several clinical studies, the development of PTH has been correlated with negative outcome in patients after TBI (Sazbon and Groswasser, 1990; Heindl and Laub, 1996; Behr et al, 1997).
Little is known about the molecular and cellular mechanisms underlying the development of PTH. Thought to be caused by injury to the hypothalamus, PTH is characterized by a lack of diurnal variation of temperature, relative bradycardia, a notable absence/lack of perspiration, and relative resistance to antipyretic medications (Cunha and Tu, 1988; Sazbon and Groswasser, 1990; Segatore, 1992; Childers et al, 1994; Powers and Scheld, 1996), all of which can help to differentiate PTH from fever in this population. Since the current empirically based pharmacologic management of PTH is ineffective in most patients and may have significant side effects such as gastric ulcers and bradycardia (Benedek et al, 1987; Horn, 1988; Meythaler and Stinson III, 1994), it is important to develop a clinically relevant model of PTH to gain a better understanding of the mechanisms underlying its development, and guide the evolution of new therapeutic interventions.
The inflammatory response to experimental brain injury includes astrocytosis and neutrophil and microglia/macrophage infiltration (Soares et al, 1995; Aihara et al, 1995; Hill et al, 1996; Knoblach et al, 1999; Hill-Felberg et al, 1999; Chatzipanteli et al, 2000). Accompanying the infiltration of inflammatory mediators into the injured brain parenchyma after TBI is an upregulation of several proinflammatory cytokines (Toulmond and Rothwell, 1995; Fan et al, 1995; Hans et al, 1999; Yatsiv et al, 2002). Cytokines, including interleukinβ (IL)-1β, IL-6, tumor necrosis factor-α (TNF-α), and interferon-γ, and prostaglandins (PGE2) activate thermosensitive neurons in the anterior hypothalamus to produce fever and the associated increase in body temperature. In response to experimental brain injury, IL-1β is upregulated in the CNS (Toulmond and Rothwell, 1995; Fan et al, 1995; Stahel et al, 2000; Morganti-Kossmann et al, 2001). The presence of IL-1β has been correlated with inflammation, cerebral edema, and breakdown of the blood–brain barrier, and is a marker for these processes (Holmin et al, 1997). Because IL-1β has been shown to be critical for stimulating fever as part of the local inflammatory response (Kozak et al, 1998) and hyperthermia after hypothalamic lesioning was diminished by blocking the action of prostaglandins, potentially released in response to IL-1 (Rudy et al, 1977; Rudy, 1980), this cytokine may serve as a potential mediator of PTH.
According to Elmquist et al (1996) and Saper (1998), regional centers within the hypothalamus involved in the febrile response to an immune stimulus include the ventromedial preoptic nucleus (VMPO) and the paraventricular hypothalamic nucleus (PVN) (Elmquist et al, 1996; Saper, 1998). The anterior perifornical nucleus (APF) receives significant input from the VMPO and is also involved in the febrile immune response via feedback to the PVN (Saper, 1998). The present study was designed to characterize the thermoregulatory responses of rats after severe lateral fluid percussion (FP) brain injury and to correlate with the thermoregulatory responses observed histopathologic alterations in selected brain thermoregulatory and circadian nuclei. We hypothesized that severe FP injury would induce, in a subgroup of animals, thermoregulatory changes similar to those seen in humans. Because PTH is also associated with a characteristic loss of diurnal variation, we examined the centers responsible for circadian rhythmicity (CR), including the suprachiasmatic nucleus (SCN) and the PVN (Reppert and Weaver, 2002; Cermakian and Boivin, 2003).
Materials and methods
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. At all times, the investigator strictly adhered to the principles enumerated in the Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (1996).
Study I: Thermoregulation and Histologic Analysis
Thermistor Implantation
A total of 92 male Sprague–Dawley rats (Harlan, Indianapolis, In, USA), weighing 350 to 400 g, were acclimatized to the animal colony for at least 1 week before surgery. Animals were maintained on a 12-hour light/dark cycle (lights on 6 a.m.) and food and water were available ad libitum. Each animal was injected with 87 mg/kg ketamine and 13 mg/kg xylazine, intraperitoneally; once a surgical plane of anesthesia was achieved, the animal was positioned on its back and a midline abdominal incision was made to expose the underlying muscle. Blunt dissection was used to achieve a 1-in opening in the peritoneum and a sterilized precalibrated thermistor (Model TA-F40, Data Sciences International, St. Paul, MN, USA) was placed in the peritoneal cavity. The abdominal wall and skin were then closed separately, bacitracin ointment was applied to the abdominal incision, and the animal was allowed to recover. Animals were administered buprenorphine 0.1 mg/kg intraperitoneally 12 hours after surgery as needed for pain management. After thermistor implantation, all animals were housed singly and provided with the same enrichment protocol for the remainder of the study.
Fluid Percussion Injury
Seven days after the thermistor implantation, animals were randomly assigned to two groups: either anesthesia and surgery without FP injury (sham-injured, n=17) or anesthesia and surgery followed by lateral FP brain injury of high severity (3.1 to 3.8 atm, n=75). Animals were anesthetized with isoflurane (induced with 3% to 3.5%, maintenance with 1.5% via nose cone) throughout the procedure. Once induced, the animal was placed in a stereotactic frame. A scalp incision was made, the scalp and temporal muscle were reflected, and a 5.0 mm craniectomy was made in the skull over the left parietal cortex, midway between bregma and lambda as originally described (McIntosh et al, 1989). A female Luer-Lok fitting was cemented into the open craniectomy site, and then used to attach the animal to the FP injury device. Lateral FP brain injury was induced using a device consisting of a Plexiglas, cylindrical, saline-filled reservoir bound at one end by a Plexiglas plunger mounted on O-rings (Virginia Commonwealth University Department of Biomechanical Engineering, Richmond, VA, USA). The opposite end of the cylinder was capped with a male Luer stub, which was connected to the animal via the female Luer-Lok fitting. A pendulum was allowed to drop, striking the plunger and producing a fluid pressure pulse of 21 to 23 ms in duration via the rapid injection of saline into the closed cranial cavity. This resulted in brief deformation of neural tissue. Twenty-six animals died from injury-related complications within the first 24 hours after injury (35% mortality rate), leaving 49 surviving injured animals for analysis. Normothermia during all surgical procedures and in the acute postinjury period was maintained through the use of a feedback-heating pad (Harvard Apparatus, Holliston, MA, USA) placed under the animal. The temperature was maintained at 37°C until the animal was able to right itself.
Temperature Assessment
Core temperature and motor activity were monitored by telemetry (Data Sciences International, St. Paul, MN, USA) while animals were housed at a mean ambient temperature of 24°C (±2°). Temperature was recorded at 15-min intervals from a sensor in the transmitter. Activity, also recorded at 15-min intervals, was determined by measuring signal strength as the animal moved about the home cage in relationship to a receiver board under the cage. Sampled data were sent to a matrix coupled to a desktop computer and then automatically stored for each animal until analysis using the Dataquest Advanced Research Technology (A.R.T., Data Sciences). The 15-min data were averaged over each hour for analysis. This method of measuring core temperature has been previously shown to have a highly significant correlation with brain temperature measurement, and can be utilized to assess brain temperature without damaging the parenchyma (Taylor et al, 2002).
Determination of Posttraumatic Hyperthermia
Comparison of baseline midline estimating statistic of rhythm (MESOR) temperature data to postinjury data occurred for each animal utilizing the Dataquest A.R.T. software system. Posttraumatic hyperthermia was diagnosed if the animal's temperature patterns after injury met the following criterion: a pattern of altered thermoregulation of hyperthermia (1°C over baseline MESOR) present without normal diurnal variation on at least 2 consecutive days (Lausberg, 1971; Cunha and Tu, 1988; Segatore, 1992; Whyte et al, 1993; Childers et al, 1994; Powers and Scheld, 1996; see also Refinetti, 1999 for a review of circadian physiology). A lack of normal diurnal variation was defined as a significant difference in daily temperature amplitude up to 1 week after injury when compared with the mean amplitude of temperature in the 3 days before injury. The extent of hyperthermia after injury was determined by subtracting baseline MESOR temperature from the maximum body temperature in the 7 days after injury.
Motor Assessment
The righting reflex was assessed immediately after injury as an indicator of severity of injury, allowing for immediate determination of adequateness of injury (Dixon et al, 1987). Any injured animal with righting reflex <5 mins was determined to have a mild injury and was excluded from the study (Schmidt and Grady, 1995). No animals in study I were excluded based on this criterion. Posttraumatic motor dysfunction was assessed at 48 hours and 1 week after injury using the composite neuroscore. A trained observer who was blinded to each animal's treatment evaluated neurologic function. Animals were scored from 4 (normal) to 0 (severely impaired) for each of the following indices: left and right forelimb flexion during suspension by the tail, left and right hindlimb flexion when the forelimbs remain on a hard surface and the hindlimbs are lifted up and back by the tail, and the ability to resist lateral pulsion toward the left and right. In addition, animals were tested for their ability to stand on an inclined plane (angle board) while facing left, right, and upward; scores (0 to 4) were assigned for each direction according to their difference from preinjury performance and then averaged for inclusion in the composite score. A composite neuromotor score (0 to 28) was generated by combining the scores for each of these seven tests, with a score of 28 indicating maximal motor function (Sanderson et al, 1999).
Histologic Analysis
At 48 hours or 1 week after FP brain injury, animals were reanesthetized with 65 mg/kg sodium pentobarbital intraperitoneally. All animals were perfused with intracardiac heparinized saline followed by 4% paraformaldehyde. Brains were removed and stored in fixative for 24 hours at 4°C. Brains were paraffin embedded and 6-μm-thick coronal serial sections were cut on a rotary microtome.
To quantify neuronal cell loss at 1 week after injury in the hypothalamus, thermoregulatory and circadian nuclei were selected for cell counting. For each animal, two nonconsecutive 6-μm sections were randomly taken from selected levels that contained the VMPO (bregma −0.3 mm), APF nucleus (bregma −0.8 mm), SCN (bregma −1.3 mm), and PVN (bregma −1.8 mm) (Paxinos and Watson, 1990; Elmquist et al, 1996; Saper, 1998).
Brain sections were selected from sham-injured animals (n=11), brain-injured animals that did not develop PTH (hypothermic) (n=11), and all brain-injured animals that developed PTH (n=11). Brain-injured animals selected for histology were matched for injury level (atm), righting reflex, and 48-hour neuroscore to control for any potential confounding effects of injury severity. Sham-injured animals were selected based on date of surgery to ensure consistency with both injured groups. Slides containing sections from the above regions were deparaffinized, rehydrated, stained with cresyl violet solution (0.3%) (Aldrich, St. Louis, MO, USA), and dehydrated in graded alcohol solutions. Sections were cleared in xylenes, coverslipped in permount, and imaged using a digital camera incorporated with an imaging analysis system (Imaging Research Inc., St. Catharines, Ontario, Canada) integrated with a light microscope.
Images of the ipsilateral and contralateral nuclei were captured at low magnification (× 80) and at higher magnification (× 400). Using the higher magnification pictures, histologic analysis was completed by a trained observer who was blinded to the experimental group after the nuclei of interest had been delineated using well-established anatomic guides (Simerly, 1995; Armstrong, 1995). The observer counted only neurons with round cell bodies and prominent nucleoli (Zhang et al, 1998) using the previously captured image, and this analysis was confirmed by a second blinded investigator. Cell counts from two nonconsecutive brain sections from each animal were averaged to give a mean cell count. Counts were made for both the ipsilateral and the contralateral nuclei.
Immunohistochemistry
For immunohistochemical labeling (n=3 injured without PTH at 48 hours, n=2 sham at 48 hours, representing all animals at 48 hours; n=6 per group at 1 week), animals were randomly selected as a subset of the histologic analysis. Slides containing sections at bregma −0.3, −0.8, −1.3, and −1.8 mm (adjacent to those used for Nissl staining in the 1 week postinjury groups) were deparaffinized and rehydrated, rinsed with Tris-buffered saline (TBS), and then treated with blocking solution (10% normal horse serum (NHS)/3% bovine serum albumin (BSA)/0.1% glycine) for 1 h at room temperature. Sections were then incubated overnight at 4°C with the following primary antibodies in 1% BSA: (a) mouse anti-rat ED-1 (Serotec, Raleigh, NC, USA, 1:50), (b) goat anti-rat IL-1β (R&D Systems, Minneapolis, MN, USA, 1:100), and (c) diluent (1% BSA as negative control). At room temperature on day 2, the slides were rinsed with TBS and then incubated overnight at 4°C with rabbit anti-GFAP (Sigma, St. Louis, MO, USA, 1:100) or diluent. At room temperature on day 3, slides were again rinsed and incubated with the following secondary antibodies in 1% BSA for 2 hours: (a) biotinylated donkey anti-mouse IgG (Jackson Immunoresearch, West Grove, PA, USA, 1:1500), (b) donkey anti-goat Alexa 488 (Molecular Probes, Eugene, OR, USA, 1:500), and (c) coumarin AMCA donkey anti-rabbit IgG (Jackson, 1:100). After this step, slides were again rinsed, incubated with streptavidin Alexa 594 in 1% BSA for 60 mins (Molecular Probes, 1:1,000) at room temperature, subjected to a final rinse, coverslipped with Fluromount G, and visualized with a fluorescent microscope (Nikon, Optical Apparatus, Ardmore, PA, USA). Once cells were visualized, cells were counted bilaterally in the nuclei of interest in a quantitative fashion, and semiquantitatively (Hill et al, 1996; Floyd et al, 2002) in brain regions (such as the corpus collosum) not directly related to thermoregulation or circadian rhythm.
Study II: Physiology
Instrumentation, Physiologic Measurements, and Fluid Percussion Injury
A separate group of 17 male Sprague–Dawley rats (Harlan) underwent thermistor implantation under anesthesia as described above. On day 7 after implantation, animals were reanesthetized with isoflurane and the tail artery was cannulated with PE-50 tubing. Once the catheter was placed and secured, baseline measurements of mean arterial blood pressure (MABP) and heart rate (HR) were obtained and the animal was placed in the stereotactic frame. Before cannulation, animals had been randomized to either sham injury (n=5) or severe FP injury (n=12) groups as described above. Mean arterial blood pressure, HR, and rectal temperature were monitored every 5 mins from the time of catheter placement until 30 mins after FP or sham injury. Baseline arterial blood gases (ABG) (pH, pO2, and pCO2) were obtained 5 mins before injury and 30 mins after injury via the tail artery catheter. Two animals died from injury-related complications immediately after injury (18%), and one injured animal was excluded because of righting reflex <5 mins, leaving nine injured animals in the final analysis. After the 30-min postinjury monitoring period, the cannula was removed; the animal was allowed to recover and was then returned to the home cage for temperature assessment as described above. At 48 hours and 1 week after injury, neurologic motor function was assessed using the composite neuroscore test.
Whole-Blood White Blood Cell Counts and Serum C-Reactive Protein Levels
To rule out infection as a cause of hyperthermia after brain injury, whole-blood samples were obtained from the 14 remaining animals from the arterial line on day 0 and the tail vein on days 2 and 7 after injury for analysis. One half of each sample from each time point per animal was centrifuged, and serum was retained for C-reactive protein (CRP) analysis. Serum samples were frozen at −80°C until all samples were ready for processing. Once thawed, semi-quantitative detection of CRP in the rodent serum was performed using a latex agglutination method with appropriate controls (BioMedica Diagnostics, Windsor, Nova Scotia, Canada). A positive reaction was indicated by agglutination of the latex particles, and semiquantitation was performed using a serum dilution series. Two controls were used in this experiment: human sera and quality control solutions provided by Biomedica Diagnostics. The other half of the whole-blood sample was sent to an accredited reference laboratory (Veterinary Hospital of the University of Pennsylvania Clinical Pathology Laboratory) for hematological analysis of white blood cell (WBC) count. After the 7-day blood draw, the animal was deeply anesthetized with 65 mg/kg pentobarbital intraperitoneally and perfused as described above.
Data Analysis
For both study I and study II, temperature and activity data were initially analyzed using the Dataquest A.R.T. system (Data Sciences). Midline estimating statistic of rhythm, waveform, amplitude, and period were analyzed for each animal to determine the presence or absence of PTH by the previously set criterion. All data were then entered into SPSS 11.5 (SPSS, Chicago, IL, USA) for analysis. Absence of normal diurnal variation was determined by comparing the mean amplitude for temperature in the 3 days before injury with daily postinjury amplitude up to 1 week, using paired t-tests corrected for multiple comparisons. Data are expressed as mean±standard deviation (s.d.) unless otherwise noted. Repeated measures one-way analysis of variance (repeated ANOVA) was used to compare measured physiologic variables among groups, including change in mean daily MESOR from baseline. Independent sample t-tests were used to examine differences between the injured groups on the following parameters: apnea time, seizure time, righting reflex, and injury level.
In study I, for neuroscore, Kruskal–Wallis one-way ANOVA and Mann–Whitney U tests were used. Two-way ANOVA was used to compare histologic cell counts among groups. One-way ANOVA was used to compare immunohistochemical cell counts among groups. For brain-injured animals developing PTH, Pearson's correlation was used to examine the relationship between extent of hyperthermia after injury and the mean number of inflammatory cells in the four thermoregulatory and circadian nuclei examined.
In study II, repeated measures one-way ANOVA (repeated ANOVA) was used to compare measured physiologic variables among groups, including MABP, HR, ABG data, CRP level, and WBC count. A significance level was set at P<0.05 for all tests, correcting for multiple measurements where appropriate.
Results
Study I
Two distinct patterns of thermoregulation predominated after severe FP brain injury. Despite maintenance of physiologic normothermia during surgery/injury, 69% (34/49) of severely brain-injured animals developed profound hypothermia followed by a slow return to baseline MESOR temperature over several hours (Figure 1A). In contrast, 13 of 49 brain-injured animals (27%) experienced PTH (loss of CR and hyperthermia) persisting for 2 or more days (Figure 1B). Profound delayed hypothermia with loss of CR was present in the two remaining animals (4%), both of which had signs and symptoms of distress requiring euthanasia before day 7 after injury. On necropsy, the abdominal organs of these two appeared to have signs of multiorgan dysfunction syndrome (petichial hemorrhages on kidneys and nonperfused bowel and small intestine) and they were excluded from the histologic and behavioral analysis. Irrespective of whether they became hypo- or hyperthermic, the majority of brain-injured animals (75%) had a significant loss of temperature CR (decrease in amplitude, P<0.05) that persisted up to 5 days after injury. This loss of temperature CR was not due to anesthesia or the surgical procedure, because sham-injured animals had a preservation of CR of temperature (Figure 2). Both a significant time effect (P<0.005) and a significant group × time effect (P<0.005) were observed among mean daily temperature patterns of brain-injured animals with PTH, brain-injured animals without PTH, and sham-injured animals, suggesting an injured-group-specific change in MESOR temperature over time (Figure 3). There was also an overall group effect (P<0.005), with animals developing PTH exhibiting significantly different temperatures from sham-injured animals (P<0.05). Specifically, the change in daily MESOR body temperature was significantly greater for PTH animals than for sham-injured animals at days 2 and 3 after injury (P<0.05) (Figure 3).
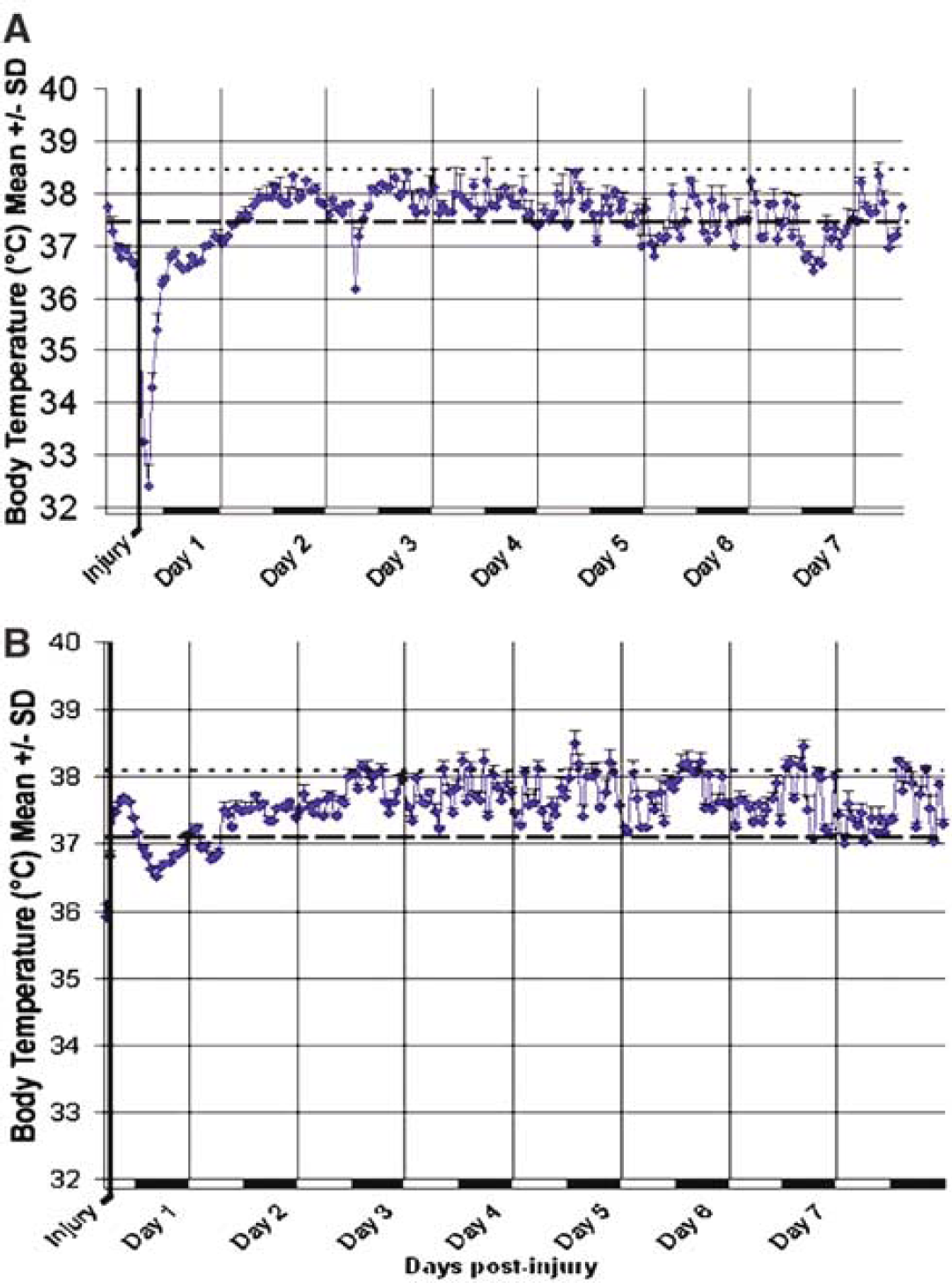
Representative temperature patterns after severe fluid percussion brain injury. (
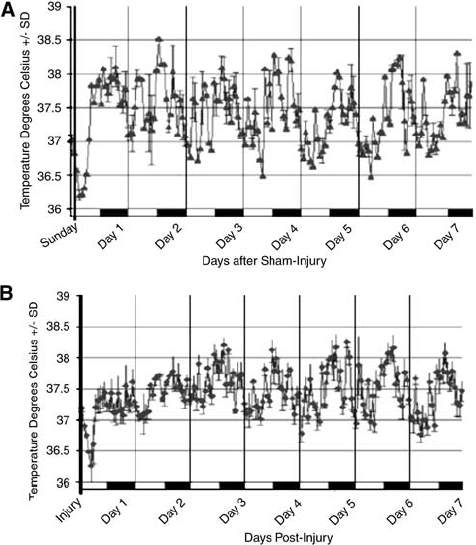
Loss of circadian rhythm (CR) of temperature after severe fluid percussion (FP) brain injury. After craniectomy without injury (sham), there is preservation of the normal amplitude (
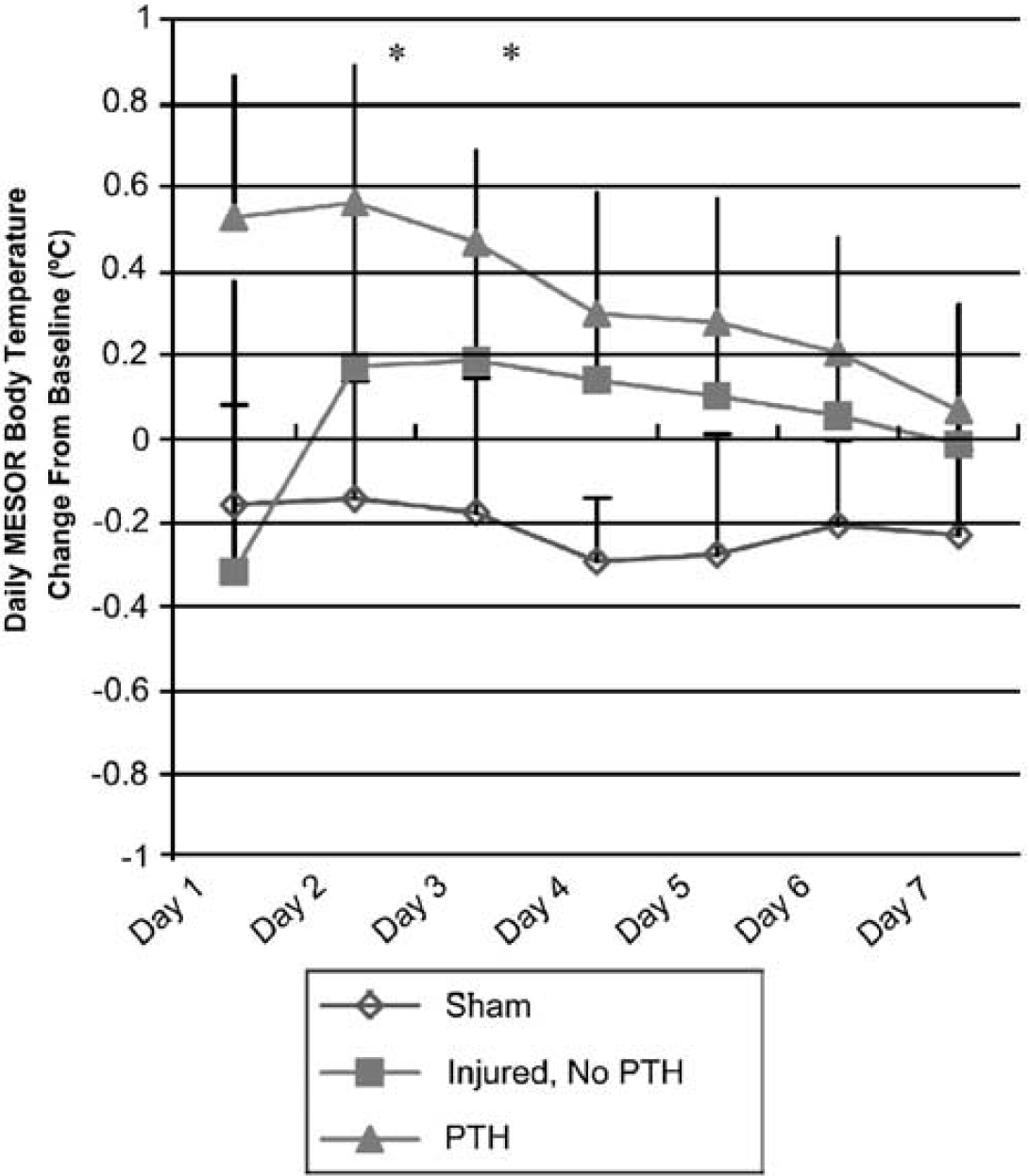
Comparison of group temperature patterns after injury. Mean daily temperature of sham-injured, injured without posttraumatic hyperthermia (PTH), and injured with PTH animals over a period of 1 week. There was both a significant time effect (P<0.01) and a group × time effect (P<0.01) via repeated measures analysis of variance (ANOVA). Temperatures of animals developing PTH were significantly different from sham-injured animals (∗P<0.05) at days 2 and 3 after injury.
Motor Function
The average righting reflex of injured animals was 15.4±0.6 mins. Brain-injured animals had a significant neurologic deficit (the median neuroscore for injured animals was 9 at 48 hours versus 27 for sham-injured animals and 13 at 1 week after injury versus 27 for sham-injured animals) (P<0.05). No significant differences were noted in brain-injured animals that developed hypothermia versus PTH after injury concerning mean apnea (44±26 versus 44±28 secs), mean length of seizure (9±15 versus 12±11 secs), mean injury level (3.40±0.24 atm versus 3.50±0.22 atm), or median 48-hour or 1-week neuroscore (data not shown).
Histologic Analysis
Cell counts. As in sham-injured controls, robust Nissl staining of neurons in the VMPO, SCN, and PVN without evidence of pyknotic cells was observed in brain-injured animals at 1 week after injury. In the APF, where the neuronal density is less than the other three nuclei, the staining was less intense, but appropriate morphology was clearly evident. A two-way ANOVA (group × hemisphere) revealed no statistically significant differences among groups or hemispheres at one week in the numbers of cells in any of the four nuclei evaluated (Table 1).
Cell counts in selected thermoregulatory and circadian nuclei
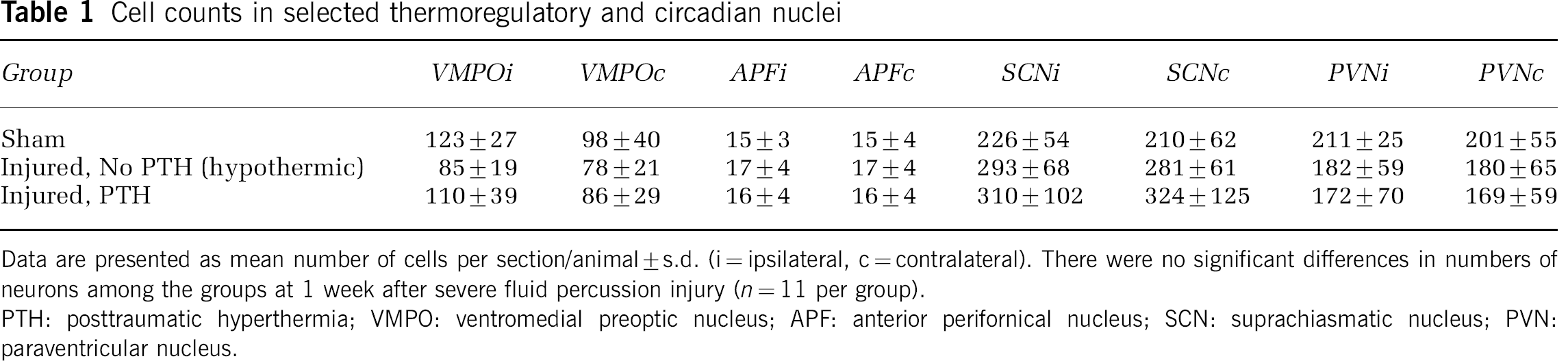
Data are presented as mean number of cells per section/animal±s.d. (i=ipsilateral, c=contralateral). There were no significant differences in numbers of neurons among the groups at 1 week after severe fluid percussion injury (n=11 per group).
PTH: posttraumatic hyperthermia; VMPO: ventromedial preoptic nucleus; APF: anterior perifornical nucleus; SCN: suprachiasmatic nucleus; PVN: paraventricular nucleus.
Immunohistochemistry. Anti-GFAP antibody was used in the present study to identify reactive astrocytes. Positive staining was seen in brain-injured but not sham-injured animals at both 48 hours and 1 week after injury in the periinjured cortex, in the ipsilateral thalamus, and bilaterally in the corpus collosum, indicating reactive astrogliosis. At 48 hours, GFAP immunopositive cells were observable bilaterally within the VMPO in the majority of injured animals (2/3) (Figure 4B). At 1 week in injured animals developing PTH, significantly increased numbers of GFAP-immunolabeled cells were evident within the PVN (Figure 4C) in comparison with sham animals (P<0.05) (Table 2). In addition, increased GFAP labeling was seen in cells in the periventricular areas of animals with PTH compared with injured animals that became hypothermic after injury (Figure 5). No colabeling of astrocytes with IL-1β was seen in any brain section.
Bilateral immunohistochemical cell counts in selected thermoregulatory and circadian nuclei
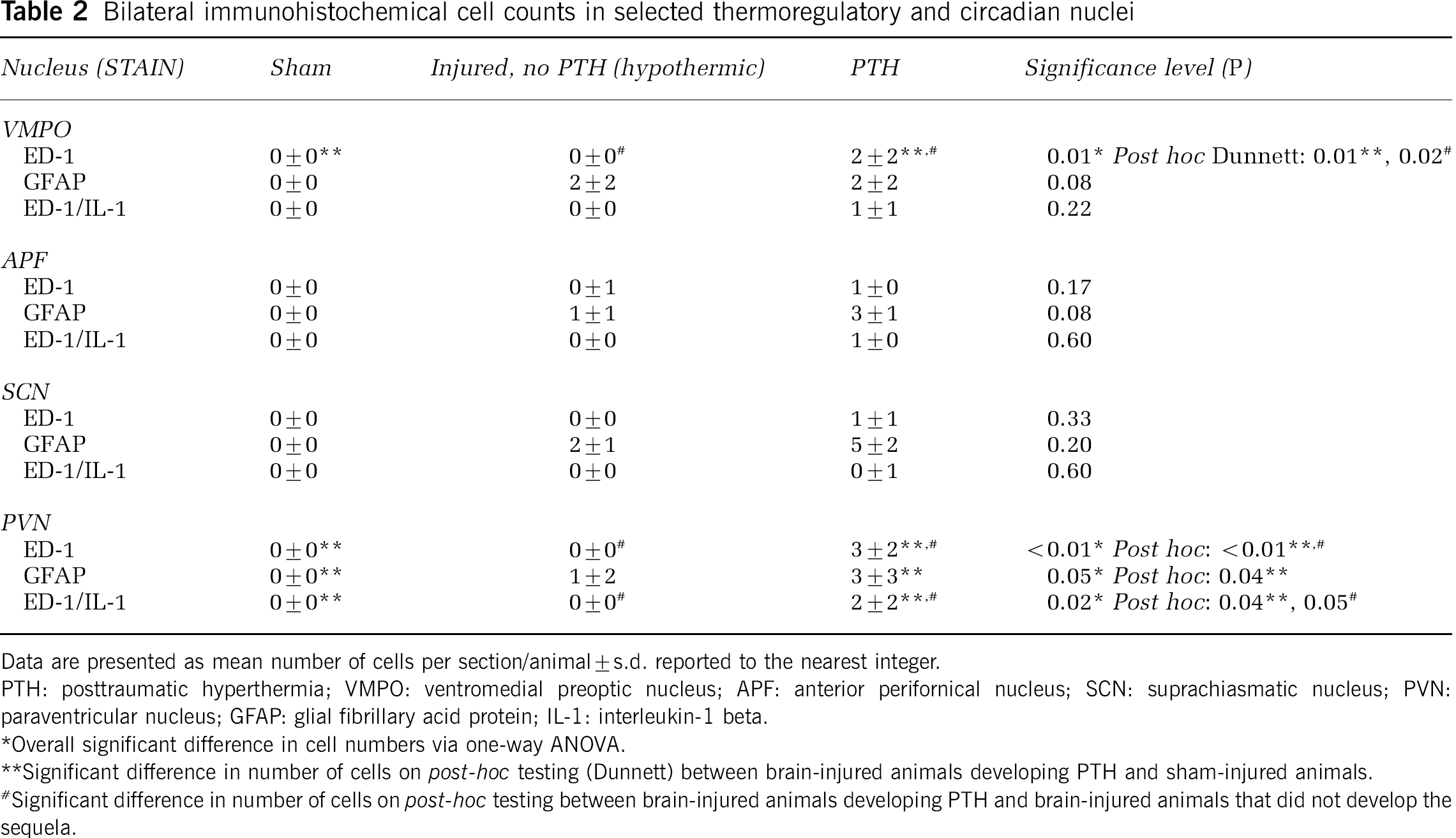
Data are presented as mean number of cells per section/animal±s.d. reported to the nearest integer.
PTH: posttraumatic hyperthermia; VMPO: ventromedial preoptic nucleus; APF: anterior perifornical nucleus; SCN: suprachiasmatic nucleus; PVN: paraventricular nucleus; GFAP: glial fibrillary acid protein; IL-1: interleukin-1 beta.
Overall significant difference in cell numbers via one-way ANOVA.
Significant difference in number of cells on post-hoc testing (Dunnett) between brain-injured animals developing PTH and sham-injured animals.
Significant difference in number of cells on post-hoc testing between brain-injured animals developing PTH and brain-injured animals that did not develop the sequela.
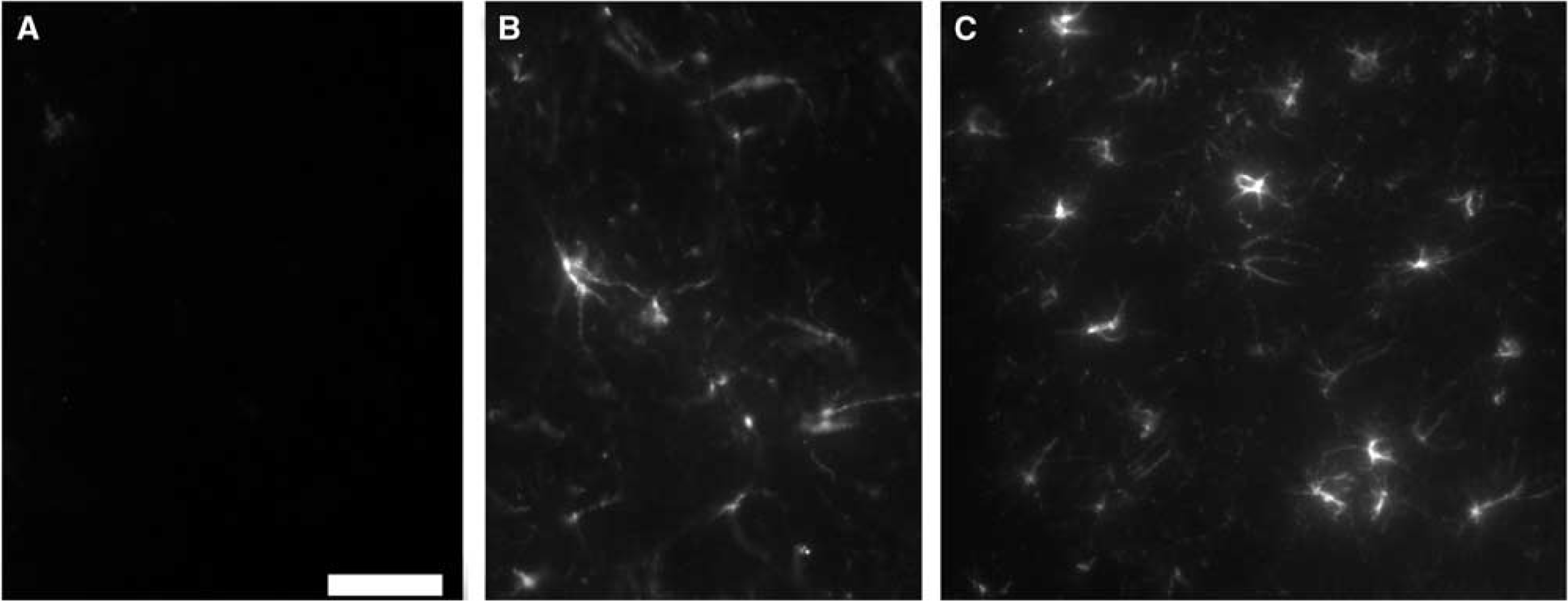
Reactive astrocytes in thermoregulatory centers. No reactive cells were seen in (
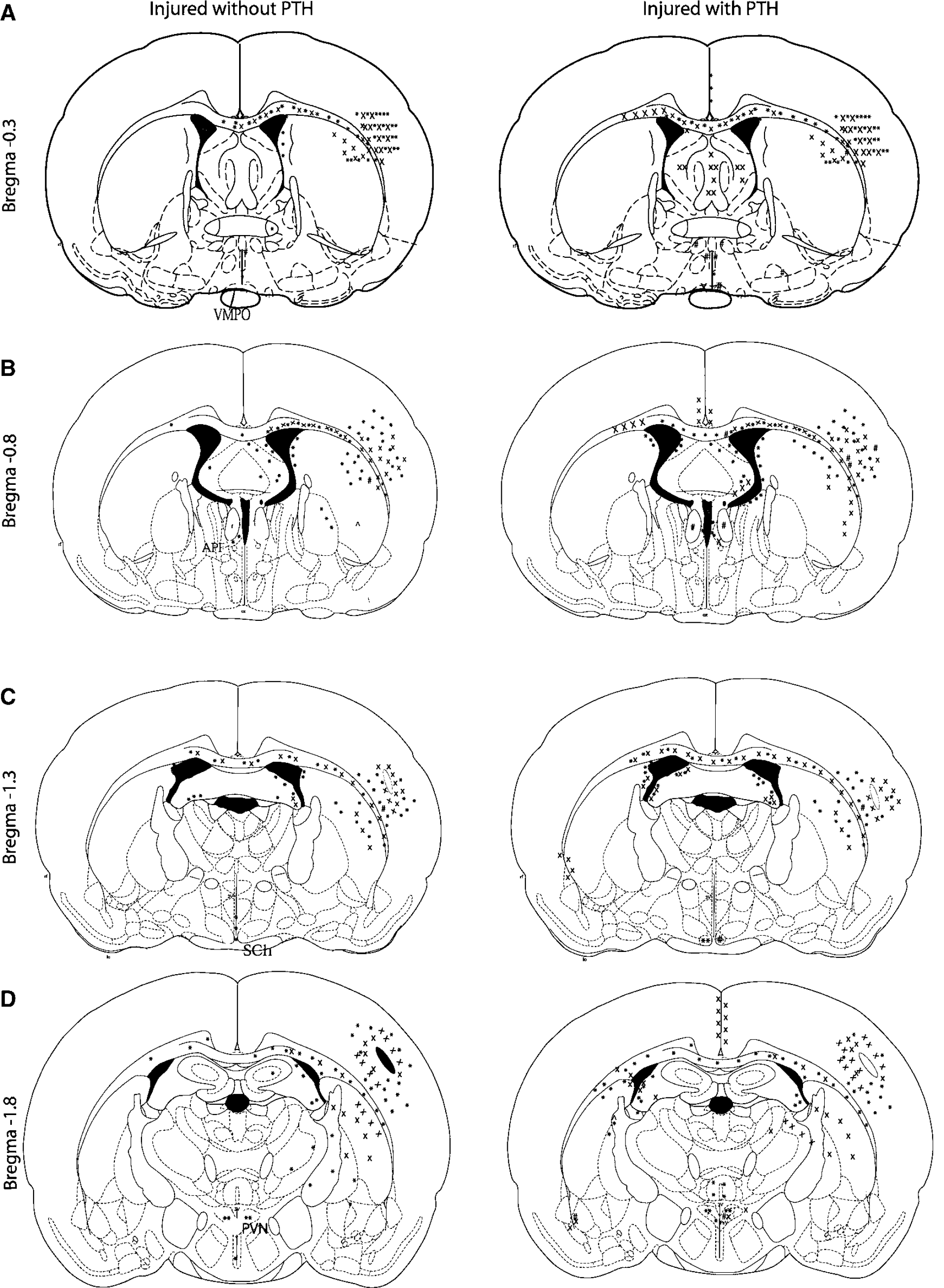
Histographs of thermoregulatory and circadian centers. Schematic representations of the typical regional distribution at (
The microglia/macrophage marker ED-1 was used to examine infiltration of inflammatory cells into thermoregulatory and circadian nuclei, and the periinjured cortex and surrounding structures. No ED-1 immunopositive cells were seen in the sham-injured brain sections examined. In both hypothermic and PTH brain-injured groups, ED-1 immunopositive macrophages and microglia were seen in the injured cortex, ipsilateral and medial corpus collosum, and ipsilateral thalamus at 1 week after injury (Figure 5). Semiquantitative analysis revealed that in rats that developed PTH, an increased number of ED-1 immunopositive cells was observed in the medial and contralateral corpus collosum, thalamic, and periventricular areas, with occasional double labeling of ED-1+/IL-1β+ cells and single labeling of IL-1β+ cells in the contralateral cortex and thalamus compared with hypothermic brain-injured animals (Figure 5). There was a significant difference among groups regarding ED-1 immunolabeling in the VMPO and PVN (P<0.05, Table 2), with animals developing PTH having significantly increased numbers of labeled microglia/macrophages at 1 week after injury in comparison with either sham or injured, non-PTH groups on post hoc testing using Dunnett's test (P<0.05, Table 2). Increased numbers of ED-1-positive cells were seen within the white matter (corpus callosum (Figure 6A), medial septum, and fornix) of animals developing PTH in comparison with their hypothermic counterparts (Figure 5). In five of six rats developing PTH, occasional ED-1+ macrophages or ED-1+/IL-1β+ microglia and macrophages were also found in the VMPO, PVN (Figure 6B), and SCN (Figure 5). In rats developing PTH, there was a significant positive correlation (r=0.788, P<0.01) between maximal body temperature after injury and the sum of the mean number of cells per section per animal positive for inflammatory markers (ED-1 and GFAP) within the four thermoregulatory and circadian nuclei examined. No cells labeling for either ED-1 or IL-1β were present in the PVN or SCN of injured, non-PTH (hypothermic) rats. No specific staining was seen in brain sections from either sham-injured or brain-injured animals after incubation in diluent without primary antibody.
Study II: Physiologic Parameters
No significant differences were observed among preinjury and 30-min postinjury pH or PO2 in any group, nor in PCO2 levels in sham-injured animals or in brain-injured animals with PTH (data not shown). There was a significant decrease from baseline in PCO2 in brain-injured animals that did not develop PTH after injury (from 41.8±3.9 to 35.4±3.1 mm Hg, P<0.05 on post hoc analysis using the Neuman–Keuls test). However, the mean PCO2 remained within normal range (35 to 45 mm Hg) at both time points. Heart rate and MABP did not significantly change over time for any group up to 30 mins after injury (Table 3), nor were there significant differences among sham-injured, brain-injured animals without PTH, and brain-injured animals developing PTH up to 30 mins after injury via one-way repeated measures ANOVA (P>0.05). Blood samples were obtained for the measurement of WBC counts and CRP levels and were assessed for each group 5 mins before and 48 hours and 1 week after FP injury. No significant differences were seen among sham-injured (n=5), TBI without PTH (n=7), or TBI with PTH (n=2) on either measure using one-way repeated measures ANOVA (data not shown).
Mean arterial blood pressure (MABP) and heart rate (HR) of rats up to 30 mins after sham-injury (n=5), severe lateral fluid percussion (FP) brain injury without posttraumatic hyperthermia (PTH) (n=7), or severe FP injury with PTH (n=2)
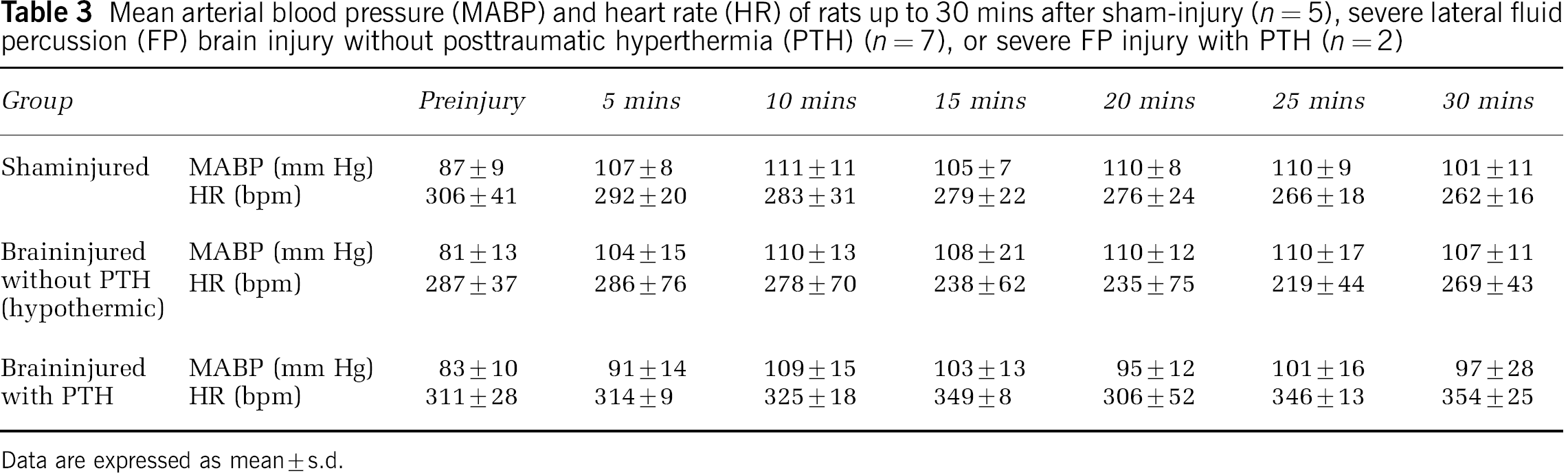
Data are expressed as mean±s.d.
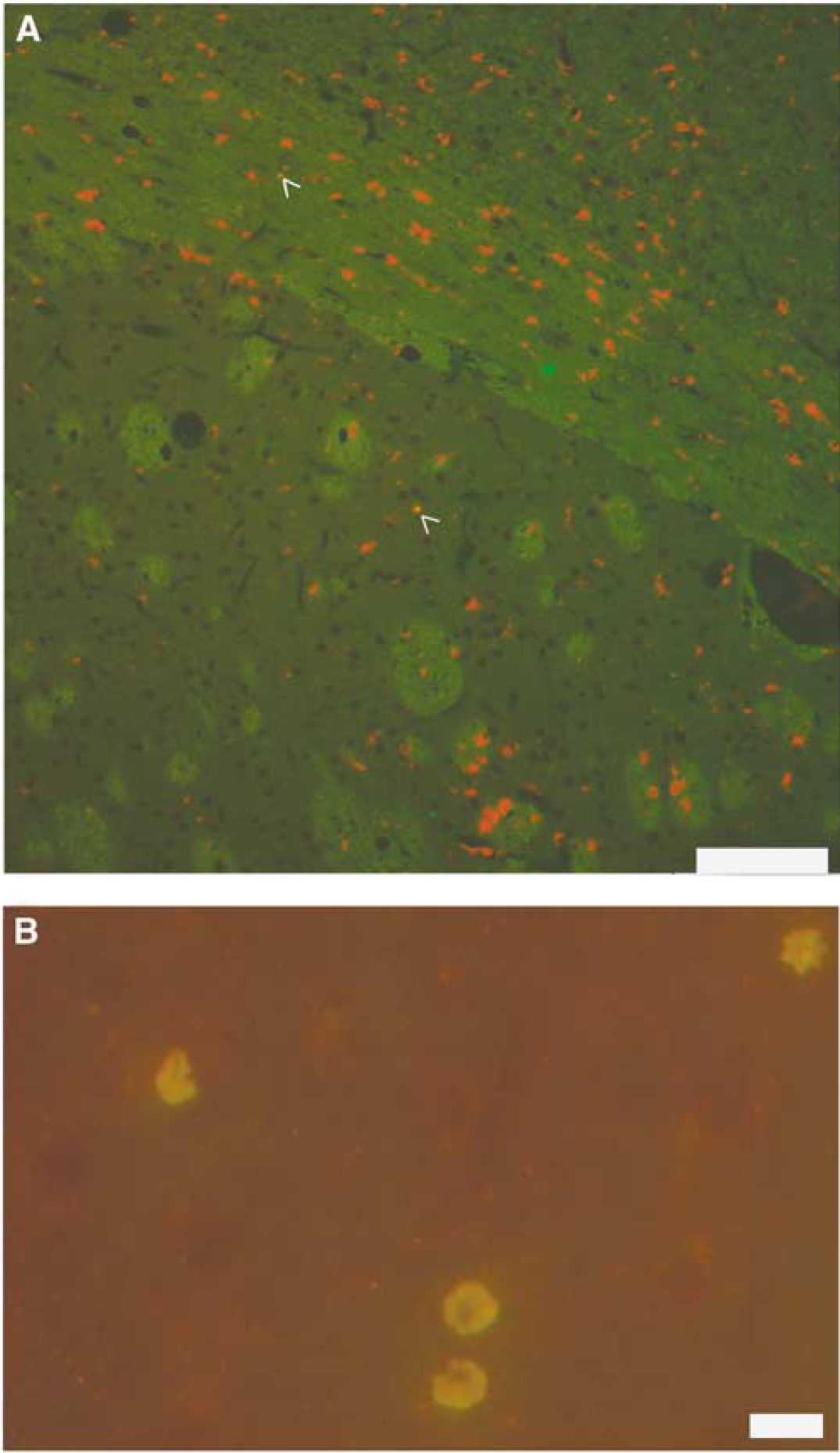
Photomicrographs of macrophage/microglia and interleukin-1 beta (IL-1β)-positive cells. Cellular infiltration into injured brain and thermoregulatory nuclei after severe lateral fluid percussion injury. ED-1 (red)/IL-1β (green)-labeled cells within (
Discussion
Our study revealed two distinct patterns of thermoregulation that emerge after severe experimental FP brain injury in the rat: acute hypothermia with slow return to baseline and PTH. To our knowledge, this is the first long-term study of temperature after FP in the rat, and the incidence of PTH associated with this model further validates its clinical relevance. A single previous study by Taylor et al (2002) reported an altered thermoregulatory pattern after controlled cortical impact (CCI) brain injury in rats, characterized by a brief period of hypothermia (maximum drop 0.3°C over 60 mins) that began 30 mins after injury, followed by hyperthermia in brain-injured animals during the light phase only (Taylor et al, 2002). In contrast to CCI, which produces a focal contusional injury, the lateral FP injury model in the rat is also associated with diffuse axonal injury (Graham et al, 2000), which has previously been associated with development of PTH (Thompson et al, 2003).
To date, no evidence is available on the incidence of hypothalamic injury after TBI in patients who survive their injuries. However, it is known that hypothalamic lesions are common after fatal TBI (Crompton, 1971), suggesting a possible correlation with severity of injury. In attempting to translate the clinical condition to an experimental model, we examined several factors when choosing the experimental TBI model. Lateral FP injury demonstrates reproducible alterations in regional cerebral blood flow (hypoperfusion), cerebral edema, metabolic dysfunction, cation dysregulation in the brain, and memory deficits (McIntosh et al, 1989; Moore et al, 2000; Hamm, 2001). Of particular interest to the development of an animal model of PTH, the lateral FP model of TBI was shown to produce a pathologic neurotransmitter response in the hypothalamus (McIntosh et al, 1994). After lateral FP brain injury, there is evidence of histopathologic damage in the cortex, thalamus, and hippocampal regions (Soares et al, 1995; Hicks et al, 1996; Bramlett et al, 1997; Pierce et al, 1998; Bramlett and Dietrich, 2002), and regional inflammatory changes including astrocytosis and microglia/macrophage infiltration (Soares et al, 1995; Aihara et al, 1995; Hill et al, 1996; Knoblach et al, 1999; Hill-Felberg et al, 1999).
A large percentage of severely brain-injured animals (70%) could not adequately thermoregulate for up to 24 hours after injury and became hypothermic despite maintenance of normothermia during surgical procedures. Similar deficiencies in the ability to self-regulate temperature have been reported in mice after middle cerebral artery occlusion, which resulted in significant differences in infarct size on analysis (Corbett et al, 2002).
This observation, coupled with preliminary observations of impaired thermogenesis in moderately brain-injured animals at 24 hours after injury in response to cold water exposure in behavioral testing (Thompson, unpublished data), points to previously unreported temperature dysregulation after experimental TBI in the rat that continues after full recovery from anesthesia. This finding of two very disparate thermoregulatory patterns with the same injury severity has several important implications for both experimental and clinical studies.
While hypothermia on admission is common in trauma patients with more severe injuries and poor prognoses (Little and Stoner, 1981; Little, 1985; Steinemann et al, 1990; Jeremitsky et al, 2003), it is currently unclear if hypothermia contributes to poor outcome or is simply correlative (Steinemann et al, 1990). We did not observe significant differences in any physiologic or neurologic outcome measures between the brain-injured animals that were hypothermic for 12 to 24 hours after injury and those that developed PTH. The brain-injured patient often has multiple injuries, including fractures, hemorrhage, etc., that may present with hypotension and hypoxia as well as hypothermia (Statler et al, 2001). Although these factors have been shown to relate to poorer outcome after TBI (Jeremitsky et al, 2003), isolated hypothermia may or may not be causative of poor outcome. Because it has also been suggested that therapeutic postinjury hypothermia is neuroprotective (Clifton et al, 1991; Dietrich 1992; Marion et al, 1997; Kinoshita et al, 2002a, 2002b), the findings of this study provide additional information to aid in the design of future preclinical neuroprotection studies. Temperature should be extensively monitored and controlled after randomization in the postinjury period to prevent the additional hypothermia as a confounding variable in the evaluation of experimental treatments.
To our knowledge, this is the first successful attempt to characterize an experimental model of PTH utilizing a clinically relevant model of impact-associated TBI. The percentage of brain-injured animals developing a thermoregulatory pattern of PTH was compatible with that reported in the clinical literature, but the duration of the effect was somewhat shorter (Meythaler and Stinson III, 1994; Childers et al, 1994). Although the present data suggest a correlation between the thermoregulatory changes observed with PTH and the inflammatory changes in hypothalamic areas such as the PVN, demonstration of a causal relationship between these two outcomes requires further study. Further experimentation, particularly examining time points earlier than 48 hours after injury, is warranted to fully explain the time course and mechanisms of this sequela. Our data suggests that the alteration in CR of temperature may be due, in part, to the presence of astrocytes and initiation of an inflammatory response in the centers responsible for CR of thermoregulation. Since IL-1β receptor antagonists are commercially available, and have previously been shown to have potential benefit in experimental TBI (Toulmond and Rothwell, 1995; Sanderson et al, 1999), targeted treatment to these nuclei in animals with PTH may show therapeutic efficacy and could be tested in the model.
Hippocampal CA3 cell loss is a major histologic feature of both the FP model (Soares et al, 1995; Zhang et al, 1998) and human TBI (Graham et al, 2000), but no data have been reported on areas such as the diencephalon after FP injury. Although we found no evidence of overt cell loss within selected diencephalic nuclei at 1 week after injury, periventricular tissue appears to be vulnerable to severe TBI as evidenced by increased inflammation in the periventricular hypothalamus. This response was observed in the subgroup of animals that developed PTH, implicating hypothalamic inflammation in this phenomenon.
The PVN appears to be particularly susceptible to damage after traumatic brain injury, with evidence of both infiltrative WBC activity and reactive astrocytosis. Interleukin-1β has previously been shown to induce astrocytosis after brain injury (Guilian and Lachman, 1985; Guilian et al, 1988), and thus the presence of both ED-1+/IL-1β-immunopositive macrophages and microglia and reactive astrocytes in the PVN is suggestive of a proinflammatory cytokine-mediated response to brain injury in this nucleus. Because the PVN is known to have roles in both maintenance of CR of temperature (Cermakian and Boivin, 2003) and thermoregulation (Saper, 1998), this is a particularly interesting observation. Additional ED-1+/IL-1β-positive microglia and macrophages were seen in the VMPO and SCN of animals with PTH, but not in animals with hypothermia, further suggesting the existence of a regional vulnerability to inflammatory changes with resulting temperature alterations observed after TBI. Of note, there was increased white matter involvement (ED-1-positive cells) in the corpus callosum, medial septum, and fornix of animals developing PTH. Since this model is known to produce diffuse white matter damage (Saatman et al, 1998; Graham et al, 2000) and diffuse axonal injury has been associated with an increased risk of PTH in the clinical setting (Thompson et al, 2003), these findings warrant confirmation with further immunohistochemical analysis. It also remains possible that the increased temperature seen in the PTH group increased the permeability of the blood–brain barrier (Kinoshita et al, 2002a), thereby increasing the subsequent inflammatory response in the periventricular area, where the thermoregulatory and CR nuclei are located. Similar patterns of inflammatory responses in the preoptic area after cerebral ischemia have also been reported in rodents (Loesch, 1983; Zhang et al, 1997; Danton and Dietrich, 2003), particularly in response to hyperthermia, suggesting that this area may be sensitive to temperature changes. The normal WBC values in animals developing PTH, coupled with the negative CRP tests, suggest that the hyperthermia observed after brain injury was not due to infection but, rather, was mediated by factors such as proinflammatory cytokines and other products of infiltrating cells.
Further investigation is required to elucidate the time course and to determine if the discrete brain regions identified in the present study are important in the development of PTH. With additional understanding of the mechanisms responsible for the development of PTH, new diagnostic tests may be evaluated in the model. Additionally, appropriate intervention strategies may be developed to attenuate the effects of PTH after TBI.
Footnotes
Acknowledgements
The authors acknowledge the expert technical assistance provided by Christopher Sparks, Asenia McMillan, David LeBold, Drs Ramesh Raghupathi, Eileen Alexy and Christian Schütz. We thank Robin Armstrong for her administrative support of our research efforts and Jeanne Marks for assistance with preparation of this manuscript.