
Introduction
Spreading depression (SD) is associated with increased cerebral blood flow (CBF). This coupling between neuronal activation and CBF during SD is inverted by combined subarachnoid application of the NO-scavenger oxy-hemoglobin (Hb) and elevated K+ so that the neuroglial depolarization wave is accompanied by an ischemic flow change (= spreading ischemia [SI]). Based on the induction of SI by red blood cell products, it has been hypothesized that SI could be the pathophysiological correlate of the widespread cortical infarcts representing the predominant lesion pattern in patients after subarachnoid hemorrhage. NMDAR antagonists reliably block SD under normal conditions. Therefore, we tested here whether NMDAR antagonists would also block SI.
Methods
A cranial window was implanted in rats under thiopental anesthesia. Artificial cerebrospinal fluid (ACSF) was brain topically superfused. CBF was measured with laser-Doppler flowmetry. Intracortical DC-potential and extracellular K+-concentration were measured with two K+-sensitive microelectrodes (measuring depth 300 μm). In group 1 (n=6), we tested the effect of intravenous (i.v.) MK-801 (5 mg/kg) on SI generated by increasing K+ concentration in the ACSF ([K+]ACSF) stepwise (3, 25, 35 mM) at 60 min intervals during continuous application of Hb. In group 2 (n=5), we increased [K+]ACSF to 130 mM at the cranial window until SDs were detected. Subsequently, the cortex was superfused with physiological ACSF again, followed by MK-801 i.v. To determine whether SD could still be elicited under this condition, [K+]ACSF was again raised to 130 mM for 60 min. In group 3 (n=5), two cranial windows were implanted over the ipsilateral hemisphere. The same protocol as in group 2 was followed for the caudal window, while the rostral window was superfused with physiological ACSF throughout the experiment.
Results
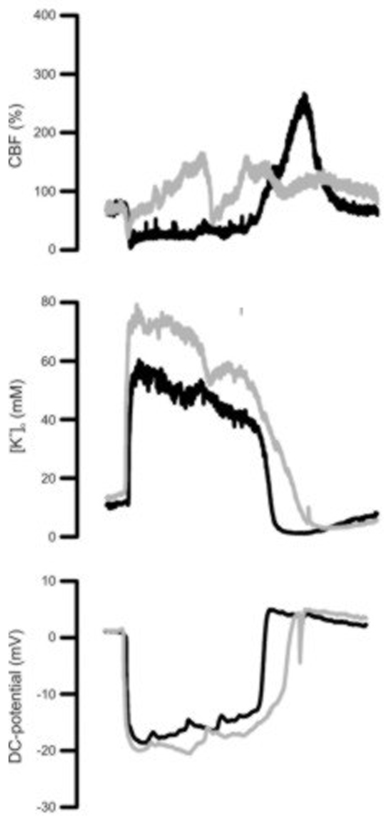
Despite NMDAR blockade, SI occurred under [K+]ACSF at 35 mM and Hb in all experiments similarly as reported previously without MK-801 (Figure 1). [K+]o gradually increased before SI occurred (8.6±2.8 mM caudally, 12.6±1.9 mM rostrally) and showed a transient peak during SI. In group 2, in response to 130 mM [K+]ACSF, [K+]o only increased to 7.7±6.8 mM (caudally) and 5.7±5.8 mM (rostrally). SD occurred in all animals. Compared to SI, DC-shift duration as well as extent and duration of the hypoperfusion were significantly smaller (P<0.001, t-test). Following MK-801, [K+]ACSF at 130 mM again induced recurrent episodes of SD in all animals. In group 3, raising [K+]ACSF at the caudal window caused a gradual rise in [K+]o (6.7±3.7 mM), whereas it remained constant at the rostral window. During 60 min superfusion with high [K+]ACSF, 5±4 SD occurred at the caudal window. 3±1 of these SD propagated into the rostral window. Following MK-801, SD still occurred caudally (8±2/h). However, SD propagation was completely blocked.
Conclusion
If SI is the pathophysiological correlate of the cortical lesions after SAH, it is unlikely that NMDAR antagonists alone are sufficient to protect the brain. Elevated baseline [K+]o reduces the efficacy of NMDAR antagonists to block SD.