
Abstract
Atrial fibrillation (AF) increases the risk and severity of thromboembolic stroke. Generally, antithrombotic agents increase the hemorrhagic risk of thromboembolic stroke. However, significant reductions in thromboembolism and intracerebral hemorrhage have been shown with the antithrombin dabigatran compared with warfarin. As thrombin has been implicated in microvessel injury during cerebral ischemia, we hypothesized that dabigatran decreases the risk of intracerebral hemorrhage by direct inhibition of the thrombin-mediated increase in cerebral endothelial cell permeability. Primary murine brain endothelial cells (mBECs) were exposed to murine thrombin before measuring permeability to 4-kDa fluorescein isothiocyanate-dextran. Thrombin increased mBEC permeability in a concentration-dependent manner, without significant endothelial cell death. Pretreatment of mBECs with dabigatran completely abrogated the effect of thrombin on permeability. Neither the expressions of the endothelial cell β1-integrins nor the tight junction protein claudin-5 were affected by thrombin exposure. Oxygen-glucose deprivation (OGD) also increased permeability; this effect was abrogated by treatment with dabigatran, as was the additive effect of thrombin and OGD on permeability. Taken together, these results indicate that dabigatran could contribute to a lower risk of intracerebral hemorrhage during embolism-associated ischemia from AF by protection of the microvessel permeability barrier from local thrombin challenge.
INTRODUCTION
Nonvalvular atrial fibrillation (AF) increases the risk and severity of thromboembolic stroke. Prevention of thrombus formation with warfarin, whereas decreasing ischemic stroke risk, is associated with increased hemorrhagic risk. The RE-LY (randomized evaluation of long-term anticoagulant therapy) trial, comparing warfarin with the direct thrombin inhibitor dabigatran, showed that dabigatran was not inferior to warfarin in efficacy (reduction of thromboembolic events), and that for prophylaxis of nonvalvular AF, there was a significant reduction in the incidence of intracerebral hemorrhage with dabigatran compared with warfarin.1,2 Although it has been suggested that the observed hemorrhagic risk reduction with dabigatran is the consequence of a higher hemorrhagic risk of warfarin, which targets multiple coagulation factors, 3 this does not explain the confinement of the risk reduction to the cerebral vasculature. This could be explained by a difference in microvascular hemostatic responses in the central nervous system (CNS) under conditions of focal ischemia. Rosenberg and Aird 4 proposed that hemostatic responses in the CNS are unique among vascular beds; 5 although the contributions that microvessel beds make to regulating hemostasis in the human CNS are not completely understood,6,7 particularly in the setting of focal ischemia.8,9
The unique structure of cerebral microvessels is related to their functions in regulating both vascular permeability and hemostasis within the CNS. First, brain capillaries consist of a non-fenestrated endothelium, basal lamina matrix, and surrounding astrocyte end-feet attached to the basal lamina. This intact endothelium–matrix–astrocyte assembly constitutes the ‘blood–brain’ permeability barrier.
10
The integrity of the permeability barrier depends on the interendothelial tight junctions (TJs), interactions between endothelial cells and astrocytes, and adhesion of endothelial cells and astrocytes to the basal lamina matrix.10,11 Second, the cerebral hemispheres contain significant quantities of tissue factor (TF), the major initiator of extrinsic coagulation, which is broadly distributed in the gray matter and surrounds noncapillary microvessels.
12
del Zoppo
Thrombin can be acutely generated in the cerebral microvessel wall during focal ischemia as indicated by fibrin deposition within the plasma column and vessel wall (see Figure 5 in del Zoppo
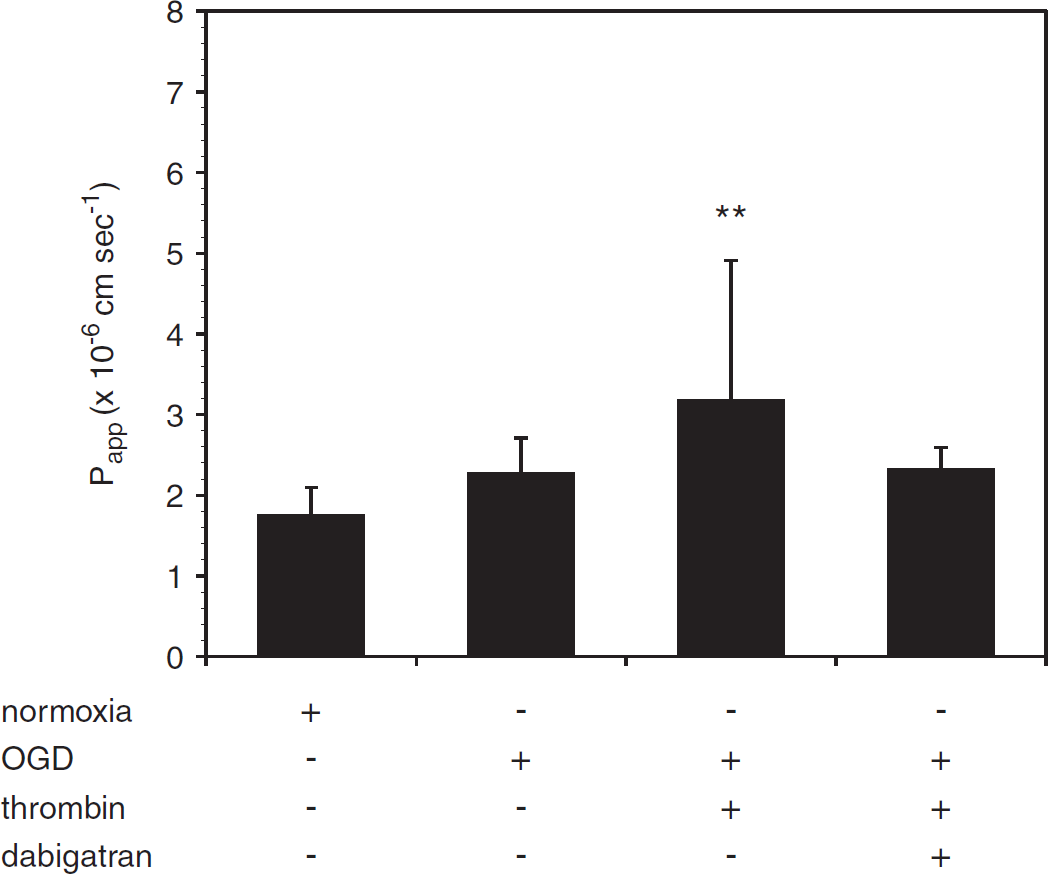
Dabigatran abrogates the effect of thrombin after oxygen-glucose deprivation (OGD). Addition of thrombin (10 U/mL, 1 h) to murine brain endothelial cell (mBEC) cocultured with astrocytes immediately after OGD (18 h) augments the effect of OGD on permeability. This additive effect is blocked by dabigatran (500 nmol/L). Data are pooled from three separate experiments (
After the onset of focal cerebral ischemia, rapid alterations in brain microvessels occur, including decreased matrix adhesion receptor expression and matrix proteins, and increased permeability.24,25 The interaction of endothelial cell β1-integrins with the extracellular matrix (ECM) stabilizes expression of the TJ protein claudin-5 and maintains permeability barrier integrity, such that direct interference with this interaction is sufficient to increase permeability. 11 Focal ischemia is associated with acute loss of β1-integrins from brain microvessels. 26 However, a causal relationship between the loss of matrix adhesion receptors in focal ischemia and increased microvessel permeability has not been established.
As thrombin has been implicated in vascular injury during ischemia, 16 we hypothesized that dabigatran could decrease the risk of intracerebral hemorrhage by direct inhibition of thrombin-induced permeability in part via the protection of β1-integrin. This study focuses on the role of the endothelial component of brain microvessels in the effects of thrombin and dabigatran on the permeability barrier under the conditions of normoxia and experimental ischemia, as from an embolic insult.
MATERIALS AND METHODS
Institutional Approvals
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Washington and conform to the standards set by the National Institutes of Health.
Reagents
Purified murine α-thrombin was obtained from Hematologic Technologies (Essex Junction, VT, USA). Murine α-thrombin was produced by the methods of Lundblad
Brain Endothelial Cell and Astrocyte Cultures
Primary brain microvessel endothelial cells (mBECs) were obtained from 2- to 3-month-old male C57 BL/6 mice (Jackson Laboratories, Sacramento, CA, USA) and prepared as described previously.
11
Brains were removed, cleaned of meninges and external blood vessels, finely minced, and dissociated in a solution containing 20 U/mL papain and 250 U/mL DNase I type IV in MEM-HEPES (minimum essential medium-4-2-hydroxyethyl-1-piperazineethanesulfonic acid) for 1 hour. The dissociated brain tissue was triturated, added to a 15 mL tube containing 22% bovine serum albumin (final concentration) and centrifuged at 1000
Endothelial cell growth media were changed the next day and the cultures were treated with puromycin (3 μg/mL) for a total of 3 days. The endothelial cell growth culture media were then replaced every 3 days thereafter. Murine brain endothelial cell were seeded onto the top surface of collagen IV-coated 24-well inserts (Greiner bio-one, Kremsmünster, Austria), without or with astrocytes seeded on the bottom surface, on day 9 or 10, and maintained for at least 48 hours before dabigatran/thrombin exposure, and/or oxygen-glucose deprivation (OGD), and permeability experiments. Alternatively, cells were seeded onto collagen IV-coated 6- well inserts for flow cytometry experiments, or onto collagen IV-coated 24-well plates for cell viability assays.
bEnd.3 cells (an immortalized murine endothelial cell line) were cultured as per the manufacturer's protocols in T75 flasks with Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal calf serum, glutamine, and antibiotics before passage to inserts as described above.
Astrocytes were cultured from the brains of neonatal (1 to 2 days old) mouse pups (Charles River, Raleigh, NC, USA) using established techniques. 29 Briefly, cerebral hemispheres were dissected from the brainstem and cerebellum, meninges, and choroid plexuses removed, and the remaining tissue minced and digested in media containing papain and DNAse. Cells were pelleted, resuspended in growth media (DMEM supplemented with fetal bovine serum, glutamine, and antibiotics) and grown to confluence in poly-D-lysine-coated flasks. Microglial cells and oligodendrocytes were removed by shaking before passage. Astrocytes typically reached confluence in 9 to 12 days, and were used in P1 only. For coculture with endothelial cells, astrocytes were passaged to the bottom surface of collagen IV-coated 24-well inserts.
Flow Cytometry
Murine brain endothelial cells were mechanically removed from the inserts, washed, blocked in 5% normal goat serum/phosphate-buffered saline, and incubated with phycoerythrin-conjugated anti-β1-integrin antibody for 1 hour on ice. Cells were permeabilized and incubated with mouse anti-claudin-5 monoclonal antibody followed by FITC-conjugated anti-mouse IgG, and then washed and suspended in 2% formaldehyde for flow cytometry analysis, as described previously. 11 The fluorescence intensity of the labeled cells was analyzed with a Becton-Dickinson FACScan (BD Biosciences), with 8,0 to 10,000 events recorded for each condition, in triplicate.
Cell Viability Assay
Cells were washed with phosphate-buffered saline, incubated with propidium iodide (PI, 33 μg/ml) for 5 minutes, and washed again before fixing with 2% paraformaldehyde. Propidium iodide-positive cells were counted on a fluorescent microscope with a x10 objective. Results are reported as the percentage of positive cells within a 0.371 mm2 grid at x200.
Permeability Measurements
The permeability of monolayers and cocultures were measured as described previously.
11
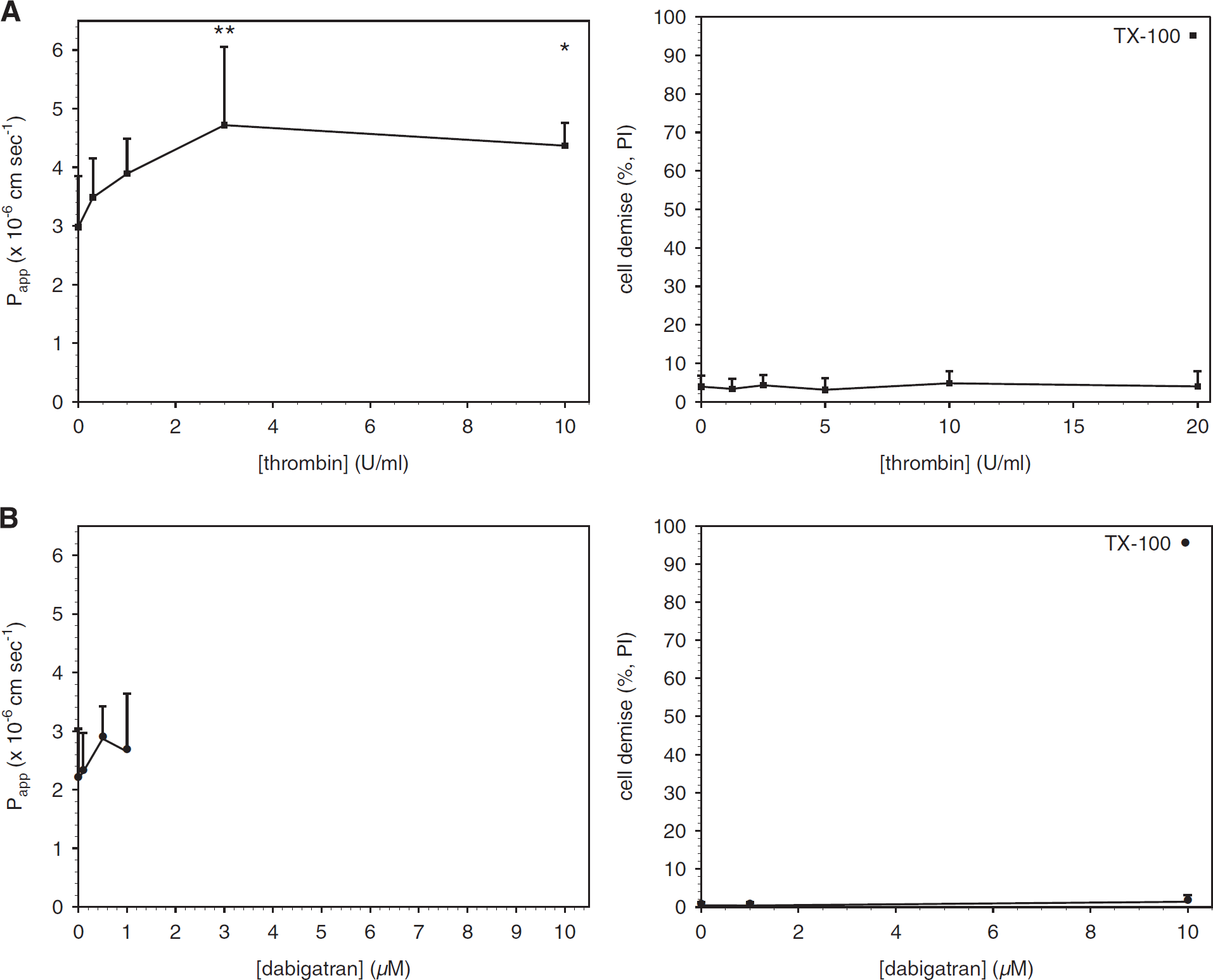
Cells were washed with phosphate-buffered saline and placed in serum-free, phenol red-free DMEM. Fluorescein isothiocyanate-dextrans (4-, 40-, or 150-kDa sizes) were added to the upper chamber (1 mg/ml, final concentration) and 50 μL samples were collected from the lower chamber at 0, 5, 10, 20, 40, and 60 minutes. The volume of each sample removed was replaced with fresh media at each time point. A 50 μl aliquot from the upper chamber was collected at the end of the experiment. Fluorescence intensity was measured (excitation 485 nm, emission 510 nm) on a fluorescent plate reader, with standard curves for FITC-dextran run in parallel. The apparent permeability coefficient (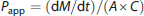
Oxygen-Glucose Deprivation
Oxygen-glucose deprivation was performed under conditions previously set in our laboratory.29–31 Serum-containing media were removed from the cell cultures by washing two times with phosphate-buffered saline before adding serum-free high-glucose medium (4.5 g/L, DMEM containing 4 mmol/L L-glutamine, penicillin, and streptomycin, supplemented with N1 medium) for normoxic controls, or low-glucose medium (1 g/L, supplemented DMEM) for OGD. Cultures containing low-glucose medium were placed in a hypoxia chamber flushed with 95% N2 and 5% CO2 for 1 hour, and then sealed for the duration of the experiment. O2 levels decreased to 0.1% to 0.4% at 4 hours, and were maintained throughout the experiment (18 hours). Normoxic controls were maintained in serum-free media under standard incubator conditions in parallel.
It was noted that endothelial cell monolayers (bEnd.3 and primary cell) showed no morphologic alterations after exposure to thrombin, dabigatran, or the combination thrombin with dabigatran under the conditions of the experiments (see Results and Supplementary Figures).
Statistical Analysis
All data are presented as mean ± s.d. with the numbers of replicates as indicated. Individual sample allocations were known by the principal author only. Normality of data sets was tested by the D'Agostino and Pearson's test. Means were compared by one-way analysis of variance (ANOVA), and significance set at
RESULTS
Thrombin Increases Murine Brain Endothelial Cell Permeability Without Endothelial Cell Toxicity
Preliminary experiments to define the conditions for the studies with mBECs were performed with bEnd.3 cells exposed to media without or with murine α-thrombin (10 U/mL) for 1 hour. Permeability was then tested with 4-, 40-, 70-, or 150-kDa FITC-dextrans. Thrombin significantly increased the permeability of bEnd.3 cell monolayers to 4-kDa FITC-dextran only (data not shown; see Supplementary Figure 1).
Acute exposure to thrombin increases brain endothelial cell (mBEC) permeability without cell toxicity. Primary mBEC grown to confluence were exposed to thrombin (
Primary mBECs were then exposed to thrombin (0 to 10 U/ml) in serum-free media for 1 hour before measurement of permeability. Thrombin had a concentration-dependent effect on mBEC permeability to 4-kDa FITC-dextran (Figure 1A), as reported previously. 21 However, thrombin concentrations up to 20 U/mL did not cause a significant increase in PI uptake (Figure 1A), indicating that the permeability changes observed were not because of cell death. Baseline transendothelial electrical resistance measurements were made in a subset of endothelial cell monolayers and reflected those reported previously. 11
Dabigatran Is Not Toxic to Murine Brain Endothelial Cell and Does Not Affect Permeability at Therapeutic Concentrations Murine brain endothelial cells were exposed to dabigatran (0 to 1 pmol/L) in serum-free media for 24 hours before the measurement of permeability to 4-kDa FITC-dextran. Dabigatran alone did not affect permeability, and did not cause a significant change in PI uptake up to 10 μmol/L (Figure 1B), a concentration ~ 20 times higher than the plasma concentration of dabigatran typically achieved in patients. 32
Thrombin Does Not Affect Endothelial Expression of β1-Integrin or Claudin-5
Exposure of mBEC to α-thrombin (1 or 10 U/mL) for 1 hour had no effect on the mean fluorescence intensity of cells labeled for β1-integrin and claudin-5 (Figures 2A and 2B) nor on the distribution of cells expressing each antigen (Figure 2C and Supplementary Figure 2). No effect on the morphology of the endothelial cells was observed under any condition.
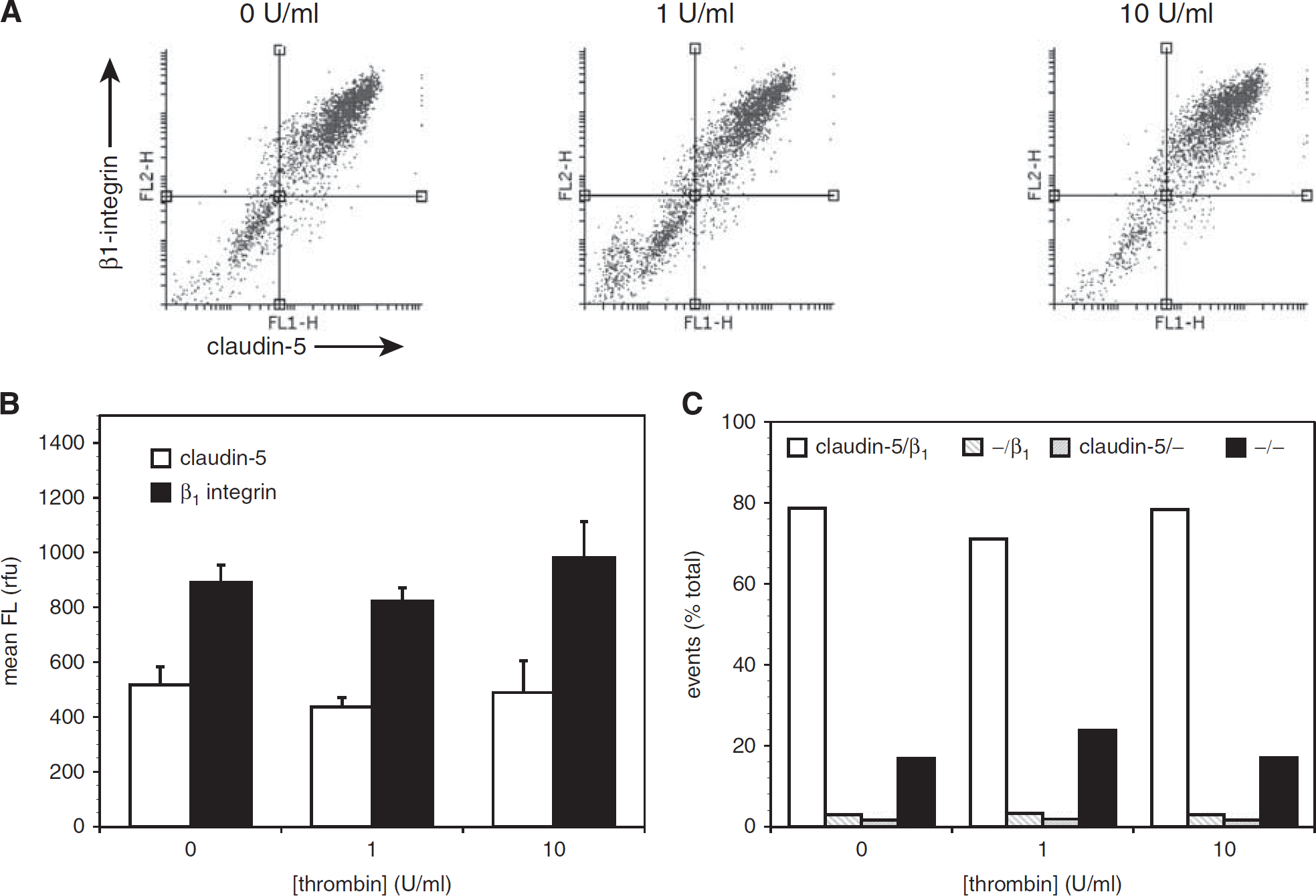
Acute thrombin has no effect on murine brain endothelial cell (mBEC) expression of claudin-5 or β1-integrin. Primary mouse brain endothelial cells were exposed to thrombin (0 to 10 U/mL) for 1 hour before labeling for claudin-5 and β1-integrin and analysis by flow cytometry. (
Dabigatran Blocks the Effects of Thrombin and Oxygen-Glucose Deprivation on Murine Brain Endothelial Cell Permeability Preincubation of mBEC with dabigatran (500 nmol/L) for 24 hours before thrombin (10 U/mL, 1 hour) exposure completely blocked the effect of thrombin on mBEC permeability in both mBEC monolayers (Figure 3A) and in mBEC cocultured with astrocytes (Figure 3B). Furthermore, the addition of 500 nmol/L dabigatran to the media of cells exposed to OGD attenuated the effect of OGD on mBEC permeability (Figure 4).
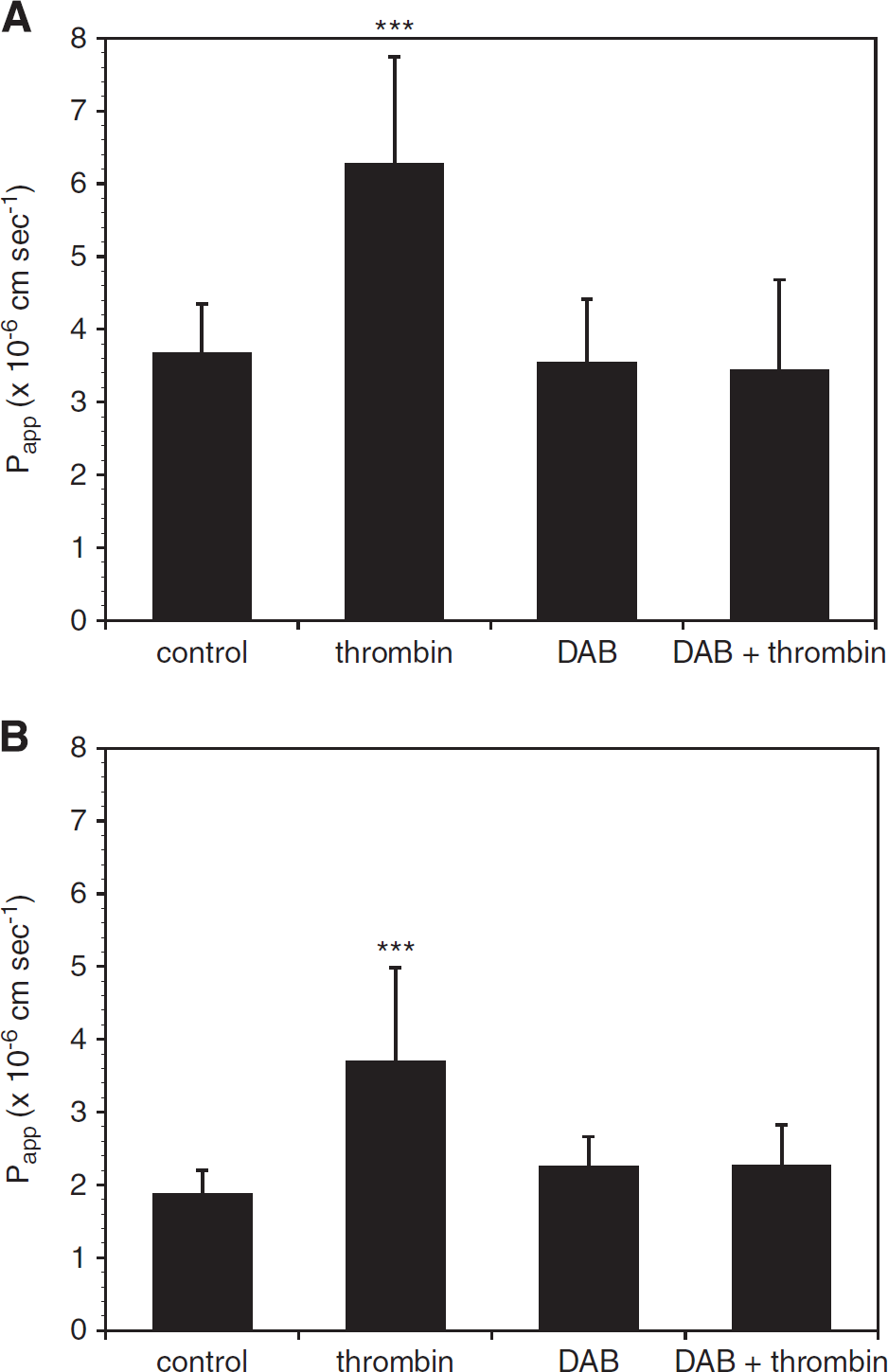
Dabigatran (DAB) inhibits the effect of thrombin on murine brain endothelial cell (mBEC) permeability. (
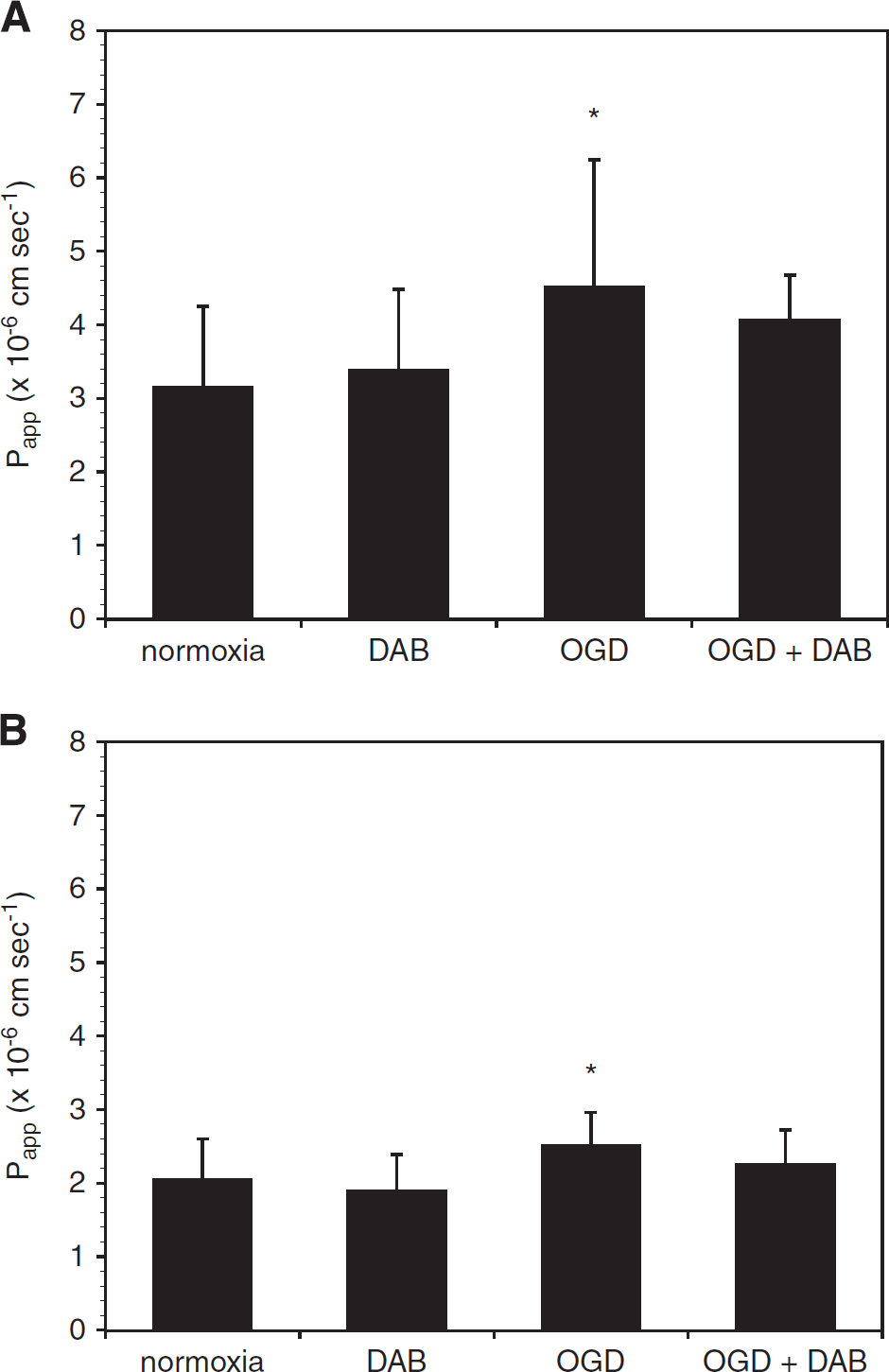
Dabigatran (DAB) abrogates the effect of oxygen-glucose deprivation (OGD) on murine brain endothelial cell (mBEC) permeability. Incubation with dabigatran (500 nmol/L) for 24 hours before and during 18 hours OGD abrogates the effect of OGD on permeability on primary brain endothelial cells grown alone (
Dabigatran Attenuates the Effect of Thrombin on Permeability in the Setting of Oxygen-Glucose Deprivation Murine brain endothelial cell cocultured with astrocytes were exposed to OGD for 18 hours, followed by thrombin (10 U/mL, 1 hour) to model the generation and extravasation of thrombin after an ischemic event
DISCUSSION
Focal cerebral ischemia induces fibrin formation within microvessels in the territory-at-risk (ischemic regions) and is accompanied by increased vascular permeability that causes edema formation and hemorrhage.8,16 Hemorrhagic transformation in the CNS is a common accompaniment of ischemic strokes, being part of the natural history, and anticoagulation is associated with an increased risk and severity of cerebral hemorrhage. 33 In a high-quality non-human primate model of middle cerebral artery occlusion, we have shown that fibrin-containing occlusions obstruct the microvasculature, and that fibrin deposits in the microvessel wall and the perivascular space occur acutely, within 60 to 120 minutes of ischemia onset. 8 This shows the acute local generation of thrombin at the abluminal interface or within the perivascular space of microvessels within the ischemic territory. Furthermore, thrombin has been implicated in barrier damage after experimental ischemia in the rodent.
In this study, we show that α-thrombin increases the permeability of primary mBEC, alone or in coculture with astrocytes, to 4-kDa FITC-dextran and that the direct thrombin inhibitor dabigatran at a clinically relevant concentration completely reverses the increased permeability (Figure 3). Previous studies investigating the effect of thrombin on the blood–brain permeability barrier have shown altered barrier function in other
The mechanism by which thrombin leads to increased endothelial cell permeability is not yet known. Thrombin can exert effects on brain endothelial cells via its interaction with the protease-activated receptors (PAR)-1 and −3,
35
and Src kinase signaling has been directly linked to thrombin-induced damage to the permeability barrier
Another potential mechanism lies in the serine protease activity of thrombin. There is sparse information concerning the impact of thrombin on ECM proteins.22,23,40,41 Among its cell-activating properties, thrombin has been shown to stimulate collagen IV synthesis from mesangial cells in culture, and to stimulate the polarized secretion of ECM components.40,41 Liotta
Another potential explanation for the observation of increased permeability caused by α-thrombin is suggested by receptor studies. Studies with bEnd.3 cells, as an
In these experiments OGD itself resulted in increased permeability that was partly reversed by dabigatran. Although these experiments were strictly serum free, the possibility that trace amounts of prothrombin and other coagulation factors adherent to the endothelial cell surface could generate thrombin locally cannot be ruled out. Tissue factor can appear on cultured endothelial cells under specific circumstances. 46 However, in bEnd.3 cells, we have observed that fetal bovine serum detectably increases endothelial cell permeability, and that this effect is not inhibited by dabigatran (Y Izawa, unpublished observations). This suggests that thrombin is not present in sufficient amounts in fetal bovine serum to account for the observed change in permeability in response to serum, or that dabigatran interacts differently with bovine thrombin. Alternatively, these observations could reflect the tightening of the barrier in response to serum-free media previously reported, 47 and indicate that factors other than thrombin are involved.
The magnitude of the effect of α-thrombin on permeability exceeds that of OGD in this system in all experiments performed. Furthermore, we found that α-thrombin and OGD additively increased endothelial cell permeability, and that dabigatran attenuates this effect to the level of OGD alone. Hence, under conditions of experimental ischemia, where thrombin is generated within the microvasculature, it is expected that the thrombin component of increased cerebral microvessel permeability can be abrogated by the antithrombin dabigatran, or other antithrombins. The action of OGD to modestly increase the
We also noted that this
There is no data regarding direct effects of a selective antithrombin on cerebral microvessel endothelial behavior during or after experimental ischemia
In the clinic, increased barrier permeability (as visualized by Gd-DTPA hyperintensities on a contrast-weighted magnetic resonance imaging) is predictive of hemorrhagic transformation in ischemic stroke patients. 25 This suggests that barrier permeability may precede the development of hemorrhage. If so, inhibition of any thrombin-dependent increase in the endothelial cell-related permeability during focal ischemia by dabigatran could contribute to the reduced risk of hemorrhage associated with this agent. It is unclear whether inhibition of other steps in extrinsic coagulation (e.g. factor Xa) might have a similar effect, as compatible studies would require the presence of plasma, which is technically not feasible.
In conclusion, we have shown that dabigatran exerts a protective effect on an
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.