
Abstract
Hypothermia is partially neuroprotective after neonatal hypoxic-ischemic encephalopathy. Blockade of connexin hemichannels can improve recovery of brain activity and cell survival after ischemia in near-term fetal sheep. In this study, we investigated whether combining delayed hypothermia with connexin hemichannel blockade with intracerebroventricular infusion of a mimetic peptide can further improve outcomes after cerebral ischemia. Fetal sheep (0.85 gestation) received 30 minutes of cerebral ischemia followed by a 3-hour recovery period before treatment was started. Fetuses were randomized to one of the following treatment groups: normothermia (n = 8), hypothermia for 3 days (n = 8), connexin hemichannel blockade (50 umol/L intracerebroventricular over 1 hour followed by 50 umol/L over 24 hours, n = 8) or hypothermia plus hemichannel blockade (n = 7). After 7 days recovery, hypothermia was associated with reduced seizure burden, improved electroencephalographic (EEG) power, and a significant increase in neuronal and oligodendrocyte survival and reduced induction of Iba1-positive microglia. In contrast, although hemichannel blockade reduced seizure burden, there was no effect on EEG power or histology (P < 0.05). There was no further improvement in outcomes with combined hypothermia plus hemichannel blockade. In conclusion, these data show that there is no additive neuroprotection with combined hypothermia and hemichannel blockade after cerebral ischemia in near-term fetal sheep.
INTRODUCTION
There is compelling clinical and experimental evidence that therapeutic hypothermia can reduce neural injury and improve neurodevelopmental outcomes after severe hypoxia-ischemia at term.1–4 However, current protocols for therapeutic hypothermia are only partially neuroprotective. 1 Attempts to further optimize cooling protocols have been largely unsuccessful. For example, extending the duration of cerebral cooling from 72 hours to 120 hours was associated with no additional improvement in electrophysiologic outcomes, and a small increase in cell death in some regions after cerebral ischemia in near-term fetal sheep. 5 Similarly, a recent clinical trial of prolonged duration and greater depth of hypothermia in neonates with hypoxic ischemic encephalopathy was suspended due to lack of benefit. 6
The partial protection found in clinical studies is likely related to the formidable clinical difficulties involved in starting hypothermia within the optimal window of opportunity. 7 A recent cohort study suggested that asphyxiated infants who were able to be cooled within 3 hours of birth had better motor outcomes than when hypothermia was started between 3 and 6 hours. 8 However, in a controlled trial, hypothermia was only able to be started in 12% of infants within 4 hours of birth. 9
There is increasing evidence that blockade of connexin hemichannels may be an important therapeutic target after cerebral ischemia. 10 Connexin hemichannels are the opposing halves of gap junctions, the intercellular channels that link the cytoplasm of adjacent cells. In the brain, Connexin43 (Cx43) is the predominant astrocytic connexin. It is upregulated in human postmortem tissue and in fetal sheep after cerebral ischemia.11,12 There is now evidence that ischemia can trigger transient unregulated opening of connexin hemichannels, leading to disruption of the resting membrane potential, release of cytotoxic levels of ATP 13 and glutamate, 14 and uptake of water resulting in cell swelling and rupture.15,16 Supporting this concept, we have recently shown that blockade of C×43 hemichannels with a specific mimetic peptide significantly improved recovery of electroencephalographic (EEG) activity, and reduced seizures and neuronal and oligodendrocyte cell death after global cerebral ischemia or asphyxia in fetal sheep. 12 , 17 , 18
These previous studies examined the effect of blocking connexin hemichannels starting 90 minutes after the end of cerebral ischemia. However, critically for potential clinical translation, the exact window of opportunity for protection with hemichannel blockade remains unknown. There is some encouraging preclinical evidence that subtherapeutic regimens of neuroprotective agents can synergistically augment hypothermic protection.19–21 Thus, in the present study we examined whether connexin hemichannel blockade was neuroprotective when started after a clinically realistic delay of 3 hours from the end of a 30-minute period of global cerebral ischemia in term-equivalent fetal sheep. We further investigated whether the combination of delayed connexin hemichannel blockade and therapeutic hypothermia could further improve EEG or histologic outcomes.
MATERIALS AND METHODS
Fetal Surgery
All procedures were approved by the Animal Ethics Committee of The University of Auckland under the New Zealand Animal Welfare Act, and the Code of Ethical Conduct for animals in research established by the Ministry of Primary Industries, Government of New Zealand. This manuscript is compliant with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. In brief, 38 time-mated Romney/Suffolk fetal sheep were instrumented using sterile techniques at 118 to 124 days gestation (term is 145 days). Food, but not water was withdrawn 18 hours before surgery. Ewes were given long acting oxytetracycline (20 mg/kg, Phoenix Pharm, Auckland, New Zealand) intramuscular injection 30 minutes before the start of surgery. Anesthesia was induced by intravenous injection of propofol (5 mg/kg; AstraZeneca Ltd, Auckland, New Zealand) and maintained using 2% to 3% isoflurane in O2. The depth of anesthesia, maternal heart rate, and respiration were constantly monitored by trained anesthetic staff. Ewes received a constant infusion isotonic saline drip (at an infusion rate of approximately 250mL/h) to maintain fluid balance.
After a maternal midline abdominal incision, the fetus was exposed and both fetal brachial arteries were catheterized with polyvinyl catheters to measure mean arterial blood pressure (MAP). An amniotic catheter was secured to the fetal shoulder. ECG electrodes (AS633-3SSF, Cooner Wire Co., Chatsworth, CA, USA) were sewn across the fetal chest to record fetal heart rate. The vertebral-occipital anastomoses were ligated and inflatable carotid occluder cuffs were placed around both carotid arteries.22,23 A 3S Transonic ultrasonic flow probe (Transonic Systems, Ithaca, NY, USA) was placed around the right carotid artery. Using a 7 stranded stainless steel wire (AS633-7SSF; Cooner Wire Co.), two pairs of EEG electrodes were placed on the dura over the parasagittal parietal cortex (10 mm and 20 mm anterior to bregma and 10 mm lateral) and secured with cyanoacrylate glue. A reference electrode was sewn over the occiput. A further two electrodes were sewn in the nuchal muscle to record electromyographic activity as a measure of fetal movement. A thermistor was placed over the parasagittal dura 30 mm anterior to bregma to measure extradural temperature and a second thermistor was inserted into the esophagus to measure core body temperature. An intracerebroventricular cannula was placed into the left lateral ventricle 6 mm anterior and 4 mm lateral to bregma for infusion of the mimetic peptide. A cooling cap made from silicon tubing (3×6 mm, Degania Silicone, Degania Bet, Israel) was secured to the fetal head. The uterus was then closed and antibiotics (80 mg Gentamicin, Pfizer, New York, NY, USA) were administered into the amniotic sac. The maternal laparotomy skin incision was repaired and infiltrated with 10 mL 0.5% bupivacaine plus adrenaline (AstraZeneca Ltd). All fetal catheters and leads were exteriorized through the maternal flank. The maternal long saphenous vein was catheterized to provide access for postoperative maternal care and euthanasia.
Postoperative Care
Sheep were housed together in separate metabolic cages with access to food and water ad libitum. They were kept in a temperature-controlled room (16±1°, humidity 50±10%), in a 12-hour light/dark cycle. Antibiotics were administered daily for 4 days intravenously to the ewe (600 mg benzylpenicillin sodium, Novartis Ltd, Auckland, New Zealand and 80 mg gentamicin, Pfizer). Fetal catheters were maintained patent by continuous infusion of heparinized saline (20U/mL at 0.15 mL/h) and the maternal catheter maintained by daily flushing.
Data Recording
Data recordings began 24 hours before the start of experiments and continued for the remainder of each experiment. Data were recorded and saved continuously to disk for off-line analysis using custom data acquisition programs (LabView for Windows, National Instruments, Austin, TX, USA). Arterial blood samples were taken for preductal pH, blood gas, base excess (Ciba-Corning Diagnostics 845 blood gas analyzer and cooximeter, MA, USA), glucose and lactate measurements (YSI model 2300, Yellow Springs, OH, USA). All fetuses had normal biochemical variables for their gestational ages.24,25
Experimental Protocols
At 128 ± 1 days gestation, ischemia was induced by reversible inflation of the carotid occluder cuffs with sterile saline for 30 minutes. Successful occlusion was confirmed by the onset of an isoelectric EEG signal within 30 seconds of inflation. The carotid occluder cuffs were not inflated in sham control experiments. Fetal blood samples were drawn just before the occlusion and 2 hours, 4 hours, and 6 hours after occlusion followed by daily sampling.
Fetuses were randomized to ischemia-normothermia (n = 8), ischemia-hypothermia (n = 8), ischemia-peptide (n = 7), ischemia-peptide-hypothermia (n = 7), or sham control (n = 8). Cooling was started 3 hours after reperfusion and continued until 72 hours after the end of ischemia in the ischemia-hypothermia and ischemia-peptide-hypothermia groups. Cooling was performed by connecting the cooling coil over the fetal scalp with a pump in a cooled water bath and circulating cold water through the cooling coil. The target extradural temperature was between 31° and 33°. In the ischemia-normothermia and ischemia-peptide groups, the water was not circulated and the cooling coil remained in equilibrium with fetal temperature.
To block C×43 hemichannels, a peptide (H-Val-Asp-Cys-Phe-Leu-Ser-Arg-Pro-Thr-Glu-Lys-Thr-OH; Auspep, Tullamarine, Victoria, Australia) that mimics the second extracellular loop of C×43 (‘Peptide 5’ reported in O'Carroll et al 26 ) was infused into the left lateral ventricle via the intracerebroventricular catheter attached to an external pump, starting 3 hours after the end of the occlusion. The ischemia-peptide and ischemia-peptide-hypothermia groups received 50 umol/kg for 1 hour followed by 50μmol/kg for 24 hours dissolved in artificial cerebrospinal fluid (KCl 5 mmol/L, NaCl 137 mmol/L, CaCl2 0.8 mmol/L, 0.1% bovine serum albumin, pH 7.4), at a rate of 1mL over the first hour, then 1 mL over 24 hours. This dosing regime has previously been shown to improve neural outcomes when started 90 minutes after the end of global cerebral ischemia in term-equivalent fetal sheep.12,17,27 The sham control and ischemia-hypothermia fetuses received infusion of the vehicle via the intracerebroventricular catheter. Seven days after ischemia, ewes and fetuses were killed by an overdose of sodium pentobarbitone (9 g, Pentobarb, Chemstock International, Christchurch, New Zealand).
Data Analysis
The total EEG power was log transformed [dB, 20 times log (power)], as this transformation gives a better approximation of the normal distribution, and then normalized with respect to the 24-hour baseline period. Spectral edge, a measure of the relative frequency of the EEG, was calculated from the power spectrum as the frequency below which 90% of total EEG power resides. Impedance increases as the temperature of the medium, through which the signal passes, decreases. Therefore, in each fetus, the slope of impedance change at the onset of hypothermia was used to correct the impedance signal: corrected impedance = impedance-(slopexΔtemperature).22,28 Seizure burden was quantified as the total duration of seizure activity per fetus in minutes.
To evaluate the effect of the two interventions on fetuses exposed to ischemia, analysis was performed using ANOVA, with hypothermia and peptide as independent variables and region or time treated as repeated measures. Post hoc analysis was used to test region or time-specific effects. The effect of ischemia-normothermia compared with sham control values was assessed separately by ANOVA, with region or time treated as repeated measures. Statistical significance was accepted when P<0.05. As seizure burden data were nonparametric, groups were compared using the Mann-Whitney test.
Immunohistochemistry
Fetal brains were perfusion fixed with 10% phosphate-buffered formalin. Coronal slices (10μm thick) were cut using a microtome (Leica Jung RM2035, Solms, Hessen, Germany) starting at the level of the dorsal hippocampus. Slides were dewaxed in xylene and rehydrated in decreasing concentrations of ethanol, then washed in 0.1 mol/L phosphate-buffered saline (PBS) for neuronal nuclear antigen (NeuN) and oligodendrocyte transcription factor (Olig2) and PBS+0.1% Triton (PBS-T) for ionized calcium-binding adapter molecule 1 (Iba1). Antigen retrieval was performed using citrate buffer (450 mL distilled water, 8mL citric acid, 42 mL sodium citrate, pH 6.5) in an antigen retrieval system (EMS Antigen 200 Retriever, Emgrid Australia Pty, Melbourne, Victoria, Australia) followed by incubation in 1% H2O2 in methanol for NeuN and Iba1 and in PBS for Olig2 to block endogenous peroxidase activity. Blocking was performed in 3% normal goat serum for NeuN and Olig2 and normal goat serum with 0.1% Triton X-100 (Scharlau Chemie, Sentmenat, Spain) for Iba1 for 1 hour at room temperature. Sections were labelled with 1:200 mouse anti-neuronal nuclei monoclonal antibody (NeuN, Chemicon International, Temecula, CA, USA) 1:400 rabbit Olig2 monoclonal antibody (Merck-Millipore, Billerica, MA, USA) or 1:200 goat anti-Iba1 (Abcam, Cambridge, England) overnight at 4°. Sections were incubated for 3 hours in biotin-conjugated 1:200 anti-mouse (NeuN), 1:200 anti-rabbit (Olig2) or 1:200 anti-goat (Iba1, Vector Laboratories, Burlingame, CA, USA) in 3% normal goat serum for NeuN and Olig-2 and 3% NHS for Iba1. Slides were then incubated in ExtrAvidin (1:200, Sigma-Aldrich Pty. Ltd, St Louis, MO, USA) in PBS for two hours at room temperature and then reacted in diaminobenzidine tetrachloride (Sigma-Aldrich Pty. Ltd). The reaction was stopped by washing in PBS and the sections dehydrated and mounted.
Neurons were imaged in the cortex of the first and second parasagittal gyri, CA1, CA3, CA4, and dentate gyrus of the hippocampus, oligodendrocytes, and microglia in the intragyral white matter of the first and second parasagittal gyri and in the periventricular white matter using light microscopy (Nikon Eclipse 80i, Scitech Ltd, Preston, Victoria, Australia). One image per area was obtained from both the left and right hemispheres from two sections per antibody (i.e., a total of four images were averaged per area per antibody for each fetus) at ×20 magnification. All sections were taken at the level at which the dorsal hippocampus becomes apparent. Cell counting was performed using ImageJ software (imagej.nih. gov). Healthy neurons were counted based on the morphologic assessment; neurons showing a pyknotic morphology were excluded. 29
Microglia showing either an amoeboid or ramified morphology were included. Imaging and analysis were performed by an investigator masked to the treatment group by separate coding of the slides.
RESULTS
Sex distribution, number of singletons/twins and post-mortem body and brain weights are shown in Supplementary Table 1. Hypothermia was associated with an independent increase in brain weight at postmortem (P < 0.05).
Blood Gas, pH, Glucose, and Lactate Measurements
There were no significant differences in baseline blood gas, pH, glucose, or lactate measurements between groups (Table 1). All groups showed a significant increase in lactate concentration after ischemia, which resolved by 6 hours, except in the ischemia-hypothermia group, which returned to baseline levels by day one. A significant increase in glucose concentration was seen after ischemia in all groups, which resolved to baseline levels by day 1 in the ischemia-hypothermia and ischemia-peptide-hypothermia groups, day 2 in the ischemia-normothermia group and day 3 in the ischemia-peptide group.
Blood gas, pH, glucose, and lactate concentrations before and after 30 minutes of global cerebral ischemia
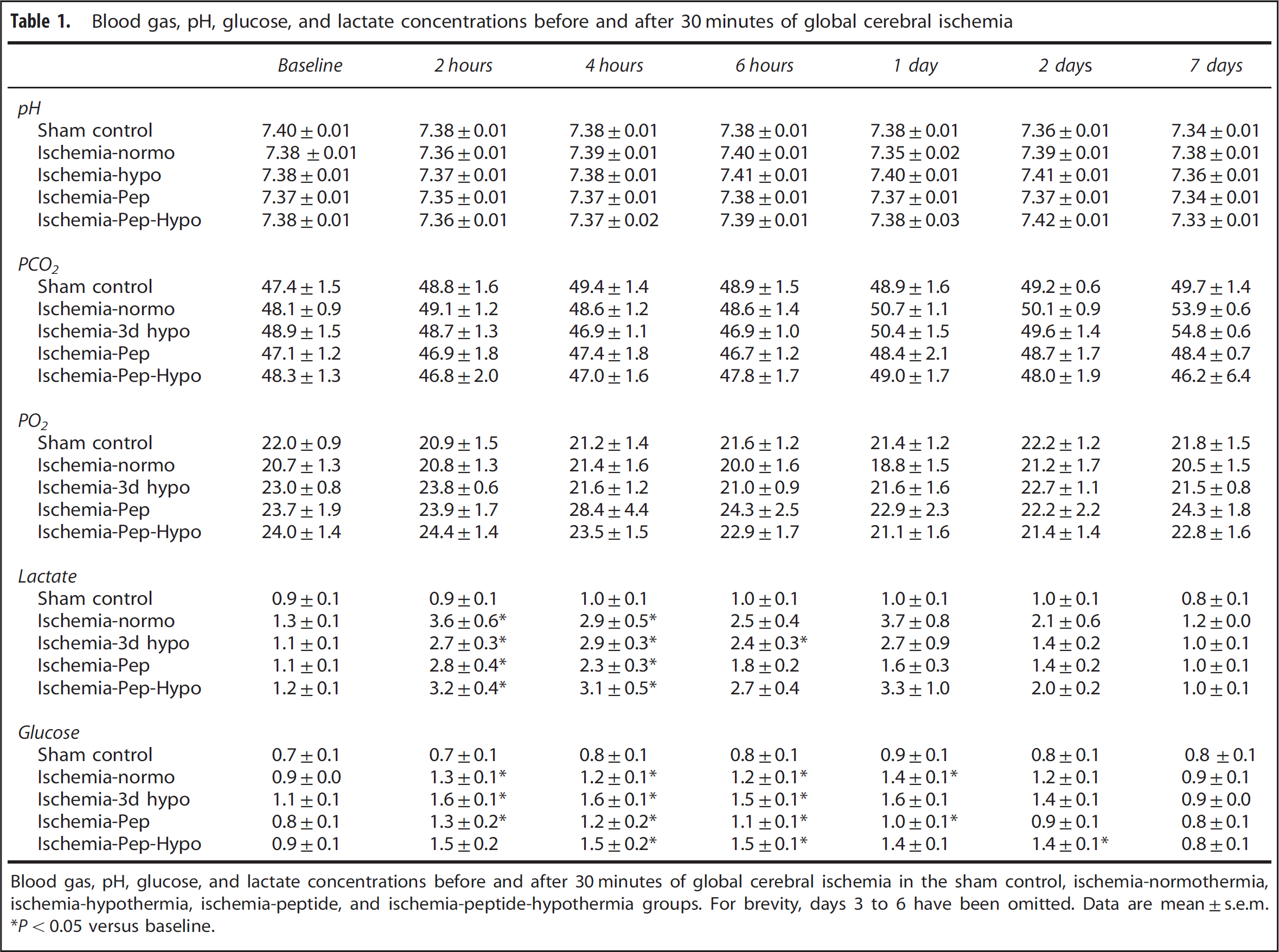
Blood gas, pH, glucose, and lactate concentrations before and after 30 minutes of global cerebral ischemia in the sham control, ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups. For brevity, days 3 to 6 have been omitted. Data are mean ±s.e.m. *P < 0.05 versus baseline.
Cardiovascular and Temperature Changes
At the onset of hypothermia extradural temperature decreased to 31.3±0.2° in the ischemia-hypothermia group and 32.1 ±0.3° in the ischemia-peptide-hypothermia group compared with 39.5±0.1° in the ischemia-normothermia and ischemia-peptide groups (P < 0.05, Figure 1). Esophageal temperature decreased to between 37° and 38° during cooling (P < 0.05, Figure 1).
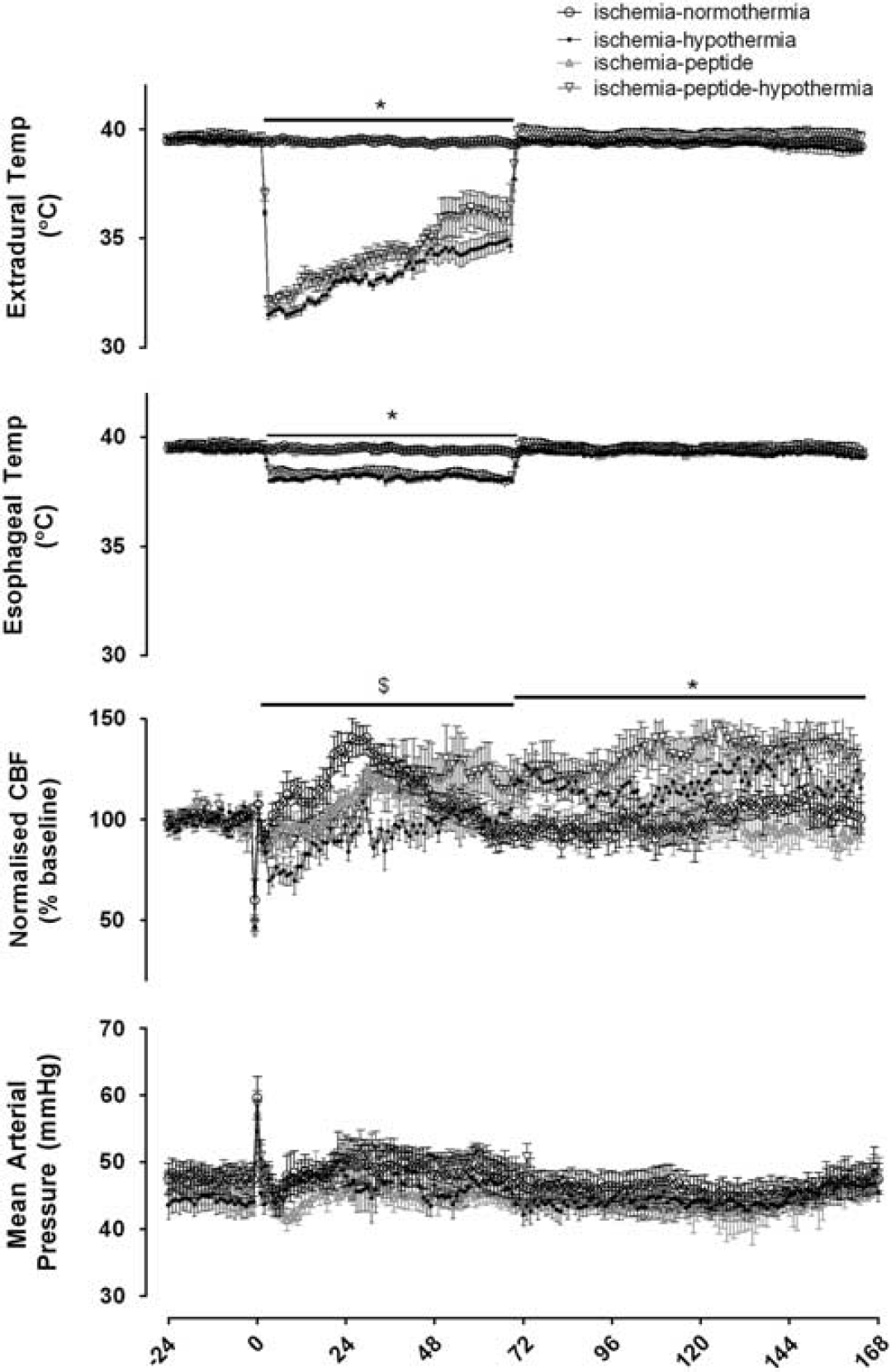
Time sequence of changes in extradural and esophageal temperature, mean arterial pressure (MAP) and carotid artery blood flow (CaBF) before, during, and after 30 minutes of global cerebral ischemia in the ischemia-normothermia, ischemia-hypothermia ischemia-peptide, and ischemia-peptide-hypothermia groups. Time zero represents the start of carotid artery occlusion. Hypothermia was associated with a significant reduction in extradural and esophageal temperature between 3 and 72 hours after ischemia (P<0.05). There were no significant differences in MAP at any time between groups. A significant decrease in CaBF was seen in the ischemia-hypothermia group over the first 72 hours after ischemia compared with the ischemia-normothermia group (P<0.05) with a significant interaction between hypothermia and peptide during this time. Between 72 and 168 hours, hypothermia was associated with a significant overall increase in CaBF. Data are mean ± s.e.m., *P<0.05 significant effect of hypothermia, $P<0.05 significant interaction between peptide and hypothermia.
Brain Activity
Ischemia was associated with rapid, profound suppression of EEG power compared with sham control (Figure 2). After release of the occlusion, EEG activity remained suppressed until the onset of a period of intense seizure activity that started approximately 8 hours after ischemia and resolved between 48 and 72 hours after ischemia. The total seizure burden was reduced from 300±119 minutes in the ischemia-normothermia group to 62 ± 22 minutes in the hypothermia group (P < 0.05) and 90 ±52 minutes in the ischemia-peptide group (P < 0.05). In contrast, the ischemia-peptide-hypothermia group showed an intermediate seizure burden (129±45 minutes), which was not significantly different from the ischemia-hypothermia and ischemia-normothermia groups (P> 0.05).
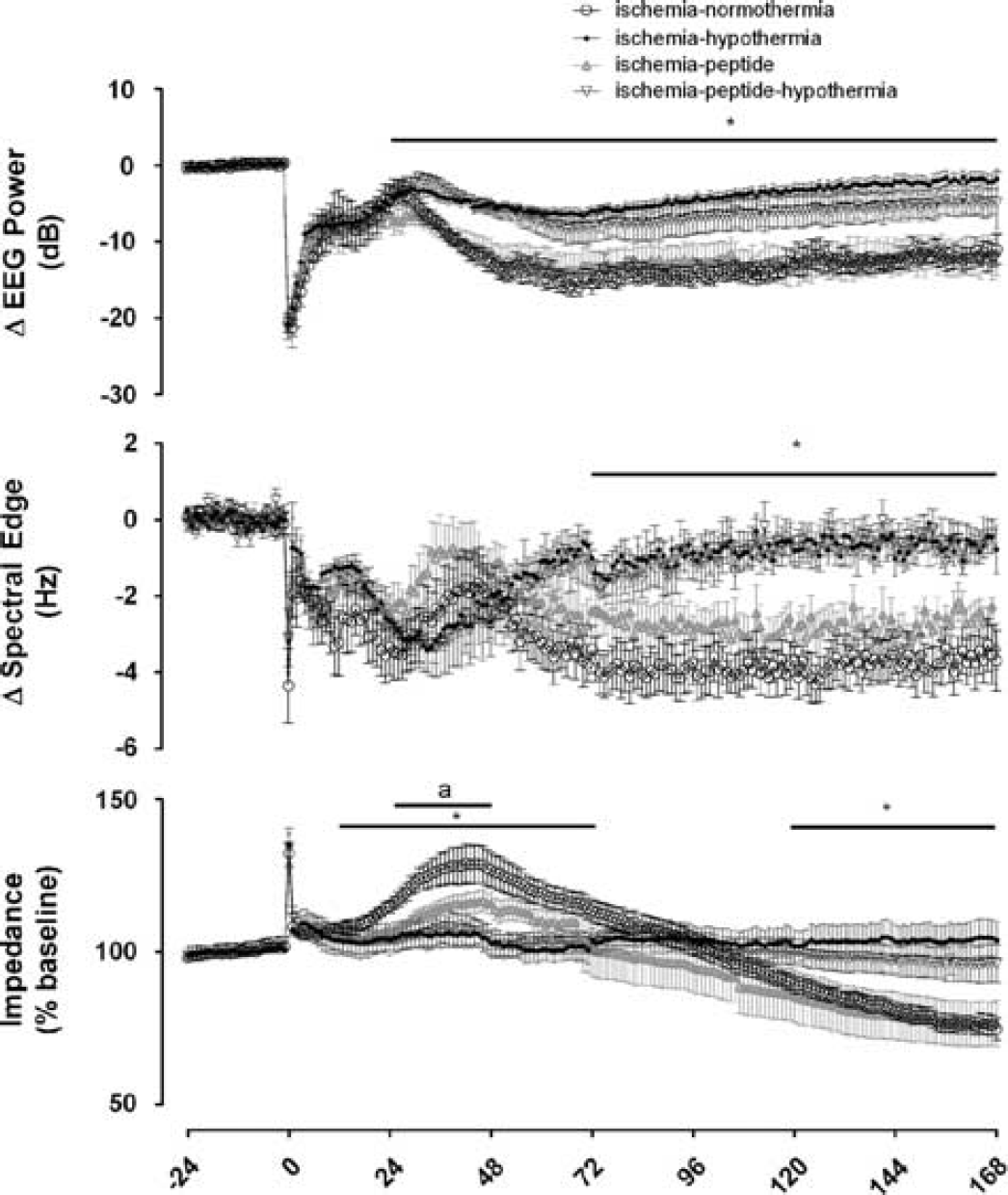
Time sequence of changes in electroencephalographic (EEG) activity, spectral edge frequency and impedance before, during, and after 30 minutes of global cerebral ischemia in the ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups. Time point zero denotes the start of ischemia. EEG activity was suppressed in all groups during ischemia. A rebound in EEG activity was seen during the seizure period between 8 and 48 hours in all groups. EEG activity in the ischemia-normothermia group was reduced for the remainder of the experiment, while hypothermia was associated with a significant overall recovery in EEG power from 24 hours onwards (P<0.05). No significant effect of peptide administration was seen on the recovery of EEG power. Spectral edge was suppressed in all groups during ischemia and remained suppressed until the end of the experiment in the ischemia-normothermia group. Hypothermia, but not mimetic peptide, was associated with a significant increase in spectral edge frequency from 72 hours onwards. Ischemia was associated with an increase in impedance in all groups which resolved after the end of ischemia. A significant rise in impedance was seen between 24 and 72 hours in the ischemia-normothermia group. Hypothermia was associated with a significant overall reduction in impedance between 12 and 72 hours with no overall effects of peptide. However, a significant reduction in impedance was seen between 24 and 48 hours in the ischemia-peptide group compared with the ischemia-normothermia group (P<0.05, repeated measures ANOVA). Data are mean ± s.e.m., *P < 0.05 significant effect of hypothermia, aP<0.05, ischemia-peptide versus ischemia-normothermia.
After seizures resolved, total EEG power fell below baseline levels in the ischemia-normothermia group. In contrast, hypothermia was independently associated with a significant improvement in the recovery of EEG power from 24 hours onwards, while mimetic peptide administration was not associated with a significant effect on the recovery of EEG power (Figure 2). Ischemia was also associated with rapid suppression of spectral edge, which remained below baseline until the end of the experiment in the ischemia-normothermia group. Hypothermia, but not mimetic peptide, was associated with a significant independent increase in spectral edge from 72 hours until the end of the experiment (Figure 2, P < 0.05). There was no interaction between hypothermia and peptide treatment for either power or spectral edge.
Ischemia-normothermia was associated with a secondary rise in cortical impedance from 12 to 72 hours after ischemia. Hypothermia was associated with an independent, sustained reduction in impedance (P < 0.05); ischemia-peptide was associated with a reduction in impedance compared with ischemia-normothermia between 24 and 48 hours (P < 0.05). Impedance fell below baseline levels from 120 hours onward in the ischemia-normothermia group, which was independently attenuated by hypothermia (P < 0.05), but not mimetic peptide.
Physiologic Parameters
A significant decrease in carotid artery blood flow was seen in the ischemia-hypothermia group during the first 72 hours after ischemia compared with ischemia-normothermia (repeated measures ANOVA, P < 0.05). There was a significant interaction between hypothermia and peptide during the first 72 hours after ischemia, such that the ischemia-peptide-hypothermia group showed greater carotid artery blood flow in this period than the ischemia-hypothermia group (i.e., delayed hypoperfusion was attenuated, P< 0.05). From 72 hours until the end of the experiment, hypothermia was independently associated with increased carotid artery blood flow (P < 0.05); there was no effect of peptide or interaction between peptide and hypothermia in this phase. There were no significant differences in MAP (Figure 1), fetal heart rate, or nuchal electromyographic between groups (data not shown).
Immunohistochemistry
Ischemia-normothermia was associated with marked loss of neurons in the cortex, CA1, CA3, and CA4, but not in the dentate gyrus, of the hippocampus, compared with sham controls after 7 days recovery (P < 0.05, Figure 3). Hypothermia was associated with an independent overall increase in neuronal survival in the cortex, CA1, CA3, and CA4 (P < 0.05). There was no effect of mimetic peptide on neuronal survival and no interaction between hypothermia and peptide.
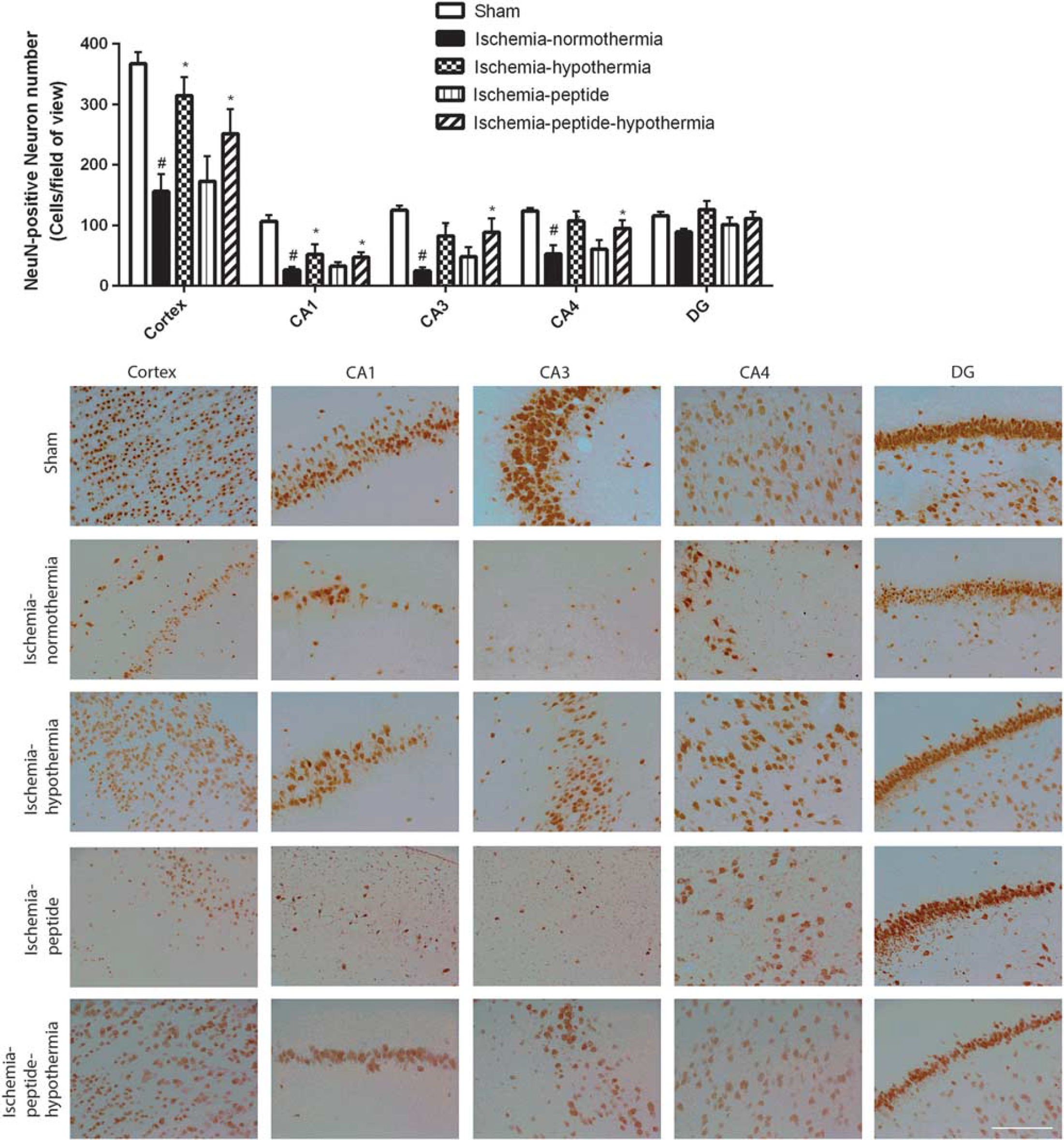
Numbers of neurons in the cortex, CA1, CA3, CA4, and dentate gyrus in the ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups 7 days after 30 minutes of global cerebral ischemia. Top: A significant decrease in neuronal number was seen in the cortex, CA1, CA3, and CA4 in the ischemia-normothermia group compared with sham control (P < 0.05). Hypothermia was associated with a significant independent improvement in neuronal number in the cortex, CA1, CA3, and CA4 (P < 0.05). No significant overall effect of peptide was seen nor any interaction between peptide and hypothermia. Data are mean ± s.e.m., #P<0.05 sham control versus ischemia-normothermia, *P < 0.05 significant effect of hypothermia. Bottom: Photomicrograph of neuronal survival in the cortex, CA1, CA3, CA4, and dentate gyrus of the hippocampus in the sham control, ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups 7 days after 30 minutes of global cerebral ischemia. Scale bar 200μm.
Similarly, ischemia-normothermia was associated with marked loss of all oligodendrocytes in the intragyral white matter of the first and second parasagittal gyri and in the periventricular white matter (P < 0.05, Figures 4 and 5). Hypothermia was independently associated with increased oligodendrocyte survival in all regions (P < 0.05), with no effect of the mimetic peptide and no interaction between hypothermia and peptide.
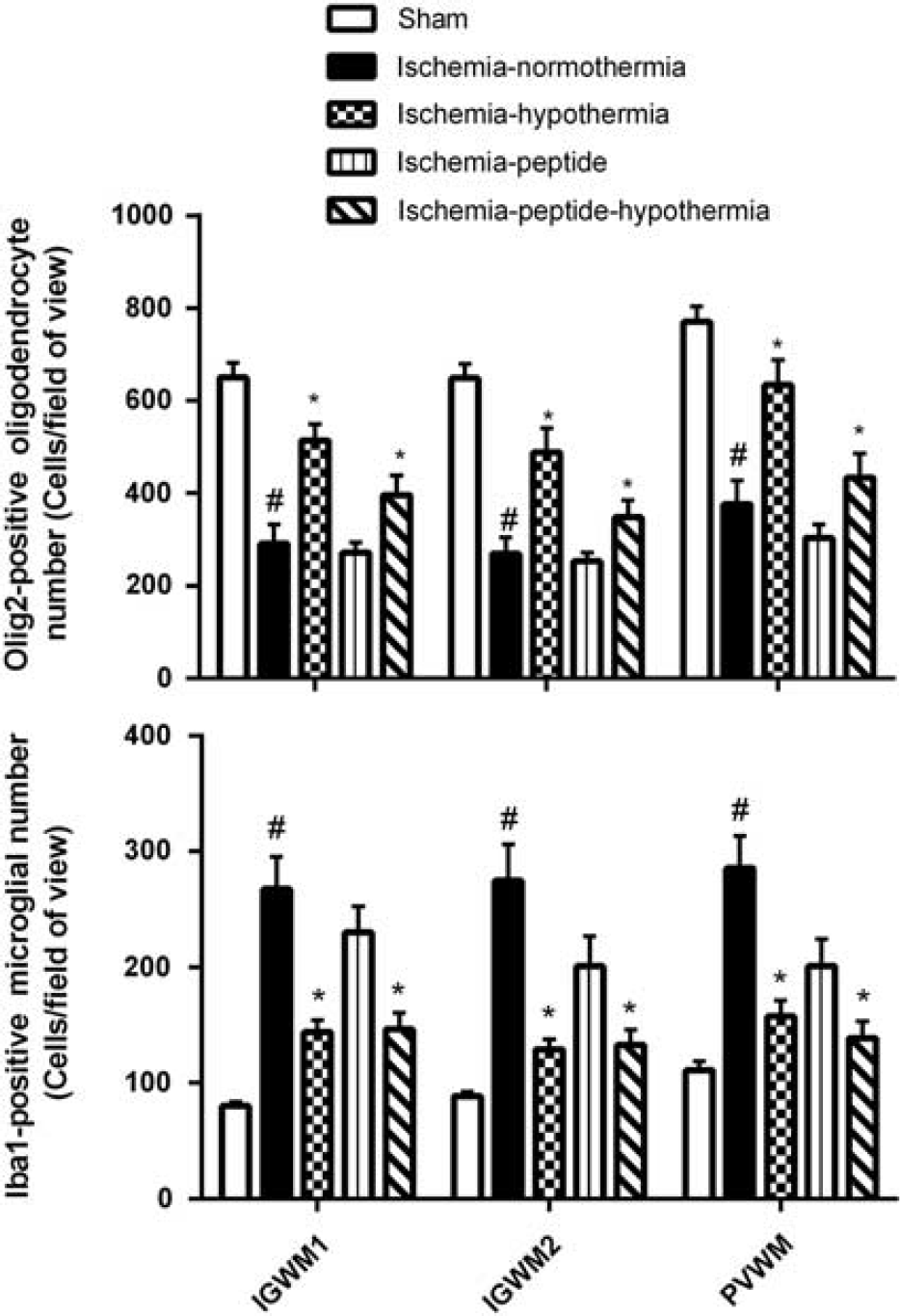
Changes in numbers of Olig2-positive oligodendrocytes and Iba1-positive microglia in the intragyral white matter of the first and second parasagittal gyri and periventricular white matter in the sham control, ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups after 30 minutes of global cerebral ischemia. In the ischemia normothermia group, a significant decrease in oligodendrocyte number and an increase in microglial number were seen in all regions. Hypothermia, but not peptide, was associated with a significant independent improvement in oligodendrocyte cell survival and reduction in microglial number. Data are mean ± s.e.m., #P < 0.05 sham control versus ischemia-normothermia, *P < 0.05 significant effect of hypothermia.
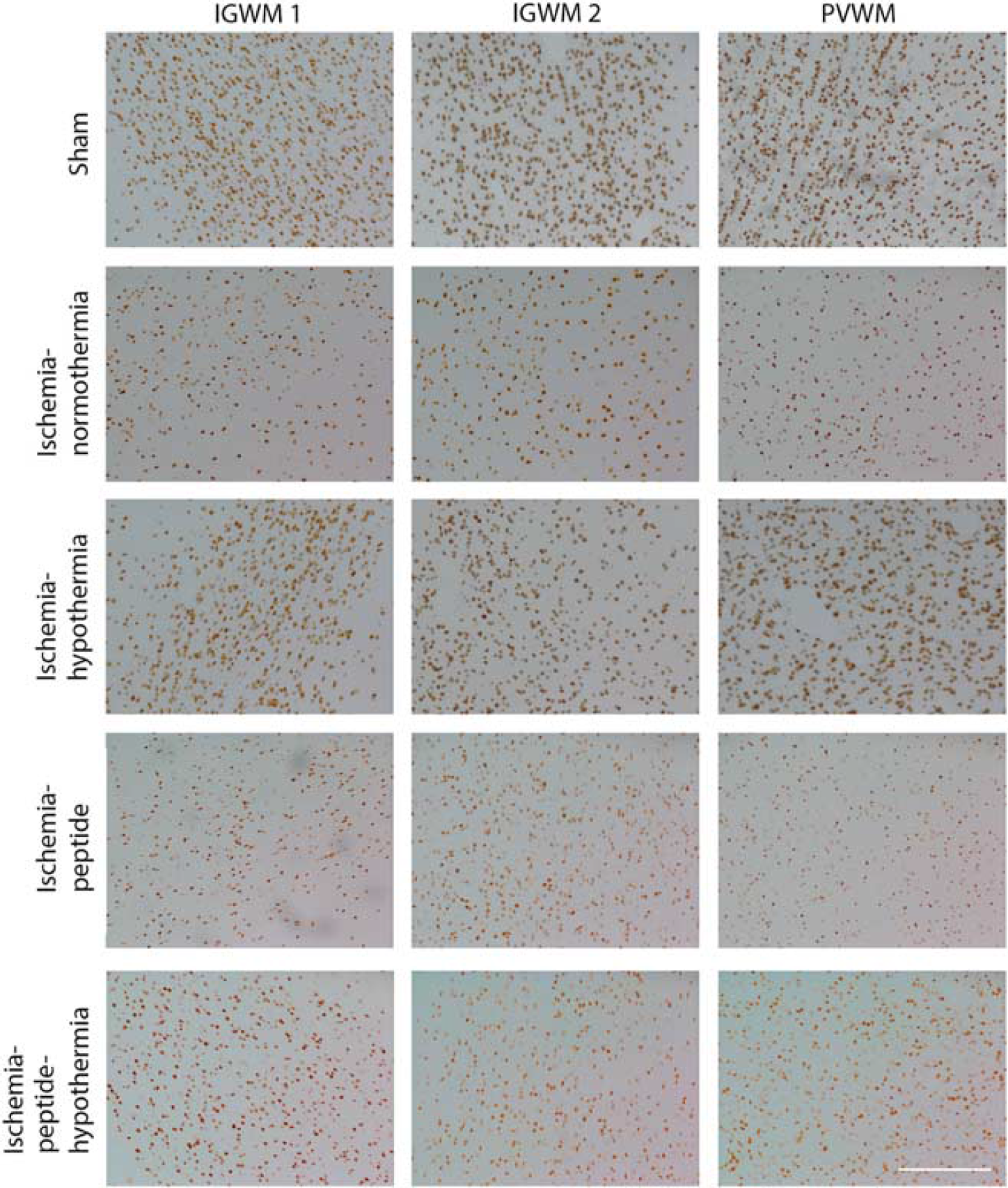
Photomicrographs of Olig2-positive oligodendrocytes in intragyral white matter of the first and second parasagittal gyri and periventricular white matter in the sham control, ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups 7 days after 30 minutes of global cerebral ischemia. Scale bar 200μm.
Ischemia-normothermia was associated with a marked increase in Iba1-positive microglia in the intragyral white matter of the first and second parasagittal gyri and in the periventricular white matter (P < 0.05, Figures 4 and 6). Hypothermia was associated with an independent reduction in numbers of microglia in all regions (P < 0.05). Peptide infusion was associated with an apparent trend to reduced numbers of microglia (P = 0.09), but no interaction with hypothermia.
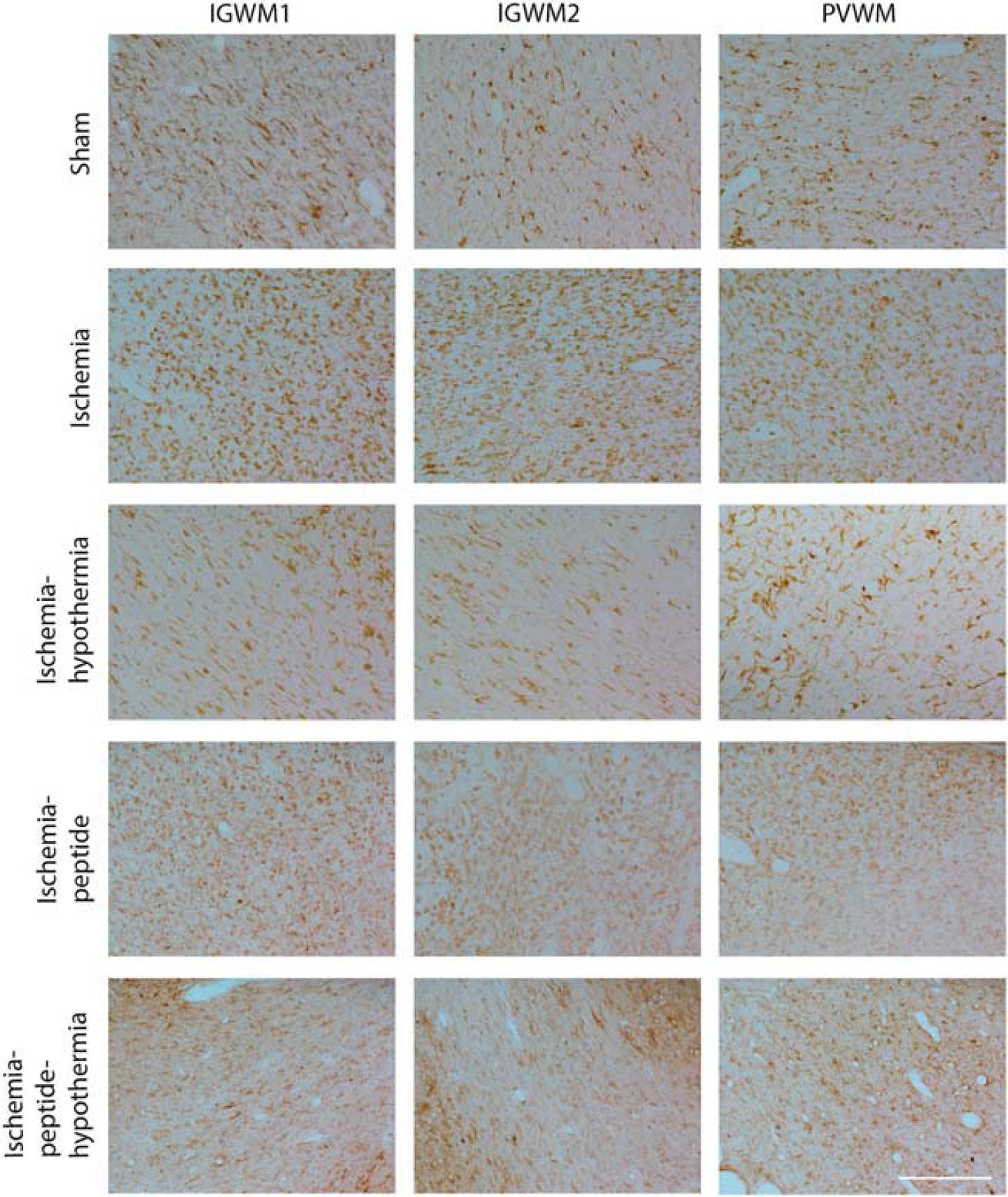
Photomicrographs of Iba1-positive microglia in intragyral white matter of the first and second parasagittal gyri and periventricular white matter in the sham control, ischemia-normothermia, ischemia-hypothermia, ischemia-peptide, and ischemia-peptide-hypothermia groups 7 days after 30 minutes of global cerebral ischemia. Scale bar 200μm.
DISCUSSION
This study has showed that the addition of connexin hemichannel blockade did not further augment neuroprotection associated with delayed but prolonged moderate head cooling in term-equivalent fetal sheep. Connexin hemichannel blockade started 3 hours after the end of ischemia was associated with a significant reduction in seizure activity and secondary cell swelling, with an apparent trend to reduced numbers of microglia, but no improvement in recovery of EEG power, neuronal or oligodendrocyte cell survival. This suggests that the window of opportunity for targeting hemichannel activity is relatively limited. Consistent with the previous findings, 5 cerebral hypothermia started 3 hours after global cerebral ischemia was associated with substantially improved recovery of EEG power, spectral edge and reduced seizure burden and secondary cell swelling. Further, hypothermia was independently associated with improved neuronal and oligodendrocyte cell survival and reduced microglial activation 7 days after ischemia. The combination of hypothermia and mimetic peptide infusion significantly attenuated postischemic hypoperfusion compared with ischemia-hypothermia, but was not associated with any additional improvement in the recovery of brain activity, cell survival or inflammation compared with hypothermia alone. The lack of further improvement in seizure burden during combined treatment raises the intriguing possibility that hypothermia and connexin hemichannel blockade may have overlapping mechanisms.
One of the most striking features of hypoxic-ischemic brain injury is that after initial recovery of cellular oxidative metabolism, there is a delayed, ‘secondary’ mitochondrial failure that spreads over many hours from the most severely damaged areas outwards, into initially intact regions. 30 This secondary failure is accompanied by transient seizure activity and secondary cytotoxic edema.22,23,30 As recently reviewed, there is increasing evidence that, at least in part, this secondary spread is related to opening of connexin hemichannels after ischemia that can mediate release of paracrine molecules that in turn propagate cell death messages. 10
Supporting this hypothesis, we have recently shown that connexin hemichannel blockade started 90 minutes after ischemia and continued for at least 24 hours markedly reduced status epilepticus in term-equivalent animals and improved recovery of brain activity and neuronal and oligodendrocyte cell survival.12,17,27 In contrast, in the current study, connexin hemichannel blockade from 3 hours after ischemia did not affect the subsequent recovery of brain activity or neuronal and oligodendrocyte survival. This suggests that connexin hemichannel involvement in the cascade leading to cell death and impaired brain function must be occurring predominantly within the first 3 hours.
The reasons for the lack of protection with delayed connexin hemichannel blockade are unclear. We have previously shown that C×43 mRNA is significantly elevated 4 hours after the end of ischemia in the near-term fetal sheep compared with sham controls. 12 This of course does not indicate whether the connexin hemichannels are still open. However, blockade of connexin hemichannels for 25 hours showed much greater neuroprotection than a short (1 hour) infusion, 12 and in the present study, blockade was associated with suppressed postischemic seizures and reduced cortical cell swelling as measured by impedance. These findings are highly consistent with ongoing opening of connexin hemichannels after the first 3 hours. We therefore speculate that lack of neuroprotection may have reflected a downstream change in response to the signals such as ATP and calcium released during hemichannel opening, such that they are no longer deleterious from 3 hours after ischemia. This is consistent with the finding of markedly reduced seizure burden during connexin hemichannel blockade despite lack of neuroprotection, suggesting that hemichannel opening contributes directly to propagation of seizures, rather than indirectly through reducing brain injury. This further supports the hypothesis that seizure activity does not necessarily potentiate injury after severe hypoxia-ischemia. 31
Prolonged mild to moderate cerebral hypothermia delayed until 3 hours after ischemia markedly improved recovery of EEG power and frequency, and white- and gray-matter cell survival, consistent with extensive previous clinical and preclinical evidence. 32 Interestingly, the only apparent interaction between hemichannel blockade and hypothermia was attenuation of the effect of hypothermia on carotid blood flow. Ischemia-normothermia was associated with a transient phase of secondary hypoperfusion, similarly to reports in many settings.33,34 The significance of secondary hypoperfusion has been controversial. Overall, evidence suggests that it is an actively mediated suppression of cerebral metabolism,35,36 leading to increased tissue oxygen levels. 37
Hypothermia was associated with marked exaggeration of this transient period of hypoperfusion during the latent phase and suppressed subsequent hyperperfusion, consistent with preclinical and clinical findings.22,38,39 The effect of hypothermia is likely mediated by hypothermic suppression of brain metabolism, of approximately 5% per 1°. 40 The mechanisms by which the combination of hypothermia and hemichannel blockade significantly attenuated hypoperfusion are unknown. Most likely, it may be a direct vascular effect, given that C×43 is prevalent in the vasculature, 41 and that hemichannel blockade reduces postischemic vessel leak after retinal ischemia. 42 Potentially, this could be mediated indirectly through altered brain metabolism, since early postischemic hemichannel blockade was associated with more rapid recovery of EEG power.12,17 However, no changes in EEG power were seen in the present study and so this is unlikely.
Preclinical studies of combination treatment for perinatal hypoxic ischemic encephalopathy have had mixed outcomes. Neither the selective NMDA receptor antagonist dizocilpine nor the insulin like growth factor-1 augmented hypothermic neuroprotection after global cerebral ischemia in the near-term fetal sheep or asphyxia in the preterm fetal sheep, respectively.43,44 However, hypothermia in combination with xenon treatment started from 5 hours after unilateral hypoxia ischemia in the P7 rat was more neuroprotective than either intervention alone. 45 Further, concurrent erythropoietin and hypothermia treatment improved outcomes more than either treatment alone after umbilical cord occlusion in nonhuman primates. 46
The lack of synergistic effect between hypothermia and connexin hemichannel blockade may reflect overlapping mechanisms of action or that blockade of connexin hemichannels with the mimetic peptide was ineffective due to the delay in treatment. If it was due to excessive delay, then earlier intervention may show greater synergism. We did not examine this possibility in the present study as we wished to test a clinically realistic delay before the intervention. Further, both hypothermia and connexin hemichannel blockade independently reduced seizure burden, with no further improvement from simultaneous treatment with both interventions. This strongly suggests that some of the effects of hypothermia are mediated through mechanisms overlapping with connexin hemichannel blockade.
A practical limitation of the current study is that connexin hemichannel blockade was achieved by intracerebroventricular administration of Peptide5, which has restricted clinical translat-ability. Further studies are needed to develop peptides with greater stability and tissue penetration to enable systemic administration.
In summary, this study has shown that blockade of connexin hemichannels delayed until 3 hours after the end of global cerebral ischemia was associated with reduced seizures and secondary cell swelling but no significant EEG or histologic neuroprotection. The combination of hypothermia and hemichannel blockade was associated with a significant attenuation of the exaggeration of secondary hypoperfusion associated with hypothermia but no further improvement in total postischemic seizure burden, recovery of brain activity, cell survival, or inflammation. This study suggests that the practical therapeutic window of opportunity for the treatment of connexin hemichannel opening may be restricted to the first 3 hours after ischemia.
Footnotes
JD, LB, CG, and AJG conceptualized and designed the study. JD and GW undertook experiments and analyzed data. AR, CY, and FZ undertook immunohistochemistry, cell quantification, analysis and preparation of figures. AJG provided overall oversight of the research. All authors critically reviewed the manuscript and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.