
Introduction
Although it seems to be clear that leukocytes contribute to brain damage after cerebral ischemia, it is still under debate how and when cells of the immune system mediate post-ischemic cell death. The aim of the current study was therefore to investigate the temporal and causal relationship of leukocyte-endothelium interactions, leukocyte migration into brain parenchyma, and neuronal cell death following reperfusion from focal cerebral ischemia.
Materials & Methods
129/Sv mice were subjected to 45 minutes of transient middle cerebral artery occlusion by an intraluminal filament. Leukocyte-endothelium interactions were assessed by intravital microscopy (IVM) 40 min, 1, 2, 3, 5, 9, and 15 hours after reperfusion. Neuronal cell death and leukocytes migrated into the brain were quantified in the same tissue volume by histology and immunohistochemistry using the pan-leukocyte marker CD45, respectively, at the same time points. For inhibition of leukocyte-endothelium interactions an anti-CD18 antibody was injected into the left common carotid artery immediately after reperfusion.
Results
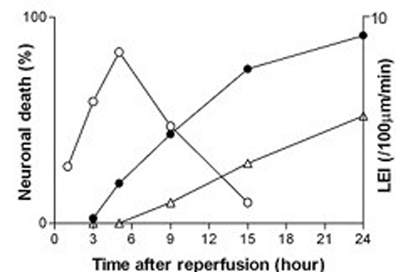
We show that interactions of leukocytes with cerebral endothelium peaked 5 hours after reperfusion following 45 minutes of transient middle cerebral artery occlusion in mice (P<0.01 vs. control). In the same area of the brain, neuronal cell death occurred between 5 and 24 hours after reperfusion while infiltration of leukocytes into the brain parenchyma was limited and started to be detectable only 9 hours after reperfusion (Figure 1). Inhibition of CD18, a leukocyte adhesion molecule, reduced adherence of leukocytes to cerebral endothelium by 60% (P<0.01) and increased the number of viable neurons by more than 5-fold (P<0.01), but did not affect the number of leukocytes in the brain parenchyma.
Summary & Conclusion
Our results show that intravascular interaction of leukocytes with cerebral endothelium seems to be sufficient for the induction of neuronal cell death after transient focal cerebral ischemia, while infiltration of leukocytes into post-ischemic brain tissue occurs too late to be responsible for this process. Despite the large number of publications dealing with the role of inflammatory cells for post-ischemic neuronal cell death, this is the first report suggesting that rather transendothelial signaling of intravascular leukocytes than physical interaction of leukocytes with neurons is responsible for cell death after cerebral ischemia. We believe that our data may help to explain previous contradicting results on the role of inflammatory cells for the pathophysiology of cerebral ischemia and may serve as a basis for future research on transendothelial signaling.