
Abstract
Excessive inflammation has been implicated in the progressive neurodegeneration that occurs in multiple neurological diseases, including cerebral ischemia, and elevated levels of the proinflammatory cytokine interleukin-1 (IL-1) have been shown to exacerbate brain damage, whereas diminishing IL-1 levels limits the extent of injury. However, to date there is no consensus regarding which receptor(s) mediates the detrimental effects of IL-1. Because we have previously demonstrated that signaling through the IL-1 type 1 receptor (IL-1R1) is necessary for microglial activation and because results from other studies have implicated microglia as effectors of neurodegeneration, we hypothesized that inactivating the IL-1R1 would decrease the extent of damage caused by a hypoxic-ischemic (H/I) insult. It is shown that a mild insult initiates progressive neurodegeneration that leads to cystic infarcts, which can be prevented by inactivating the IL-1R1. The IL-1R1 null mice also show preserved sensorimotor function at 1 month's recovery. The mild insult induces multiple proinflammatory cytokines and activates microglia, and these responses are dramatically curtailed in mice lacking the IL-1R1. Importantly, the neuroinflammation precedes the progressive enlargement of the infarct, suggesting that the inflammation is causal rather than a consequence of the brain damage. These findings show that abrogating the inflammation consequent to a mild H/I insult will prevent brain damage and preserve neurological function. Additionally, these data incriminate the IL-1R1 as a master proinflammatory cytokine receptor.
Introduction
Hypoxic/ischemic (H/I) brain damage is the third leading cause of death and the number one cause of adult disability in developed countries (American Heart Association, 2002). Despite substantial research on this injury few treatments are available to preserve brain cells after H/I. There is accumulating evidence that neuroinflammation contributes to the wave of secondary damage that follows H/I (Tan et al, 2003), which develops as a consequence of the initial activation of endothelial cells, endogenous microglia, and resident perivascular/parenchymal macrophages, followed closely by the mobilization and recruitment of peripheral inflammatory cells to the site of damage (Barone et al, 1991; Feuerstein et al, 1998; Stoll et al, 1998). Activated microglia are known to produce a number of proinflammatory mediators, including interleukin-1 (IL-1), that increase the competence of the cerebral endothelium to recruit peripheral leukocytes into the damaged brain and activated microglia have been imaged within the damaged human brain from 5 days to 7 months after cerebral ischemia (del Zoppo et al, 2000; Gerhard et al, 2000; Pappata et al, 2000).
The rapid release of the proinflammatory cytokine, IL-1, in and around areas of damage is a common sequelae subsequent to a wide range of CNS injuries, including cerebral ischemia (Basu et al, 2002; Griffin et al, 1989; Legos et al, 2000; McGuinness et al, 1997; Minami et al, 1992; Touzani et al, 1999; Touzani et al, 2002). A large body of research has shown that increased IL-1 exacerbates the damage due to injury, while factors that diminish the IL-1 response limit the damage (Loddick and Rothwell, 1996; Yamasaki et al, 1995). A more detailed knowledge of IL-1 signaling in the CNS could, therefore, lead to better, more efficacious treatments for cerebral ischemia and a number of other neurodegenerative diseases.
Two ligands, IL-1α and IL-1β (cumulatively referred to as IL-1), stimulate an inflammatory response by binding to the type I IL-1 receptor (IL-1R1), and possibly to other incompletely characterized receptors. There is also a second receptor, the Type II receptor, that can bind these ligands but does not transduce a response and is thought to act as a decoy, or sink, to inhibit IL-1 activity (Boutin et al, 2003; Sims, 2002). Signaling by IL-1 ligands also is regulated by the naturally occurring antagonist, IL-1ra, which competitively inhibits ligand binding to the IL-1R1. Interleukin-1α and IL-1β have similar biologic activities, but IL-1β is upstream of IL-1α and IL-1β is the preferentially secreted form in the CNS (Boutin et al, 2001; Yao et al, 1992). Binding of IL-1α or IL-1β to the IL-1R1 leads to a signaling cascade that involves multiple second messengers (O'Neill and Greene, 1998). Inhibiting IL-1 activity, either through neutralizing antibodies, exogenously added IL-1ra, or inhibiting caspase 1 ameliorates the damage in a number of animal models of neurodegenerative diseases, including H/I, brain trauma, multiple sclerosis, and Alzheimer's disease (Jacobs et al, 1991; Loddick et al, 1996; Relton and Rothwell, 1992; Yamasaki et al, 1995). The clearly beneficial aspects of exogenously added IL-1ra suggest that therapies designed to interfere with IL-1 signaling may have clinical relevance in limiting the damage caused by these CNS injuries. However, IL-1ra does not gain easy access to the CNS and it also antagonizes binding of the IL-1 ligands to the human type 2 receptor (Arend, 1993); therefore, there is a clear need to identify the IL-1 receptor that is responsible for exacerbating damage after cerebral ischemia and to develop specific small molecule inhibitors for that receptor or essential downstream signaling components.
Methods
Mice and Genotyping
Four- to six-month-old male IL-1R1 (–/–) mice that had been backcrossed 9 times onto a C57BL/6 background were interbred to generate the IL-1R1 null mice used in these studies. Age-matched C57BL/6 mice were bred to generate the wild type (WT) mice used. Interleukin-1 type 1 receptor null mice were originally provided by the Immunex Corporation (Seattle, WA, USA). All mice were bred and maintained at the Penn State College of Medicine by the Department of Comparative Medicine, an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility. Animal experimentation was in accordance with research guidelines set forth by the Pennsylvania State University and the Society for Neuroscience Policy on the use of animals in neuroscience research.
Polymerase chain reaction (PCR) analysis of tail DNA was used to confirm the identify of the mice used for these studies. DNA was isolated from the mice by phenol chloroform extraction. Three different sets of primers were used for the PCR reaction: 5′ GAGTTACCCGAGGTCCAG 3′; 5′ GAA GAAGCTCACGTTGTC 3′; 5′ GCGAATGGGCTGACCGCT 3′. The PCR reaction was performed under the following conditions: 94°C for 30 secs, 53.5°C for 1 minute, and 72°C for 1.5 mins. Thirty-five cycles were performed. The PCR products were resolved and visualized on a 1% agarose gel containing ethidium bromide and run in 1 × TAE buffer. The expected finding for a type I IL-1R WT (+/+) is a single band at 1150 bp. A heterozygote (+/–) had two bands present at 1150 bp and 860 bp. An IL-1R1 null mouse (–/–) had a single band at 860 bp. Experiments were performed either with homozygous WT (+/+) or with IL-1R1 null (–/–) mice.
Induction of Unilateral Cerebral Hypoxia-Ischemia
Hypoxia-ischemia was induced in adult mice by a modification of a procedure developed in the immature rat (Levine, 1960; Rice et al, 1981; Vannucci et al, 1996) and, as described previously, in adult mice (O'Donnell et al, 2002). On the morning of the experiment, the animals were anesthetized with halothane (4% induction, 1.5% maintenance in room air), and the right common carotid artery was isolated and double ligated with 4-0 surgical silk. The incision was sutured, and the animals were allowed to recover with access to food and water for 3 hours. To induce hypoxia, each animal was placed in a 500 mL glass jar partially submerged in a temperature-controlled water-bath and exposed to 8% O2/balance N2 for 22 mins. The water-bath temperature was maintained at 35.5°C, which in previous experiments was shown to maintain the animals' core body temperature at 37.5°C to 37.7°C throughout the hypoxic interval. Animals were allowed to recover in room air for 30 mins and then returned to their cages with free access to food and water.
Magnetic Resonance Imaging and Infarct Volume Measurements
Magnetic resonance (MR) imaging was performed at 24 hours after H/I with 33 WT mice and 28 IL-1R1 null mice. Imaging was also performed at intervals including 7 days, 1 month and 2 months after HI on 14 WT and 11 knockout animals. Magnetic resonance imaging was performed on a 3.0 T MRI spectrometer (Medspec S300, Bruker Instruments, Ettlingen, Germany) with a gradient coil (9.5 cm aperture and gradient strength 1.1 T/m). Each mouse was imaged using a 2.5 cm slotted-tube resonator. Before imaging, the mice were anesthetized with xylazine and ketamine (2 mg/kg of xylazine, 15 mg/kg of ketamine, intraperitoneal). For the damage assessment 10 continuous coronal slices, 0.5-mm thick, were acquired (effective TEeff = 69.4 ms, TR=3,000 ms, slice thickness 0.5 mm, field of view 2 × 2 cm2, matrix 256 × 256 and 8 averages) in 6 mins, using the rapid acquisition with relaxation-enhancement (RARE) imaging sequence. The first slice was positioned 2 mm posterior to the rhinal fissure. To quantify transverse relaxation time (T2) changes for the longitudinal studies, each mouse was imaged with a T2-weighted multiecho-spin-echo sequence in addition to RARE. The T2-weighted spin-echo sequence (TE = 10 to 152 ms, N = 15 echoes, TR = 3,000 ms, 128 × 128 matrix size, 13 mins acquisition time and 2 averages) had the same slice thickness and position as that in RARE imaging. Image analysis was accomplished using the CCHIPS/IDL software (Schmithorst et al, 2001). Slices were segmented by setting an intensity threshold and infarction areas were determined using a semiautomated routine on the RARE scans. Infarct volume was calculated based on the damaged area in each slice times the distance between the slices, and corrected for brain edema as described previously (Swanson et al, 1990). Infarct volumes were calculated as percent of the hemisphere. T2 maps were calculated on a pixel-by-pixel basis from the multiecho images.
Immunohistochemistry and In Situ Hybridization
Animals used for immunocytochemistry for lectin and thionin staining were perfused with culture medium containing 7 U/mL heparin followed by a fixative containing 2.5%. (cryostat section) or 4% (paraffin section) paraformaldehyde in phosphate buffer, pH 7.35. The brains were processed for cryostat and paraffin sectioning, and processed for lectin histochemistry and in situ hybridization as previously described (Basu et al, 2002). The plasmid for ionized calcium-binding adaptor molecule-1 (Iba-1) was provided by Dr Yoshinori Imai (National Institute of Neuroscience, Japan).
Cerebrovascular Assessment
The cerebrovasculature was examined using intracardiac injection of carbon black ink. WT and IL-1R1 null mice were anesthetized with xylazine/ketamine and the left cardiac ventricle was perfused with 4% paraformaldehyde for 5 mins, followed by perfusion with carbon black ink diluted in 4% paraformaldehyde. The brains were carefully removed, left in fixative, and the Circle of Willis and major arteries were carefully examined under a dissecting microscope.
Immunoblotting
Animals used for protein isolation were euthanized at day 2 and perfused with culture medium containing 7 U/mL heparin to remove circulating leukocytes. Cortical regions from both hemispheres (contralateral control and H/I side) were dissected and placed in 1.5 mL microfuge tubes with 150 μL of homogenization buffer (20 mmol/L Tris, 1 mmol/L EDTA, 255 mmol/L sucrose with aprotinin, leupeptin, pepstatin, and AEBSF). Samples were homogenized with teflon-glass homogenizer and centrifuged at 1000g for 10 mins. Supernatant was further sonicated and then protein concentrations were determined using the Pierce BCA Protein Assay Kit according to the manufacturer's instructions. All tissue samples were stored at –80°C until needed. Ten micrograms of each sample was electrophoresed on a NuPAGE 3% to 8% Tris-Acetate gradient gel (Invitrogen, Carlsbad, CA, USA) and transferred to a nitrocellulose membrane. The membrane was then blocked in 2% nonfat dry milk in 1 × PBS-Tween-20 for 1 hour at room temperature with gentle agitation. After blocking, the blots were incubated with anti-COX-2 (cyclooxygenase-2), purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) diluted 1:1,000 in 1% BSA diluent (diluent composed of 1 mg/mL bovine serum albumin (BSA) dissolved in 1 × PBS-Tween-20) overnight at 4°C with gentle agitation. After extensive washes in 1 × PBS-Tween-20, blots were incubated with donkey anti-goat horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at a dilution of 1:20,000 in 1% BSA diluent for 1 hour, with agitation. The blots were rinsed again in 1 × PBS-Tween-20. The Renaissance chemiluminescence reagent from New England Nuclear (Boston, MA, USA) was used according to the manufacturer's instructions. The blots were exposed to film for 20 to 30 secs. The blots were stripped (30 mins at 50°C in 62.5 mmol/L Tris-HCl pH 6.8, 2% sodium dodecyl sulfate (SDS), 100 mmol/L 2-mercaptoethanol) and reprobed with anti-β-tubulin to determine whether the samples were loaded equivalently. The dilution of anti-β-tubulin (Santa Cruz Biotechnology) was 1:1,000 in 1% BSA diluent. Optical density measurements were made using a UVP Chemi-Imaging system (Upland, CA, USA). We had previously optimized the antibody concentration for Cox-2 so that the band intensities produced for Cox-2 were within the linear response range for the chemiluminescence method (Basu et al, 2002).
RNase Protection Assay
Animals used for RNA isolation were euthanized at 1 and 3 days after H/I, perfused with media containing 7 U/ml heparin, and then with Diethylpyrocarbonate-treated PBS. Cortical regions from both hemispheres (contralateral control and H/I side) were dissected and snap frozen in 1.5 mL microfuge tube. Total RNA was purified from homogenized tissue using Trizol reagent (Life Technologies, Invitrogen Corporation, Carlsbad, CA, USA) following the manufacturer's instructions. Multiprobe DNA templates for cytokines (TNFα, IL-1β, IL-12 p40, IL-10, IL-1α, M-CSF, IL-18, IL-6, IFN-β, MIF) and the housekeeping genes, L32 and GAPDH, were all purchased from BD Biosciences (San Diego, CA, USA). RNase protection assay was performed according to the manufacturer's protocol. Briefly, the DNA templates used to synthesize antisense riboprobes were labeled with [α-32P]UTP (Perkin-Elmer Research Inc., Boston, MA, USA) using T7 polymerase. Labeled probes were hybridized with 5 μg of total RNA at 56°C for 16 hours. Samples were then digested with RNase A and T1, and treated with proteinase K. The remaining RNase-protected RNA duplexes were extracted with phenol/chloroform/isoamyl alcohol (25:24:1) and resolved on 5% denaturing polyacrylamide gels. Undigested labeled probes were loaded in the gels to serve as size markers. Mouse control RNA (provided by manufacturer) and yeast tRNA were loaded in the gels to serve as positive and negative controls for the assay. Dried gels were visualized by autoradiography and quantified using a Phosphorimager (Molecular Dynamics, Sunnyvale, CA, USA) after an exposure of 15 to 20 hours. The exposed phosphor screen was then captured by laser scan, and individual bands were quantified using the ImageQuant software program supplied with the scanner. In addition, steps were taken to limit experimental variability, including the running of duplicate reactions.
Behavioral Test
The corner test was performed before and after 7 and 30 days of recovery from H/I on both WT and IL-1R1 null mice. In the home cage, a mouse was placed between two boards each with a dimension of 30 × 20 × 1 cm3 attached at a 30° angle with a small opening along the joint between the two boards. The mouse was placed half-way between the two angled boards facing the corner. The turns in one versus the other direction were recorded from 20 trials for each test. Turning movements that were not part of a rearing movement were not scored. Scoring was performed in a masked manner to avoid bias toward any one group of animals. The corner test has been shown to be sensitive to chronic sensorimotor and postural symmetries and is highly predictive for infarct volume measured from 1 week to 90 days after H/I (Zhang et al, 2002). The nonischemic mouse turns either right or left randomly, while the ischemic mouse preferentially turns toward the injured, ipsilateral side. Wild-type and IL-1R1 null mice were tested before H/I insult to obtain a baseline measure of performance and then tested 1 week and 1 month after insult.
Statistical Analyses
Statistical significance of group differences between WT and knockout mice with respect to cell counts was assessed using the two-sample t-test. Group differences between WT and knockout mice with and without H/I insult with respect to densitometric analyses was assessed using two-way analysis of variance including multiple comparison adjusted post hoc analyses based on the Tukey approach. Data from the sensorimotor test were analyzed using repeated measures analysis of variance to assess differences between WT and knockout mice after H/I insult. The validity of all these analyses was assessed visually using box-plots. All analyses were performed using the SAS statistical software system version 8.2 (SAS Institute Inc., Cary, NC, USA).
RESULTS
To assess the role of IL-1 signaling in the outcome from ischemic brain damage, we used mice lacking the gene for the IL-1R1 and compared their vulnerability to WT mice. We assessed damage in the two mouse populations to a mild H/I insult at the global level using MR imaging and a sensorimotor behavioral test, at the cellular level using immunohistochemical and in situ hybridization analyses, and at the molecular level using RNase protection assays and Western blot analyses to measure mRNA and protein changes for a number of inflammatory mediators. In agreement with a previous study on these mice, an analysis of the gross anatomy of the cerebrovasculature did not reveal any differences between WT and IL-1R1 null mice (Boutin et al, 2001).
Mice Lacking IL-1R1 Show Reduced Damage in Response to Hypoxic/Ischemic Insult
To determine whether decreased IL-1 signaling through the IL-1R1 would decrease the extent of brain damage after H/I, adult WT mice and mice lacking the IL-1R1 were compared using MRI for damage after 22 mins of H/I. Hypoxia/ischemia was induced by ligating the right carotid artery, allowing the animals to recover for 2 hours, then exposing the animals to 8% oxygen at 35.5°C for 22 mins. The rationale for using this paradigm is that it reproducibly produces mild damage in the region of the middle cerebral artery (MCA) in the hemisphere ipsilateral to the ligation (O'Donnell et al, 2002). An analysis of core body temperature using a rectal probe did not reveal any differences in core temperature between the WT and null mice before insult (37.0±0.05; 37.1±0.41, respectively) and at 3 hours (36.8±0.5; 37.1±0.06, respectively) and 10 hours (36.5±0.7; 37.0±0.6, respectively) of recovery.
Magnetic resonance imaging allowed the extent of damage to be repeatedly assessed in individual mice over 2 months of recovery. A total of 61 mice were analyzed by MR imaging at 24 hours of recovery. An additional 25 mice were analyzed at intervals including 1, 7, 30, and 60 days of recovery. Figure 1 shows a representative comparison of three WT and three IL-1R1 null mice 24 hours after the H/I insult. The area of hyper-intense signal on T2-weighted MRI, which represents vasogenic edema, is clearly increased in the cerebral cortex and in the striatum of WT mice compared with the IL-1R1 null mice.
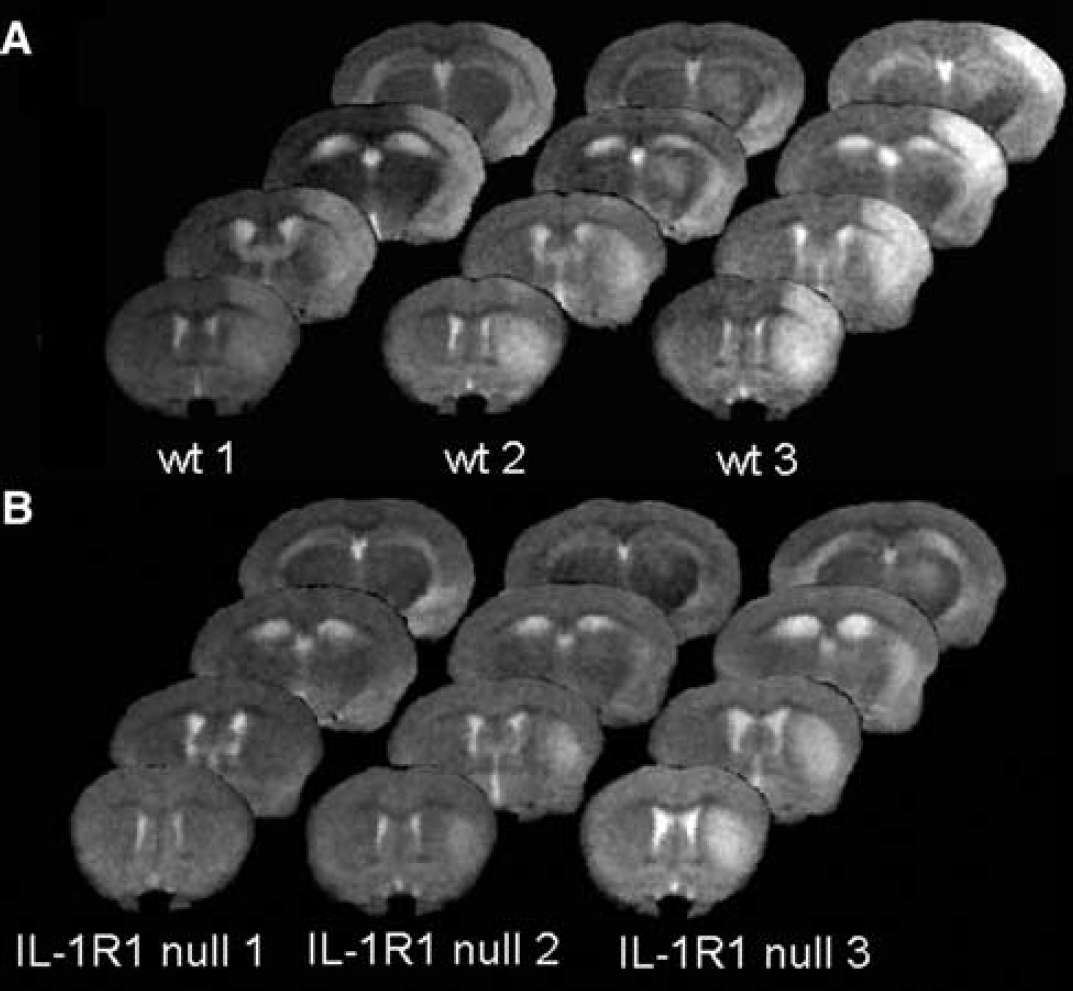
Vasogenic edema in wild-type (WT) versus IL-1R 1 null mice at 24 hours after hypoxia/ischemia. Panels (
Rapid acquisition with relaxation enhancement images were used to calculate infarct volume at the times examined. Figure 2 depicts the differences in infarct volume 24 hours after insult for both groups of animals. Infarct volume was reduced by approximately 60% overall in the IL-1R1 null versus WT and was reduced by approximately 50% when the cerebral cortex was analyzed separately. These animals were then followed at longer recovery intervals to examine whether the protection was temporary or long-lasting. Figure 3 shows T2-weighted MRI images for 3 mice in each group at 24 hours, 7 days, and 1 month. The early protection from H/I seen in the receptor null mice was maintained with time. Strikingly, the size of the core infarct expanded over time in 8/11 of the WT mice, whereas the area of hyper-intense T2-weighted signal decreased in size over time in the null animals. In 8/8 IL-1R1 null animals assessed repeatedly over 2 months, infarcts did not develop.
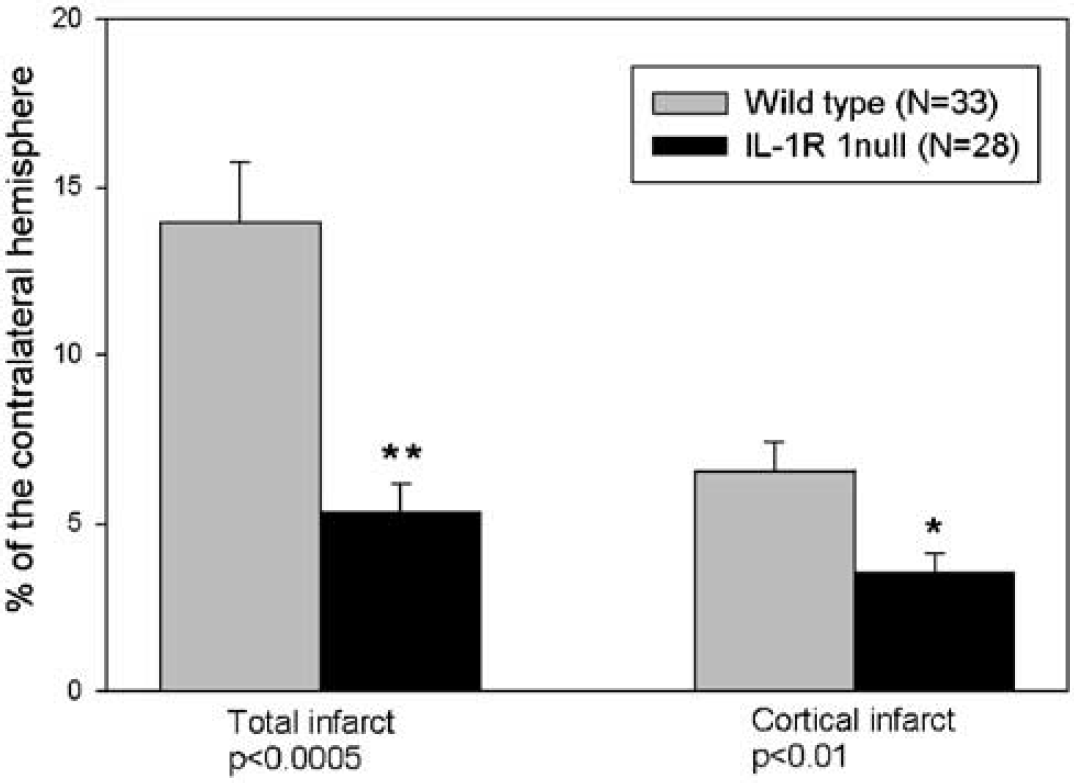
Total and cortical infarct volume for wild-type (WT) and IL-1R1 null mice 24 hours after hypoxia/ischemia (H/I). Mean total and cortical infarct volumes were calculated from the T2-weighted magnetic resonance (MR) images at 24 hours after H/I by the summation of lesion areas multiplied by slice-to-slice distance. Infarct volumes were corrected for brain edema and expressed as the percentage of the contralateral hemisphere. Compared with WT animals (N = 33), total infarct volumes were ˜60% smaller in IL-1R1 null mice (n = 28) P<0.0005 (**) at 24 hours after HI. Interleukin-1 type 1 receptor null mice had significantly reduced, P 0.01 (*), cortical infarct volumes at 24 hours after HI. Statistical significance was assessed using a two-sample t-test.
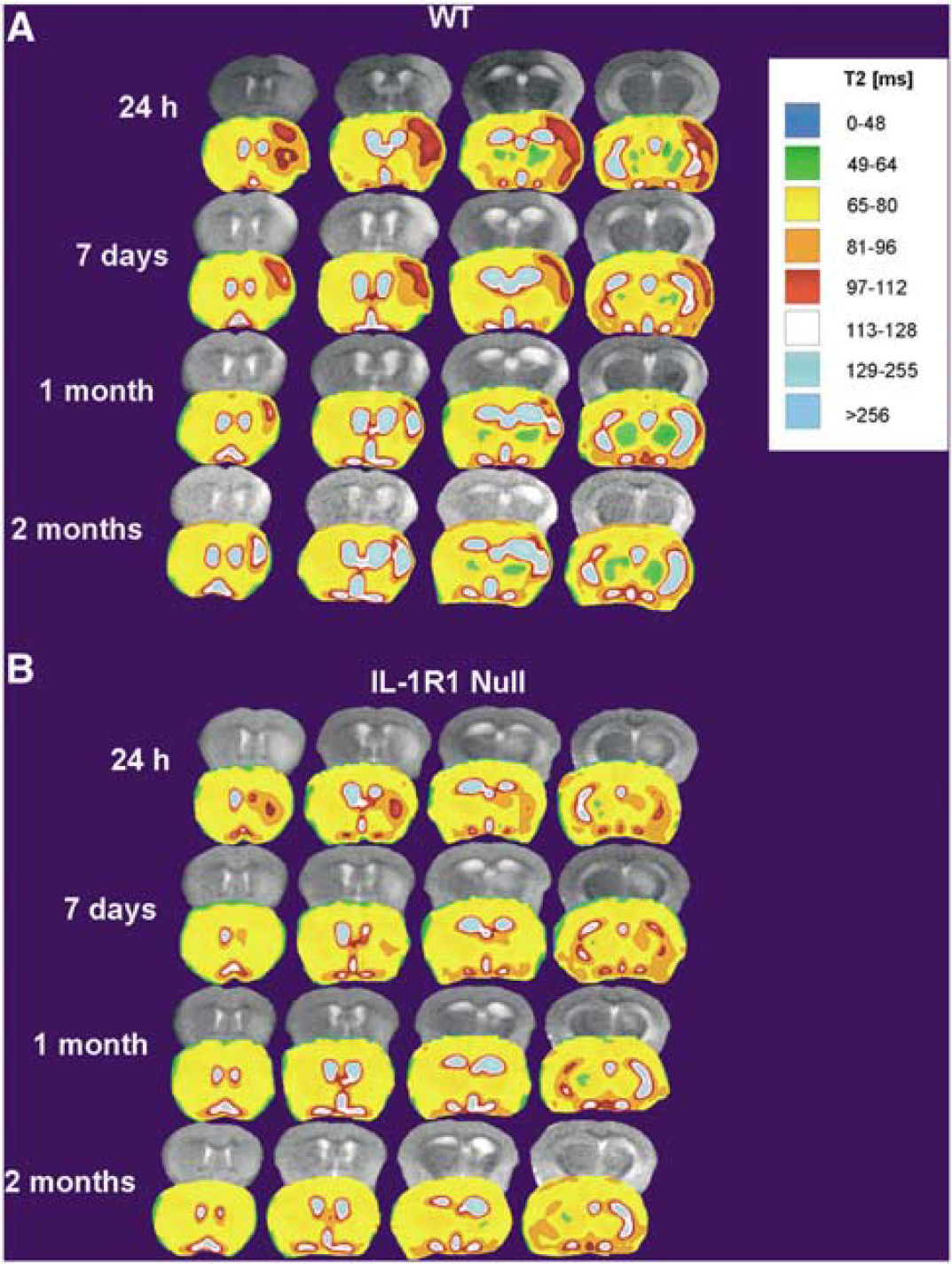
Evolution of brain damage in wild-type (WT) versus IL-1R1 null mice from 24 hours to 2 months after hypoxia/ischemia. T2 maps of brain tissue at 24 hours, 7 days, 1 month, and 2 months with corresponding RARE images in one WT mouse (
Sensorimotor Function is Preserved in Mice Lacking IL-1R1 After Hypoxic/Ischemic Insult
The functional recovery of the brain after H/I-induced damage was assessed in WT and IL-1R1 null mice using the corner test. Figure 4 shows the results of their test scores. The IL-1R1 null mice did not show a preference in their turning at any time point examined, suggesting relatively little neurological functional deficits as a result of the H/I insult. WT mice, however, showed a statistically significant increase in turning behavior toward the damaged side at the 1 month testing time, indicating a deficit in sensorimotor function.
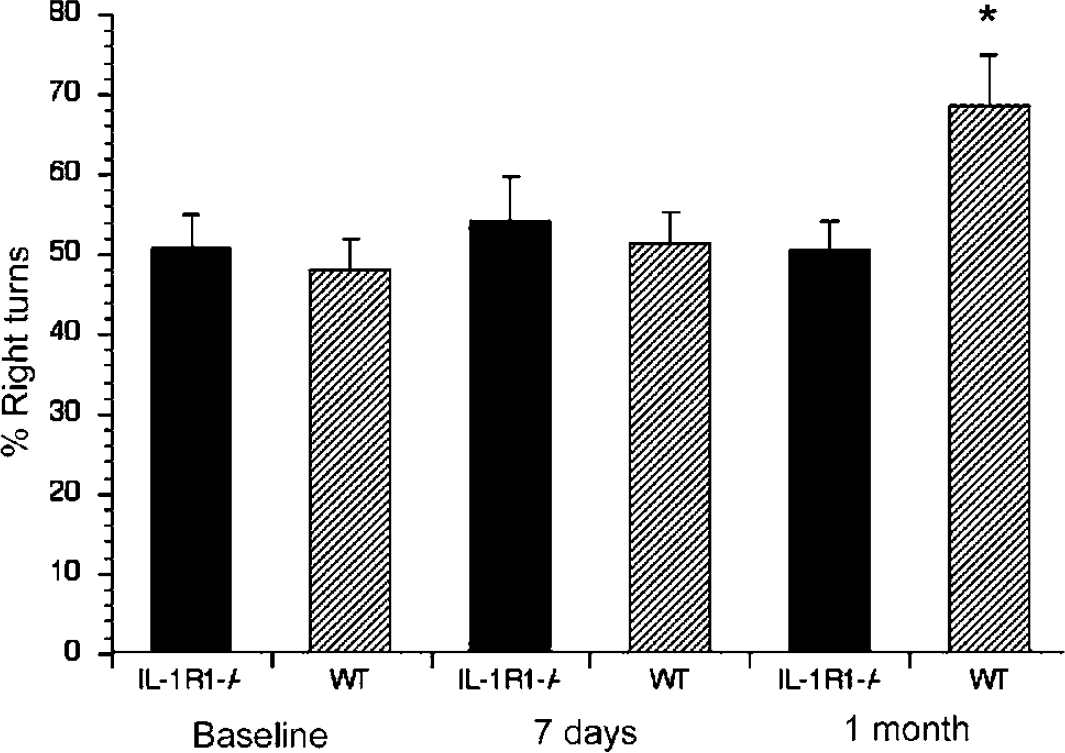
Sensorimotor function is preserved in IL-1R1 null mice after hypoxia/ischemia (H/I). The corner test was used to assess sensorimotor function. Wild-type and IL-1R1 null mice were tested before H/I to obtain a baseline measure of performance and then tested 1 week and 1 month after H/I. Wild-type mice, however, showed a statistically significant increase in turning behavior toward the damaged side at the 1 month testing time, indicating a deficit in neurological function. *P<0.05 by repeated measures ANOVA.
Mice Lacking IL-1R1 Show Increased Neuronal Survival 2 Months After H/I
To confirm the MRI analysis we assessed the extent of damage at the cellular level. Two months after H/I, brains from WT and receptor null mice were processed for histology. Thionin staining was used to assess the degree of neuronal survival in the damaged hemisphere. Figure 5 shows a representative field from both WT and IL-1R1 null mice. Neurons present in the territory affected by the insult in the IL-1R1 null mice are more numerous as well as healthier in appearance than those in the affected hemisphere of WT mice, indicating that the lack of the IL-1R1 preserves neurons after an H/I insult.
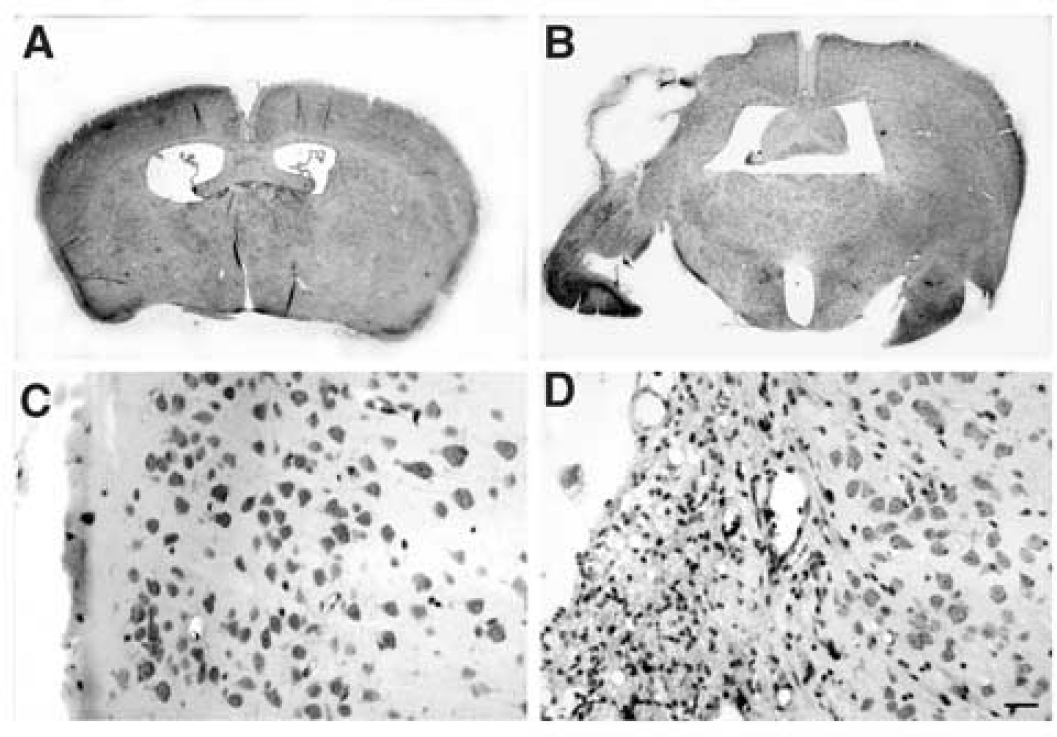
Cortical neurons are preserved at 2 months after hypoxia/ischemia. Thionin staining was performed after 2 months of H/I in both WT (
Mice Lacking IL-1R1 Show Reduced Levels of Phagocytes After H/I
Microglia and invading leukocytes are a major source of IL-1 in the CNS after cerebral ischemia. Therefore, we determined whether the lack of the IL-1R1 affected the number and activational state of the microglia/macrophages in the damaged CNS. After H/I, brains from WT and IL-1R1 null mice were stained using the GSA-IB4 lectin. Using this histochemical stain, resting or quiescent cells exhibit long thin processes, whereas the processes shorten and thicken as the microglia become activated towards amoeboid phagocytes. As illustrated in Figure 6, WT and IL-1R1 null mice had similar numbers of quiescent and activated microglia; however, IL-1R1 null mice had substantially fewer phagocytes, suggesting a decreased level of neuronal death in these animals after H/I insult, that there was a reduction in macrophage infiltration, or both. In situ hybridization for Iba-1 also was performed. Iba-1 is a calcium binding protein specific for microglia, and its expression increases during microglial activation (Ito et al, 1998). As illustrated in Figure 6 (panels D and E), Iba-1 expression clearly increases in the affected hemisphere of the WT mouse while little increase is seen in the affected hemisphere of the IL-1R1 null mouse, again suggesting that microglial activation is reduced in the IL-1R1 null mice after H/I insult.
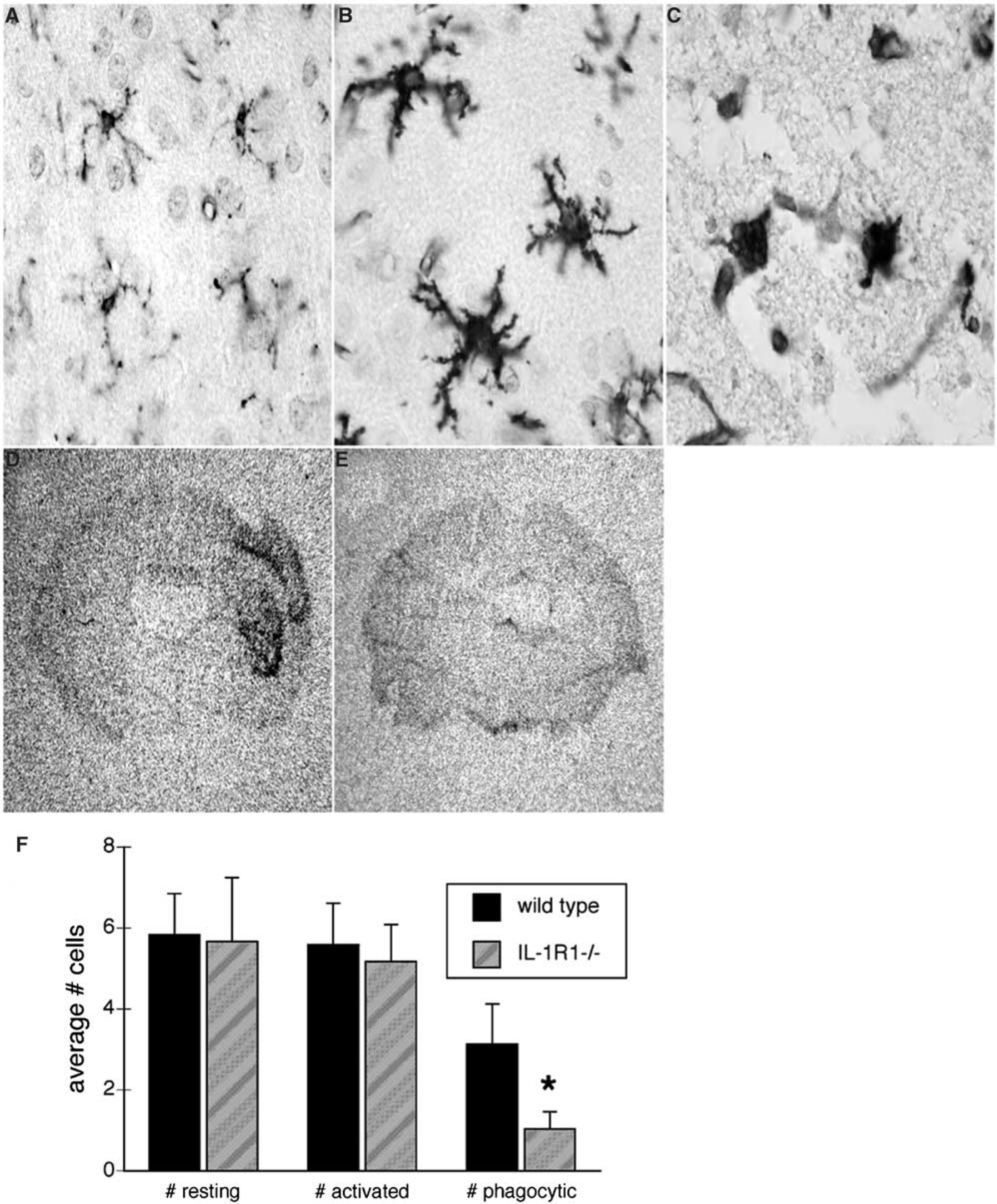
Neocortical microglia are less activated in the IL-1R1 null mice after hypoxia/ischemia (H/I). Panels (
Mice Lacking IL-1R1 Show Reduced mRNA Levels of Inflammatory Mediators After H/I
To determine whether the absence of the IL-1R1 would reduce the subsequent expression of proinflammatory mediators, the mRNA levels for several key proinflammatory cytokines were examined by RNase protection assay. WT and IL-1R1 null mice were subjected to H/I and at 18 or 72 hours after the insult, the animals were killed, their brains removed and RNA isolated from both the contralateral and ipsilateral hemispheres. As illustrated in Figure 7, the damaged hemisphere of WT mice contained induced levels of IL-1β, IL-1α, IL-6, and TNFα. These cytokines were also increased in IL-1R1 null mice as a result of H/I; however, their levels rarely reached levels higher than those seen in the undamaged WT brain. Interestingly, the basal levels of these cytokines are lower in the knockout animals, suggesting that IL-1 signaling influences normal levels of a number of inflammatory cytokines. The reduced levels of these cytokines in the damaged hemisphere of receptor null mice indicate reduced neuroinflammation in response to the H/I insult. Decreased levels of cytokines were not observed uniformly as levels of MIF and M-CSF showed little or no increase as a result of H/I and also did not differ significantly between WT and IL-1R1 null mice.
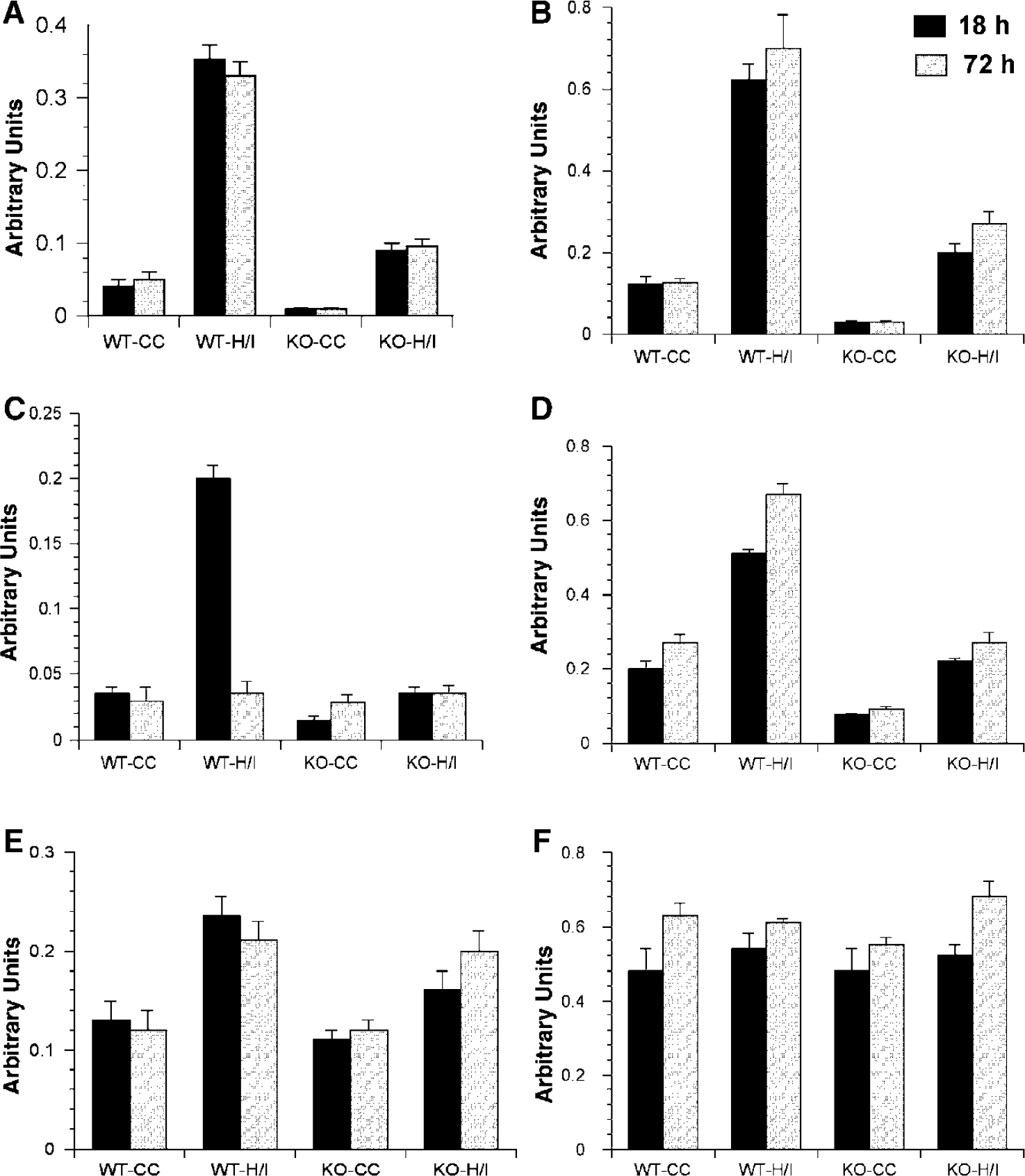
Abrogated response of proinflammatory cytokines in IL-1R1 null mice after hypoxia/ischemia (H/I). (
Mice Lacking IL-1R1 Show Reduced Levels of COX-2 Protein After H/I
COX-2 is an enzyme involved in prostaglandin synthesis and has been used by numerous investigators as an index of inflammation. COX-2 protein levels were assessed using Western blot analysis on protein homogenates isolated from damaged and undamaged hemispheres of mice after an H/I insult. As depicted in Figure 8, COX-2 protein levels mirrored the changes obtained from the analysis of cytokine mRNA levels. COX-2 was dramatically increased in the WT brain as a result of H/I. In the IL-1R1 null mice, the basal levels of COX-2 were lower than those of the WT animals and although COX-2 levels increased as a result of H/I, they did not increase substantially higher than the levels seen in the undamaged WT brain.
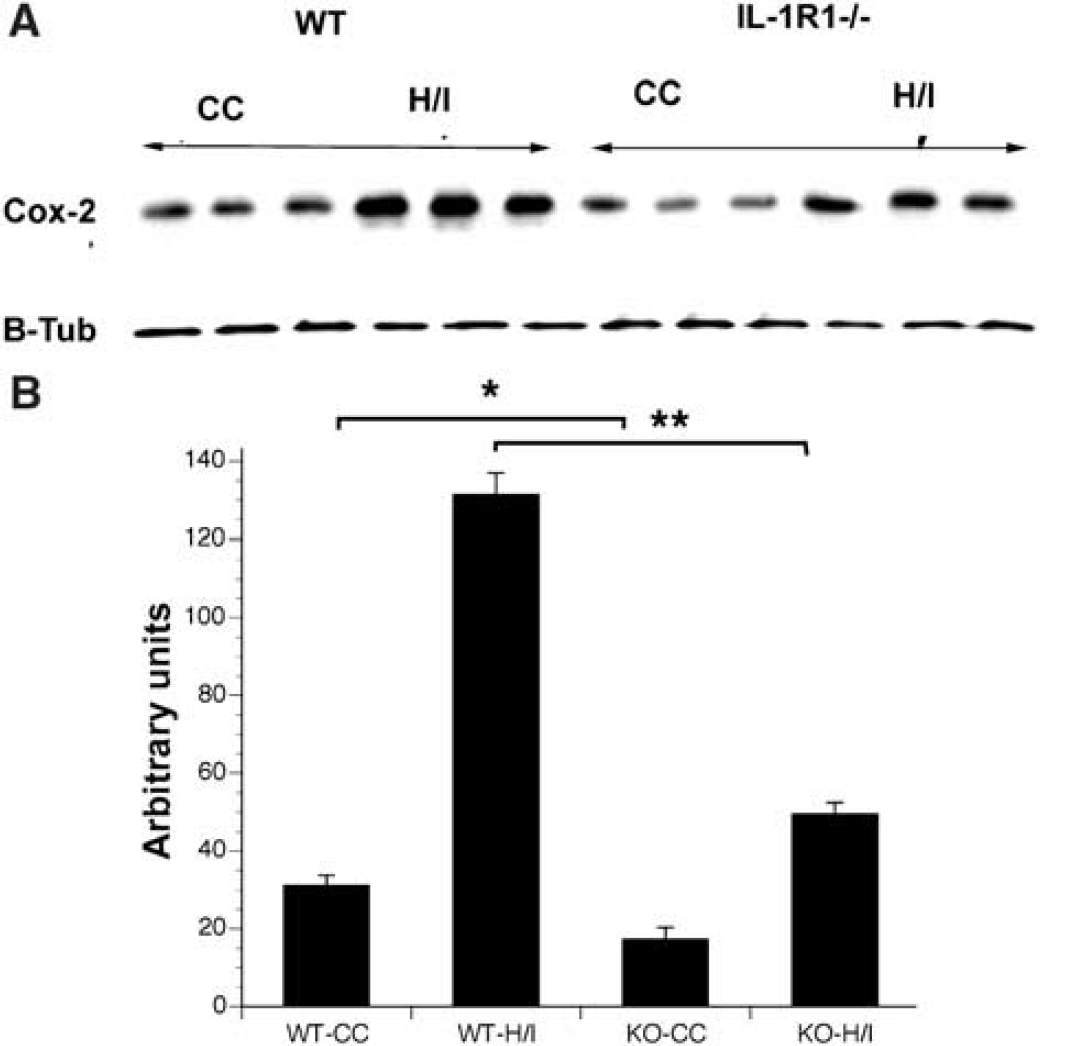
Abrogated response of cyclooxygenase-2 in (Cox-2) IL-1R1 null mice after hypoxia/ischemia (H/I). (
DISCUSSION
Recent studies have implicated neuroinflammation in the etiology of brain damage subsequent to cerebral ischemia; however, to date there is no consensus regarding which inflammatory mediators represent key targets for pharmacological intervention. The results reported here show that mild H/I initiates a slow and progressive neurodegenerative process that leads to cystic infarcts, which can be prevented by inactivating the IL-1R1. We show that inactivating the gene for IL-1R1 decreases the production of numerous downstream proinflammatory mediators that significantly decrease the amount of neuronal damage observed after an H/I insult. The observation that the perifocal area enlarges in WT mice but does not increase in the IL-1R1 null mice directly implicates IL-1R1 as a cause of the progressive neurodegeneration that ensues after a mild H/I insult. Importantly, neuroinflammation precedes the progressive enlargement of the infarct, suggesting that the inflammation is causal rather than a consequence of the brain damage. Thus, therapies designed to block signaling by this receptor will likely prevent secondary neuronal damage subsequent to cerebral ischemia. Furthermore, because IL-1R1 null mice are essentially normal and IL-1 is not essential for maintaining normal brain functions, therapeutic interventions targeting the IL-1R1 will likely have few serious side effects or complications. Unfortunately, there are no chemically synthesized small molecules that cross the blood-brain barrier presently available to antagonize this receptor.
Neuroinflammation in cerebral ischemia is thought to develop as a consequence of two sequencial processes. The first process is the activation of endothelial cells, microglia, and resident perivascular/parenchymal macrophages followed closely by the mobilization and recruitment of peripheral inflammatory cells into the site of damage (Barone et al, 1991; Feuerstein et al, 1998; Stoll et al, 1998). The activated microglia, are known to produce a number of proinflammatory mediators that increase the competence of the cerebral endothelium to recruit peripheral leukocytes, primarily polymorphonuclear leukocytes and monocyte/macrophages, into the damaged brain (del Zoppo, 1994). This inflammatory response was originally believed to be delayed and a reaction to necrosis; however, recent studies in animal models have identified leukocytes in microvessels as early as 30 mins after an MCA occlusion (Garcia et al, 1994). In the same animal model, necrosis was not observed until 72 to 96 hours after the MCA occlusion (Garcia et al, 1995, 1993).
A number of mechanisms have been postulated to explain how leukocytes may damage the brain. These include: (1) obstructing microvessels and thus hindering cerebral blood flow (CBF) to the ischemic region after reperfusion (del Zoppo et al, 1991); (2) production of vasoconstrictive mediators, such as superoxide anions, thromboxane A2, endothelin-1, and prostaglandins that can alter cerebral artery vasoreactivity (Hamann and del Zoppo, 1994; Hartl et al, 1996); (3) release of cytotoxic enzymes, free radicals, and nitric oxide (NO) (Hartl et al, 1996); and (4) release of proteolytic enzymes that can damage endothelial membranes and the basal lamina, thus altering the blood-brain barrier and contributing to the formation of edema (Hamann et al, 1996, 1995). Support for the view that leukocytes can damage viable tissue during acute cerebral ischemia comes from data showing that infarct volume is reduced in neutropenic animals compared with normal controls (Bednar et al, 1991; Connolly et al, 1996; Matsuo et al, 1994). Furthermore, in humans, a correlation between the degree of leukocyte accumulation and infarct volume has been observed by CT studies (Akopov et al, 1996). The infiltration of leukocytes into the ischemic brain is a multistep process (Furie and Randolph, 1995) whereby they first marginate in the venules, adhere to endothelial cells and finally migrate into the parenchyma. At each one of these steps, their functions are regulated by inflammatory mediators such as cytokines and chemokines that are produced soon after the onset of cerebral ischemia.
Interleukin-1 actions in ischemia and other acute brain damage have been well documented in rodents. Numerous studies have shown early increases in IL-1β expression in response to ischemia (Bhat et al, 1996; Hagberg et al, 1996; Legos et al, 2000; Liu et al, 1999; Rothwell and Relton, 1993; Szaflarski et al, 1995; Yamasaki et al, 1995). Furthermore, administering exogenous IL-1β exacerbates ischemic damage (Yamasaki et al, 1995), while neutralizing antibodies to IL-1β or administering the natural antagonist IL-1ra have been shown to block or reduce ischemic brain damage (Loddick and Rothwell, 1996; Relton and Rothwell, 1992; Yamasaki et al, 1995). The receptor responsible for the damaging effects of IL-1 after cerebral ischemia has to date been unresolved. In this report, we show that mice lacking the IL-1R1 have a reduced level of damage after an H/I insult. Our results further show that IL-1R1 null mice have (1) a diminished activation of microglia in response to H/I, (2) an attenuated level of COX-2 expression in response to H/I, (3) attenuated levels of proinflammatory cytokines, (4) preservation of sensorimotor dysfunction after H/I, and (5) protection from progressive neurodegeneration subsequent to H/I. Based on these results, we conclude that IL-1 signaling through the IL-1R1 is critical for initiating and propagating neuroinflammation and subsequent neurodegeneration observed after an ischemic insult.
The protection obtained is likely a result of diminishing the production of a number of inflammatory mediators rather than inhibiting any one signal. Interleukin-1 increases the expression of endothelial cell adhesion proteins and the subsequent infiltration of leukocytes to sites of neurodegeneration. In an earlier study, we showed that the induction of V-CAM is significantly reduced in the IL-1R1 null mice compared with WT mice after a traumatic brain injury (Basu et al, 2002). The absence of this receptor on the endothelium would decrease the extent of inflammation. Furthermore, we observed reduced recruitment of leukocytes into the damaged brain in the IL-1R1 null mice. Interleukin-1 is also known to increase the expression of other proinflammatory mediators such as IL-6, TNFα, and COX-2 from microglia and other cell types. This suggests that IL-1 is at the apex of the pyramid of cytokine signaling in the CNS. While H/I damage still induced expression of these mediators in the receptor null mice, their levels rarely reached the basal level seen in WT animals. These results suggest that a critical level of expression of some or all of these proinflammatory mediators may be necessary to induce neuroinflammation and that in the absence of IL-1 signaling these levels are not reached.
The protection obtained is not likely due to differences in CBF. Boutin et al (2001) demonstrated that mice lacking both forms of IL-1(α and β) exhibited dramatically reduced ischemic infarct volumes (70% reduction) compared with WT mice, and in that communication the investigators used laser Doppler flowmetry to monitor CBF during and after insult. Doppler monitoring showed that changes in CBF during or subsequent to transient forebrain ischemia were similar in all strains. Additionally, Touzani et al (2002) used laser Doppler flowmetry to measure CBF during and after ischemia in IL-1R1 null mice. They did not find any significant differences in CBF between WT and IL-1R1 null mice during or after transient forebrain ischemia. These studies provide evidence that the absence or presence of IL-1 or the IL-1R1 receptor does not have a measureable effect on CBF and, hence, on the outcome of insult. Moreover, we have previously documented decreased neuroinflammation subsequent to a stab-wound injury, where there should be no contribution of CBF to the extent of injury and here, we show that there is progressive neurological damage in the WT mice that correlates with the neuroinflammation. This progressive degeneration does not occur in the IL-1R1 nulls. Thus, while we did not directly measure CBF herein, there are multiple lines of evidence to reject the hypothesis that increased CBF is responsible for the neuroprotection observed.
Studies have suggested that receptors other than the type 1 receptor exist within the brain and that these other receptors are responsible for exacerbating damage after cerebral ischemia. Using MCA occlusion on IL-1R1 null mice Touzani et al (2002) failed to observe any difference in infarct size between WT and null mice. Furthermore, addition of exogenous IL-1 to the IL-1R1 null mice exacerbated the damage seen after experimental cerebral ischemia. Taken together, their results suggested that other receptors for IL-1 exist within the brain. There are two likely explanations for the discrepant results between their study and ours: (1) We assessed neuroprotection after a more mild insult than was used by Touzani et al (2002); thus, as a consequence of a more severe insult other tissue damaging mechanisms might have masked the protection afforded by deleting the IL-1R1. (2) Touzani et al (2002) used IL-1R1 null mice on a C57BL/6/SV129 mixed background whereas we used mice that had been backcrossed nine generations onto a C57BL/6 background. Because the SV129 mouse strain is known to be one of the most resistant strains of mice to ischemic damage, achieving damage in this strain likely required a more severe insult, which, again, could have masked the contribution of IL-1β to the insult (Fujii, 1997).
Progressive neurodegeneration after cerebral ischemia is not a new phenomenon; however, our data strongly implicate neuroinflammation in the second wave of cell death. Du et al (1996) used a mild focal adult ischemia model and reported that the infarct volume increased over time. Similar results have been reported by using a perinatal model of H/I (Du et al, 1996; Geddes et al, 2001). In neither study was the cause of the progressive degeneration identified. Supporting a role for IL-1 in this progressive degenerative process is the observation that levels of caspase 1 and of IL-1β increase for weeks subsequent to an H/I insult (Bhat et al, 1996; Hedtjarn et al, 2002). The observation that the perifocal area enlarges in WT mice but does not increase in the IL-1R1 null mice directly implicates IL-1R1 signaling as a cause of progressive neurodegeneration after cerebral ischemia. Importantly, because the neuroinflammation preceded the progressive enlargement of the infarct, this result suggests that the inflammation is causal rather than a consequence of the brain damage.
To determine whether the preservation of brain tissue correlated with a preservation of neurological function, we used the corner test as a measure of sensorimotor function. We observed significant differences in the behavior of WT versus IL-1R1 null mice in this test after an H/I insult. While no differences were observed at early times after insult, clear differences were seen 1 month after insult and these differences correlated with the increase in infarct size observed over time. These data correlate well with those of others who noted that significant differences could only be seen 60 to 90 days after the insult (Zhang et al, 2002). By linking the extent of damage seen by MR imaging to a behavioral test that measures sensorimotor function, we were able to show that the disruption of IL-1 signaling through the IL-1R1 led to a functionally relevant sparing of neuronal damage after H/I insult.
The critical role of IL-1 in a number of neurodegenerative diseases including cerebral ischemia is well documented. While the exact nature of IL-1's actions in these disorders is not clearly understood, most of the disorders include a neuroinflammatory component. The results of our studies show that disrupting IL-1 signaling through the IL-1R1 downregulates a number of proinflammatory mediators, limits microglial activation, and reduces the neuronal damage observed after a mild H/I insult. Therapies designed to inhibit signaling by this receptor will likely prove efficacious in preventing secondary neuronal damage subsequent to cerebral ischemia. Owing to the neuroinflammatory component in a number of other neurodegenerative diseases, it is likely that such therapies also would prove useful in these disorders.
Footnotes
Acknowledgements
These data were presented at the International Society for Cerebral Blood Flow and Metabolism 2003 annual meeting. This article is dedicated to Vaunceil Brazel.