
Abstract
Neuroinflammation following traumatic brain injury (TBI) is increasingly recognized to contribute to chronic tissue loss and neurologic dysfunction. Circulating levels of S100B increase after TBI and have been used as a biomarker. S100B is produced by activated astrocytes and can promote microglial activation; signaling by S100B through interaction with the multiligand advanced glycation end product-specific receptor (AGER) has been implicated in brain injury and microglial activation during chronic neurodegeneration. We examined the effects of S100B inhibition in a controlled cortical impact model, using S100B knockout mice or administration of neutralizing S100B antibody. Both interventions significantly reduced TBI-induced lesion volume, improved retention memory function, and attenuated microglial activation. The neutralizing antibody also significantly reduced sensorimotor deficits and improved neuronal survival in the cortex. However, S100B did not alter microglial activation in BV2 cells or primary microglial cultures stimulated by lipopolysaccharide or interferon gamma. Further, proximity ligation assays did not support direct interaction in the brain between S100B and AGER following TBI. Future studies are needed to elucidate specific pathways underlying S100B-mediated neuroinflammatory actions after TBI. Our results strongly implicate S100B in TBI-induced neuroinflammation, cell loss, and neurologic dysfunction, thereby indicating that it is a potential therapeutic target for TBI.
INTRODUCTION
Traumatic brain injury (TBI) represents a major public health problem, with >1.7 million new cases annually reported in the United States alone, according to the Centers for Disease Control and Prevention. TBI causes cell death and neurologic dysfunction through both physical disruption of the tissue (primary injury), as well as delayed molecular and cellular mechanisms that cause progressive white and gray matter damage (secondary injury) and may potentially continue for months to years. 1 Such secondary injury mechanisms, such as chronic microglial and astrocyte activation, contribute to pathogenesis of brain injury, exacerbating neurologic dysfunction. 2 Over the years, we have demonstrated the progressive and sustained nature of neuroinflammation through microglial and astrocyte activation, particularly in the cortex, using multiple experimental models of TBI.3,4
One of the biochemical responses following glial activation induced by brain injury is increased circulating serum and cerebro-spinal fluid levels of the calcium-binding protein S100B, which is primarily expressed in the astrocytes and Schwann cells. 5 The mechanisms of action of S100B as an intracellular regulator are different from those as an extracellular factor. As an intracellular regulator, S100B stimulates cell proliferation and migration, inhibits apoptosis and differentiation, and activation of astrocytes, which may have implications for brain repair after central nervous system (CNS) injury. 6 In contrast, as an extracellular factor S100B interacts with advanced glycation end product-specific receptor (AGER) to exert beneficial or detrimental effects, or pro-proliferative or pro-differentiative consequences based on its concentration and microenvironment.7,8 In addition to its expression in astroglia and Schwann cells, S100B has also been found in adipocytes, chondrocytes, cardiomyocytes, lymphocytes, bone marrow cells, and melanocytes where it exerts intracellular and extracellular functions. 6 Therefore, both CNS and circulatory S100B exert the same functions that are dependent on their concentration and may serve as important biomarkers of CNS injury. Intracellular S100B interaction with Src kinase activates phosphoinositide 3-kinase signaling and downstream RhoA/Rack-dependent stress fiber formation as well as glycogen synthase kinase 3β/Rho-dependent stellation. 9 In addition, S100B is also expressed in oligodendrocytes and cultured microglia/microglial cell lines, 10 but S100B-regulated intracellular events in these cells have not been elucidated. More importantly, the concentration of S100B and the state of the cell are two major variables that determine whether extracellular S100B exerts trophic or toxic effects. The overproduction of S100B by activated astrocytes after brain injury increases microglial and astrocyte activation, as well as neuronal cell death. 9 The release of S100B can stimulate the generation of oxidative stress-related enzymes and pro-inflammatory entities, such as inducible nitric oxide synthase (iNOS), tumor necrosis factor-alpha (TNF-α), interleukin-1 β (IL-1β), and IL-6. 9 Therefore, S100B is regarded as a biomarker of activation of pathologic mechanisms after brain injury and neuroinflammatory/neurodegenerative disorders.11,12 The elevated levels of S100B in neurodegenerative models have been found to be associated with impairments in learning and memory function.13,14 In addition, recently an association between elevated circulating S100B levels and impaired neurologic outcomes has been observed after clinical brain injury.5,15
Studies suggest that cellular signaling resulting from the interaction of S100B with AGER may be responsible for its pro-inflammatory effects. 16 AGER is a member of the immunoglobulin (Ig) family of cell surface molecules that recognizes multiple ligands, including AGE, amphoterin, amyloid-β-peptide and β-fibrils, and S100B. Extracellular S100B has been observed to exhibit trophic effects on neurons by interaction with AGER,7,8 which has been implicated in neuroprotection and neurodegeneration as well as in inflammatory responses. 17 S100B at high concentrations via acute stimulation of AGER causes neuronal apoptosis by excessive extracellular signal-regulated kinase 1 (ERK1)/2 activation and overproduction of reactive oxygen species (ROS). 7
In our study, we evaluated the effects of S100B inhibition using either a knockout model or administration of a neutralizing S100B antibody in a mouse model of controlled cortical impact (CCI). Furthermore, we investigated the potential interaction of S100B with AGER in CCI, as well as the effects of S100B on primary microglial cultures or a mouse microglial cell line after activation with lipopolysaccharide (LPS) or interferon gamma (IFNγ). Our study highlights the importance of S100B not only as a biomarker for TBI-induced injury but also as a potential therapeutic target.
MATERIALS AND METHODS
Controlled Cortical Impact
Our custom-designed CCI-injury device3,18 consists of a microprocessor-controlled pneumatic impactor with a 3.5-mm diameter tip. Male mice were anesthetized with isoflurane (4% induction, 2% maintenance) evaporated in a gas mixture containing 70% N2O and 30% O2 and administered through a nose mask. The mouse was placed on a heated pad and a core body temperature was maintained at 37 °. A 10-mm midline incision was made over the skull, the skin and fascia were reflected, and a 4-mm craniotomy was made on the central aspect of the left parietal bone. Moderate injury was induced using an impactor velocity of 6 m/s and deformation depth of 2 mm, as previously detailed.3,18 Sham animals underwent the same procedure as injured mice except for the impact.
Genetic intervention: The S100B−/− line (CD1 background) was a generous gift of Dr Alexander Marks (University of Toronto). 19 Genotyping was performed as previously described, 20 and all experiments used male S100B−/− and S100B+/+ littermates (20 to 25 g, 3 months old).
Neutralizing anti-S100B antibody treatment: Male C57Bl/6 mice (20 to 25 g, 3 months old) were randomized into six groups and administered 10 mg/kg anti-S100B (BL356, Bethyl Laboratories, Montgomery, TX, USA) or an isotype matched IgG (Catalog no. P50-1000, Bethyl Laboratories) antibody at 24 hours and 14 days after injury, via the intraperitoneal route, based on preliminary pharmacokinetic data.
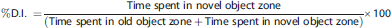
Microglia Cultures
Primary cortical microglia were obtained from postnatal day 2 mouse pups and cultured as described before. 23 The BV2 murine microglia were grown and maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) at 37 ° in a humidified incubator under 5% CO2. S100B (1 μmol/L; Sigma-Aldrich, St Louis, MO, USA) was added to microglia alone and for 1 hour before LPS (Sigma-Aldrich, indicated concentrations in μg/mL) or recombinant mouse IFNγ (R&D Systems, Minneapolis, MN, USA, indicated concentrations in ng/mL) stimulation.
Statistical Analysis
The number of animals per group for each assessment was based on our prior studies using the CCI model3,18,21 and satisfied power requirements. Quantitative data were expressed as mean ± s.e.m., and
RESULTS
Genetic Ablation of S100B Provides Neuroprotection
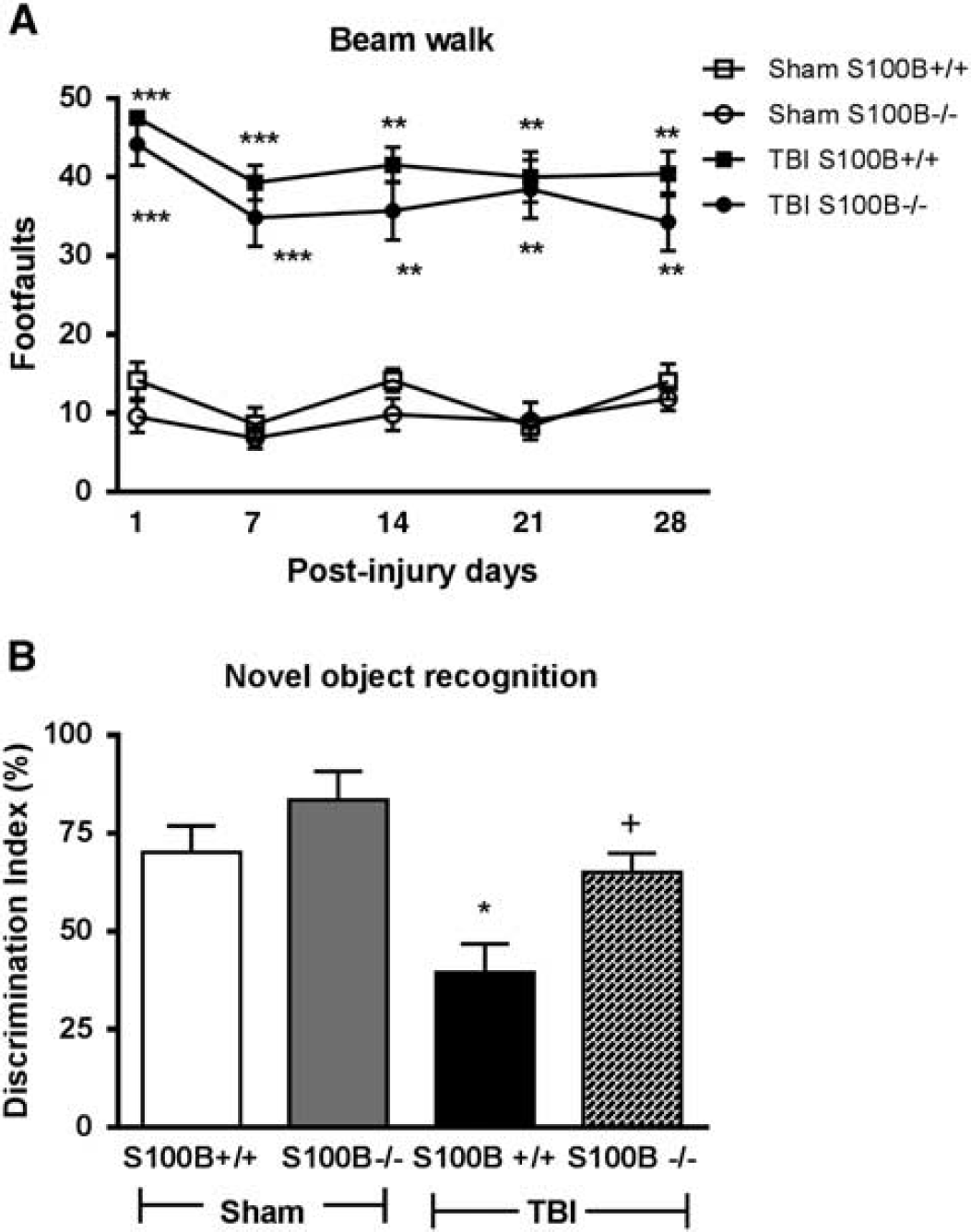
S100B−/− mice had improved retention memory function after traumatic brain injury (TBI). (
Spatial learning and reference memory were assessed using the acquisition phase and probe trial of the MWM test, respectively.
TBI resulted in spatial learning impairments on PIDs 16 and 17 (
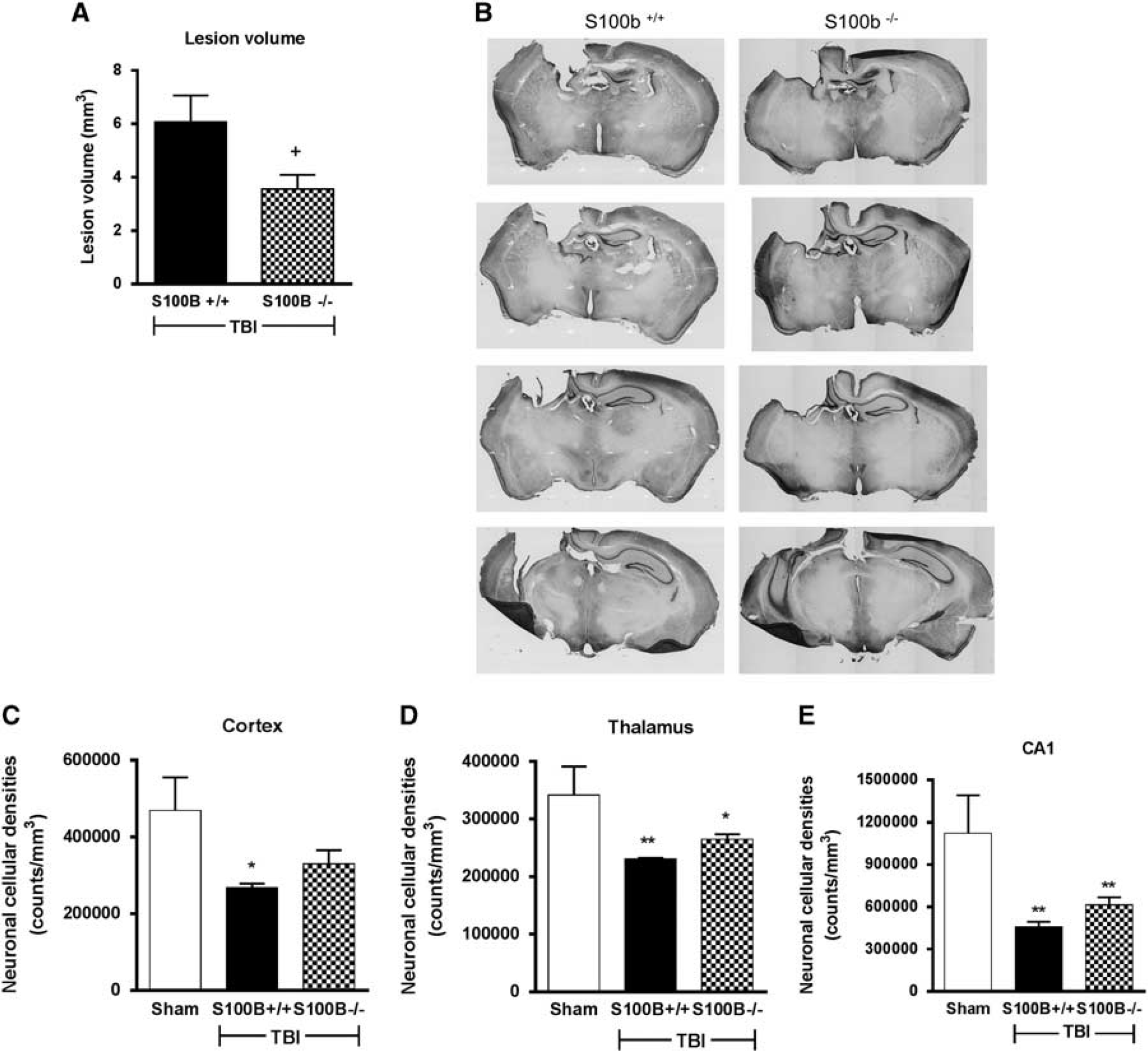
S100B−/− mice had reduced lesion volume but no reduction in neuronal cell loss after traumatic brain injury (TBI). (
Stereological assessment of the surviving neurons (
Brain injury results in a switch in microglial phenotype from a quiescent form displaying ramified cellular morphologies to more activated forms displaying hypertrophic or bushy morphologies (Figure 3A), with the latter being the most reactive. Stereological assessment of microglial cell number and activation phenotype (
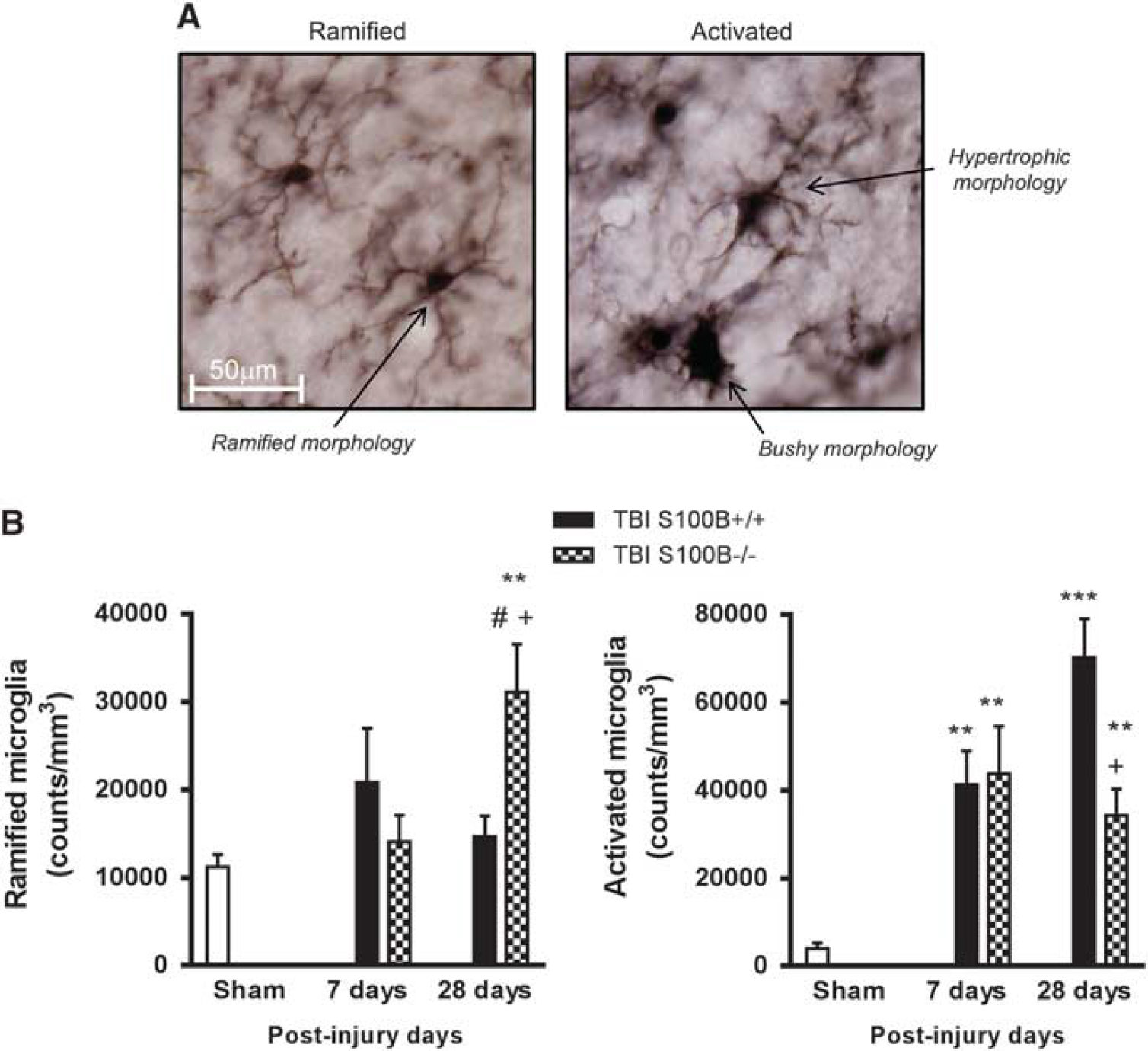
S100B−/− mice had reduced microglial activation in the injured cortex after traumatic brain injury (TBI). Stereological assessment of microglial cell number and activation phenotype was performed in the cortex at 7 and 28 days after TBI. (
Systemic Treatment With a Neutralizing S100B Antibody Provides Neuroprotection
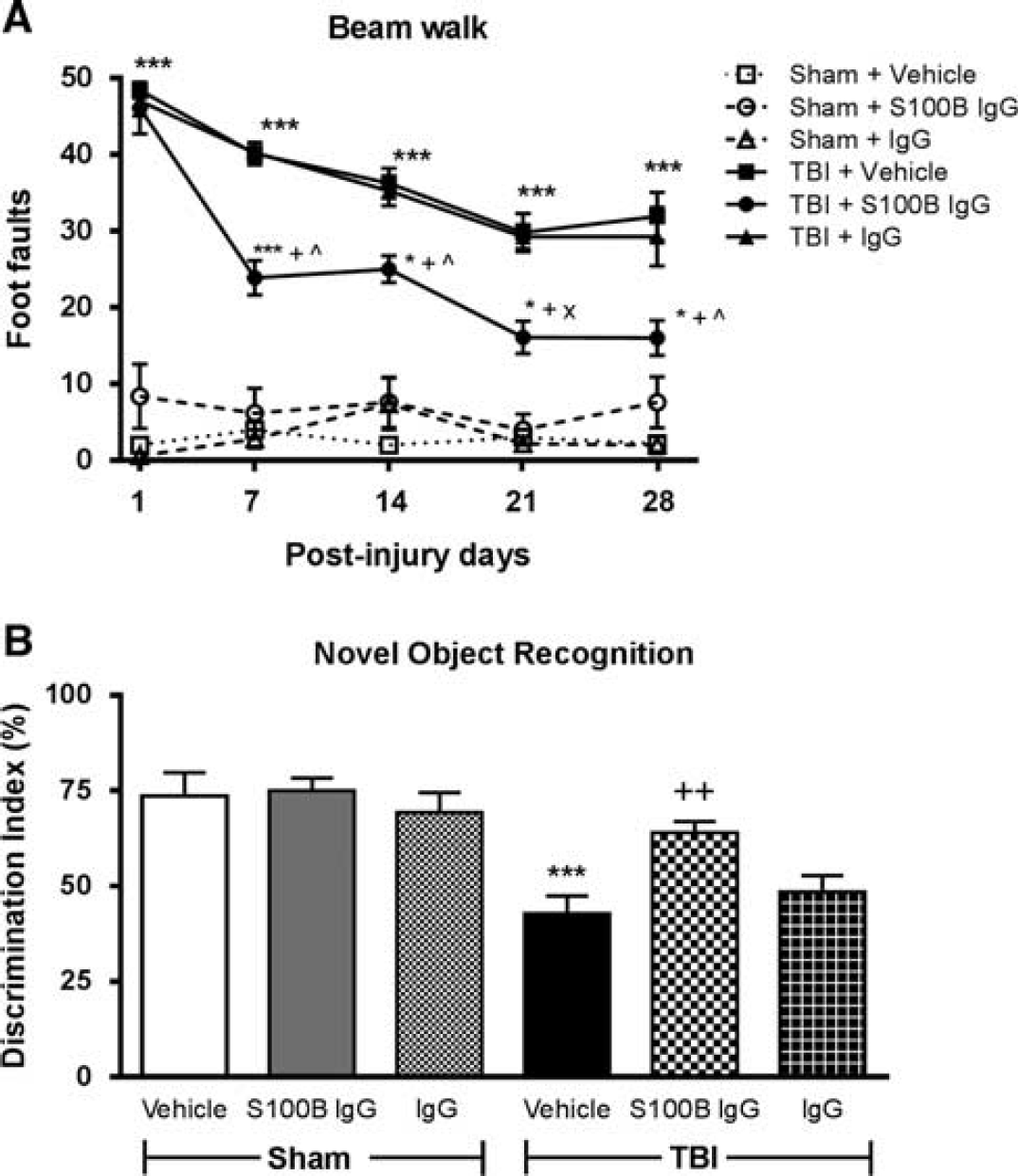
Systemic administration of a neutralizing S100B antibody improved functional outcomes following traumatic brain injury (TBI). (
Spatial learning and reference memory were assessed using the acquisition phase and probe trial of the MWM test, respectively. TBI resulted in spatial learning impairments on PID 17 (data not shown;
Retention or intact memory was evaluated using the NOR test and statistically analyzed by one-way ANOVA followed by Tukey's
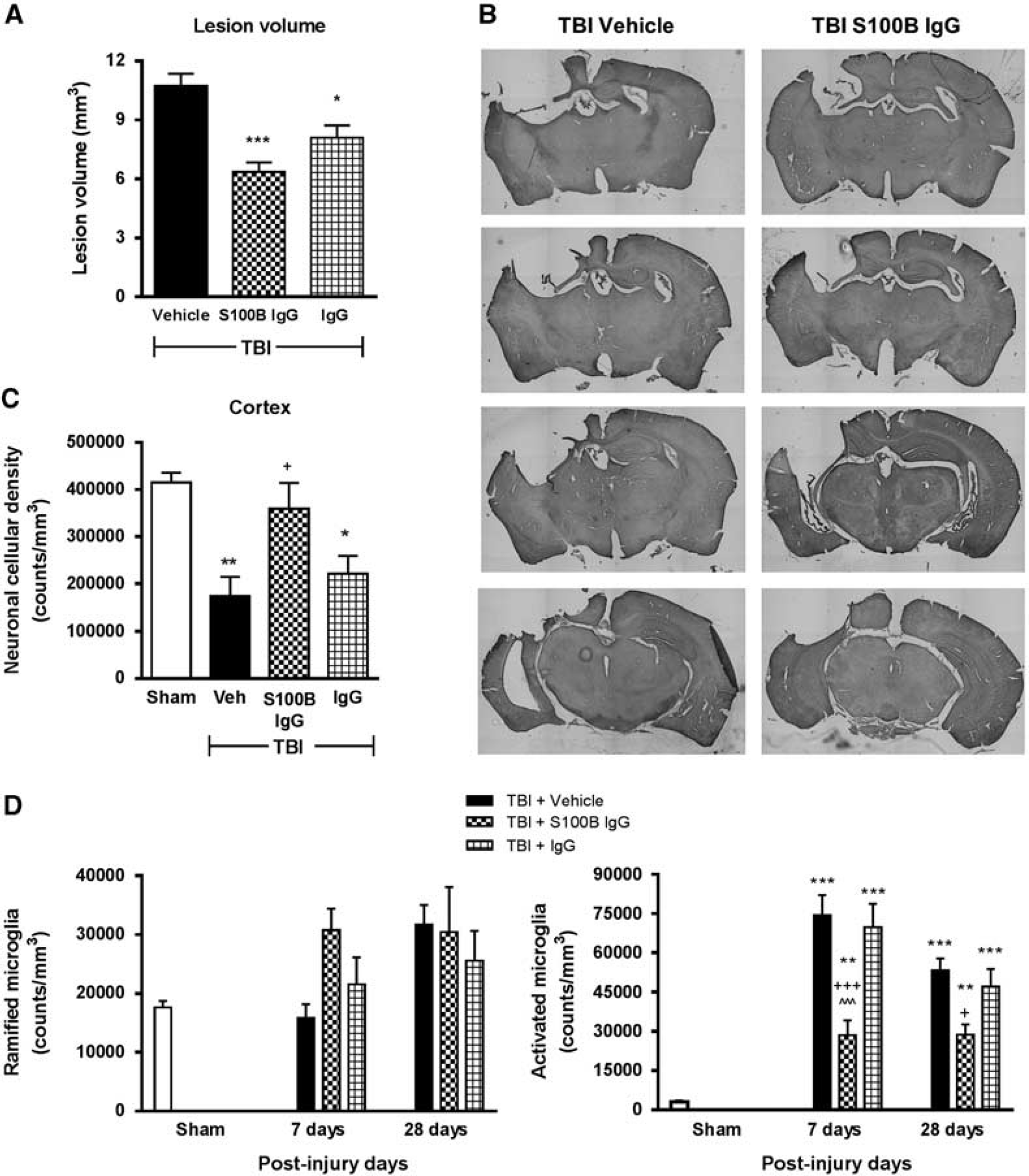
Systemic administration of a neutralizing S100B antibody reduced lesion volume, improved neuronal survival, and attenuated microglial activation in the cortex after traumatic brain injury (TBI). (
Stereological assessment of surviving neurons (
Stereological assessment of microglial cell number and activation phenotype (
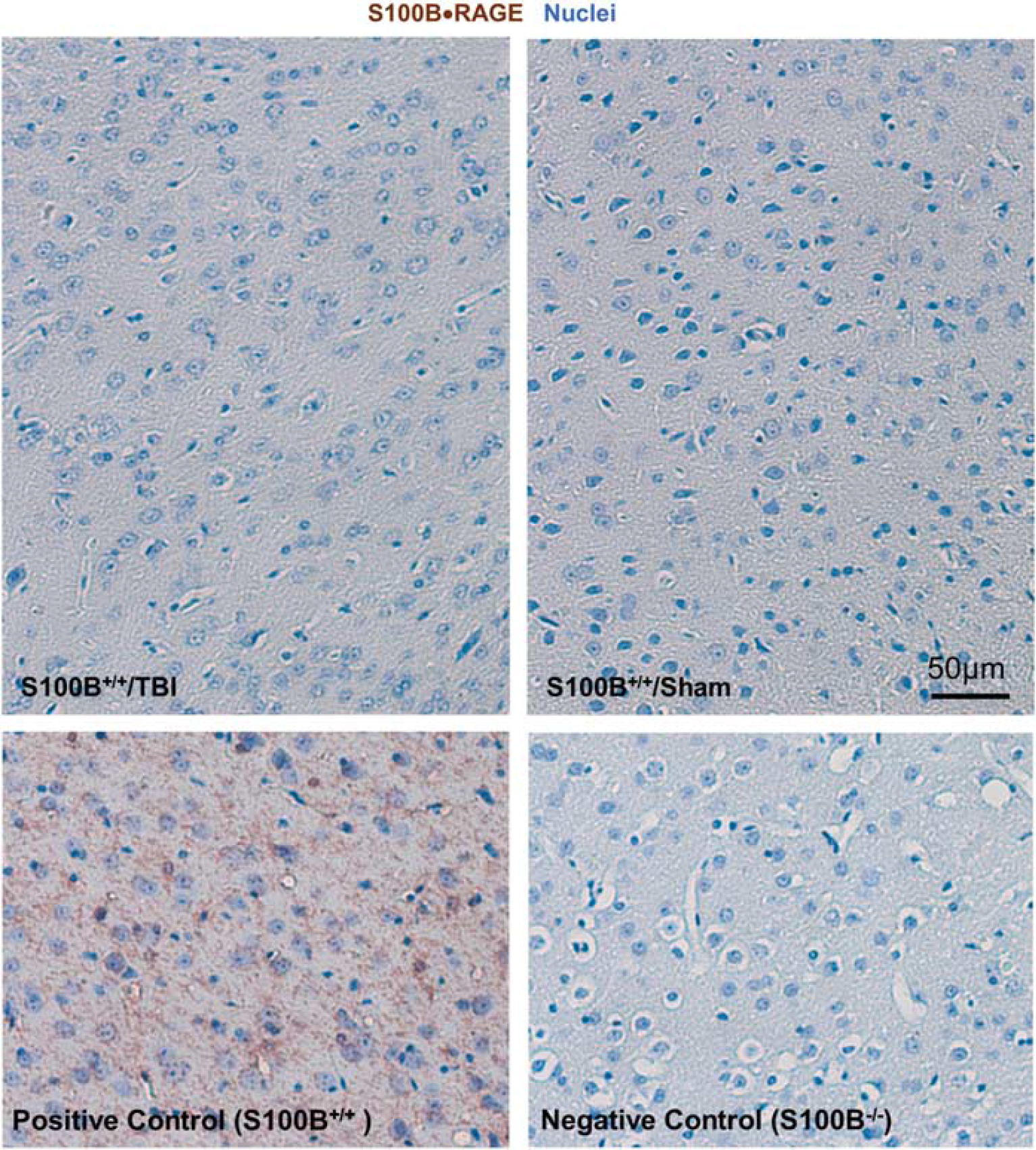
S100B–AGER complexes are not detectable in traumatic brain injury (TBI). Representative micrographs of ipsilateral cortical regions from TBI/sham sections 7 days after injury and positive/negative control sections processed for proximity ligation assays using S100B+AGER primary antibodies (brown) and counterstained with hematoxylin to visualize nuclei (blue).
S100B does not Influence Key Microglial Activation Markers In Vitro
To determine whether S100B can modulate microglial activation, we examined its effects on NO release and/or ROS production, classic markers of activation using two
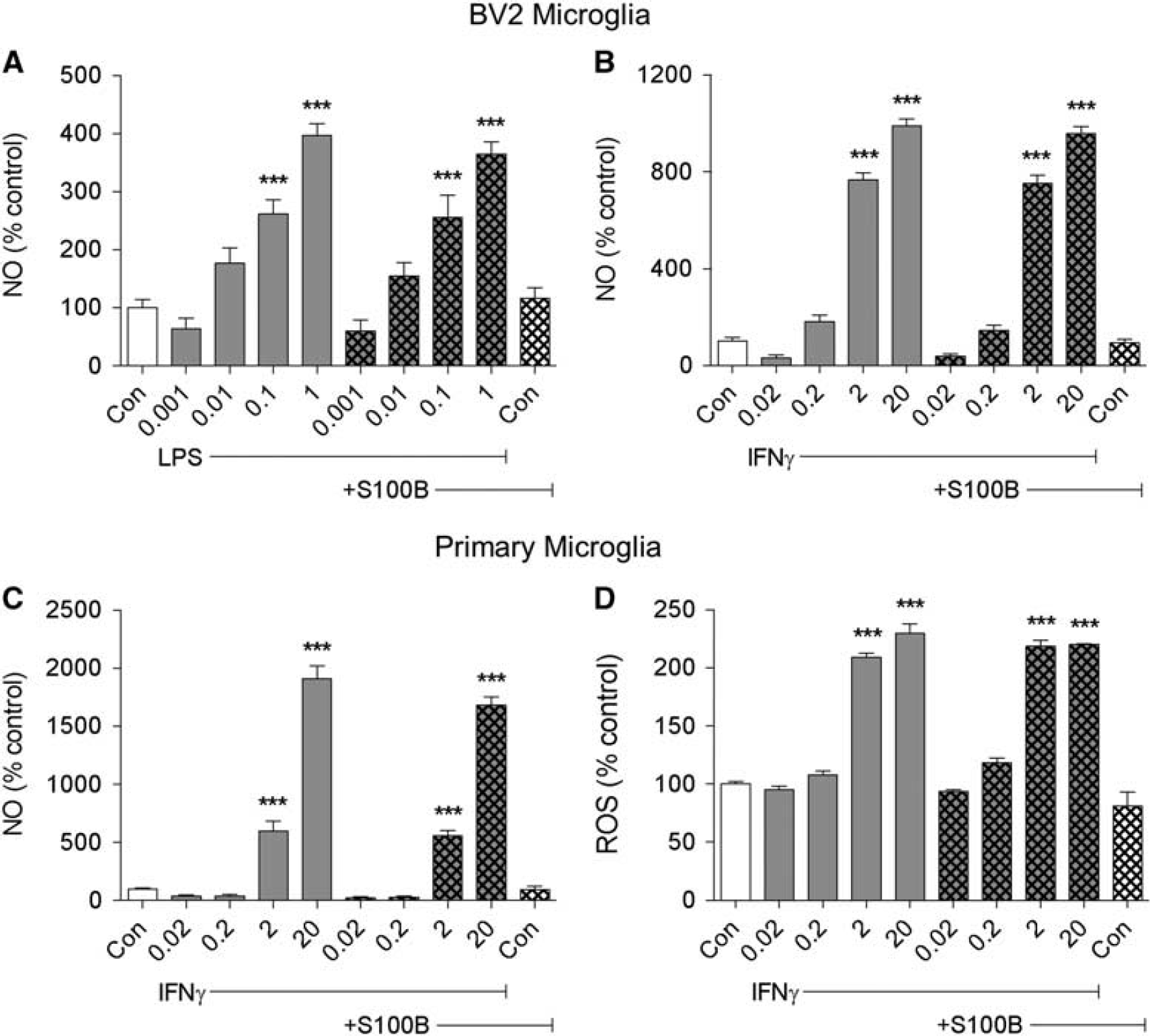
S100B does not influence microglial activation markers
DISCUSSION
S100B belongs to the S100/calmodulin/troponin C superfamily of proteins that function as calcium-modulated proteins and exert their effects on the CNS in a concentration- and calcium-dependent manner. 24 Extracellular S100B promotes neurite extension outgrowth and enhances cell maintenance in cultures of embryonic chick cerebral cortex neurons 25 and stimulates proliferation of primary rat astrocytes. 26 Increased intracellular calcium by S100B protein has been reported to decrease neuronal cell death and mitochondrial dysfunction in rat hippocampal neurons may provide neuroprotection for CNS neurons at nanomolar concentrations. In contrast, other studies have reported neurotoxic, particularly neuroinflammatory effects of S100B, via activation of iNOS-causing astrocytic death at micromolar concentrations. 27 S100B has long been considered an important marker of brain injury after TBI, with elevated serum levels being correlated with poor outcomes both experimentally and clinically.14,15 Multiple experimental TBI models have shown significant increases in S100B levels in the brain early after TBI 28 and persisting up to 5 days.29,30 Intraventricular infusion of S100B resulted in a significant improvement in memory function after lateral fluid percussion-induced brain injury, 29 but higher serum levels of S100B were correlated with impaired cognitive performance. The latter finding is in agreement with many clinical observations and was thought to reflect activation of secondary injury mechanisms.
There is a transient increase in blood–brain barrier permeability after CCI in rodents, 31 which may be associated with functional changes in the blood–brain barrier rather than mechanical disruption of cerebrovascular barrier. Blood–brain barrier changes and permeability to albumin and other high-molecular weight proteins has been observed within 4 to 6 hours after TBI and persists for several days. This may be due to increased paracellular permeability of endothelial barrier associated with changes in expression, distribution, and function of tight-junction proteins and from modified transport of pinocytic vesicles across blood-brain barrier. Several experimental studies have demonstrated that there is a surge in the production of pro-inflammatory molecules, including S100B 32 by brain parenchymal cells. Several
studies have shown that overproduction of S100B by activated astrocytes after brain injury further increases microglial and astrocyte activation, as well as neuronal cell death. 9 Release of S100B can contribute to the generation of pro-inflammatory factors such as iNOS, TNF-α, IL-1β, and IL-6 that worsen neuroinflammation and may contribute to long-term neurologic dysfunction. 9 The objective of our study was to further examine the role and potential mechanisms by which S100B can modulate secondary injury. We evaluated the therapeutic potential of S100B inhibition via genetic as well as pharmacological means against TBI-induced microglial activation and behavioral deficits using the well-established CCI mouse model, which recapitulates certain pathophysiologic features of clinical TBI and results in long-term neurologic deficits.3,18,21 In our study, S100B knockouts or delayed systemic administration of a neutralizing S100B antibody IgG were used to inhibit S100B. Anti-S100B IgG was administered systemically at two delayed time points of 24 hours and 14 days after TBI to simulate a clinically relevant treatment paradigm.
CCI led to long-term behavioral impairment in terms of sensorimotor, spatial, and retention memory functions. CCI-induced sensorimotor deficits were significantly reduced by systemic administration of a neutralizing S100B antibody, which also significantly reduced lesion volume as compared with untreated injured animals. The extent of locomotor impairment after injury to the sensorimotor cortex is often correlated with lesion size. 21 However, knocking out S100B reduced lesion volume but did not cause significant improvements in sensorimotor outcomes. Interestingly, treatment with an IgG control after TBI reduced the size of the lesion. The latter finding is consistent with previous studies showing that IgG treatment can improve function after experimental TBI, possibly reflecting its antiinflammatory actions.33,34 The non-specific IgG effect on the reduction of lesion volume could likely be attributed to an inflammatory or acute phase reaction to the IgG that either counteracts or modulates TBI-induced inflammation in favor of repair or recovery. There is also a possibility of the oncotic colloidal pressure associated with hemodynamic changes and/or edema after TBI being altered after control IgG administration.33,34 However, the molecular mechanisms underlying this effect have not been established to date and need to be investigated.
To further evaluate treatment effects, neuronal cell loss was quantitatively assessed in the cortex and thalamus using unbiased stereological techniques. CCI caused neuronal loss in both cortex and thalamus. Acute S100B inhibition with a neutralizing S100B antibody, but not IgG treatment, significantly improved neuronal survival in the cortex but not in the thalamus. In contrast, chronic S100B inhibition using genetic intervention did not cause a statistically significant attenuation of CCI-induced neuronal loss in any of the assessed brain regions, though we observed a trend towards increased neuronal survival in the cortex of injured S100B−/− mice. Improvement in neuronal survival in the cortex may not be directly correlated with the reduction in the size of the cortical lesion, as the cortical neuronal counts represent changes surrounding the lesion and not just at the site of the lesion, whereas the lesion volume is determined by the missing tissue at the site of the lesion.
To determine the effect of S100B inhibition on memory function, different cognitive tasks were used. Improved cognitive performance in the MWM of spatial learning and reference memory serves as a potential indicator of reduced hippocampal damage. CCI induced significant cognitive deficits in acquisition phase of the MWM task, indicating impairment in spatial learning (data not shown). In contrast, our data from the probe trial of the MWM test after both interventions indicated that injured animals did not spend significantly greater amount of time in the target quadrant when compared with sham groups and therefore did not show statistically significant impairment in reference memory function in this test. Neither genetic ablation nor neutralizing antibodies altered MWM performance after CCI (data not shown). Improved memory function in the MWM test is demonstrated by a strong correlation between cognitive improvement and increased neuronal survival in the CA3 and DG hippocampal subregions.18,21 CCI caused significant neuronal cell loss in CA1, CA2/3, and DG hippocampal subregions, which was not modified by S100B inhibition. Retention memory can be assessed using the NOR test, which serves as a measure of object discrimination/exploration based on retention memory function as well as locomotor activity. Performance in the NOR test reflects both cortical and hippocampal function.18,21 CCI caused significant impairment in retention memory based on object discrimination/exploratory behavior. Either knocking out S100B or administration of a neutralizing antibody significantly improved functional outcomes in the NOR test. Together, these findings suggest that effects of S100B inhibition on retention memory function in the NOR test likely reflect neuroprotective effects in the cortex.
Chronic inflammation after CNS trauma may provide a mechanistic link between early and chronic neurodegeneration. 3 Previous studies have suggested that sustained microglial activation after TBI may contribute to chronic neuronal cell loss and associated neurologic dysfunction.3,4,21 We have previously demonstrated using stereological assessment and 3D reconstruction analysis in multiple models of TBI3,4,21 that the process of microglial activation after TBI is slow to start as indicated by the low numbers of microglia displaying activated (hypertrophic and bushy) morphologies at 24 hours, followed by a statistically significant elevation in the numbers of activated microglia at 7 days after injury, which is the peak in activation in these models. Although these numbers decline at later time points compared with peak levels at 7 days after injury, microglia are chronically activated up to 12 months after injury, such that there are significantly increased numbers of activated microglial cells at 12 months after injury when compared with sham-injured control levels. 35 Interestingly, our previous pharmacological intervention studies using cell cycle inhibitors demonstrated that the 7- and 28- day time points were ideal to evaluate treatment effects on posttraumatic microglial activation, both at the peak and at a more chronic time point.4,21 Therefore, based on our previously published findings on the time course and response of an intervention to microglial activation after TBI, we performed a quantitative assessment of microglial activation after TBI at 7 and 28 days, respectively. Microglial activation status was based on morphologic features using unbiased stereology, as described previously.3,21 CCI significantly increased the numbers of activated microglia at 7 days when compared with sham controls, with increases sustained at 28 days. Knocking out S100B also significantly attenuated the numbers of activated microglia and increased the numbers of ramified microglia at 28 days. Neutralizing antibodies significantly attenuated the numbers of activated microglia at 7 and 28 days after CCI. Unlike the lesion size, treatment with control IgG did not cause attenuation of cortical microglial activation after TBI. The administration of control IgG did not attenuate sensorimotor deficits, improve neuronal survival, and reduce microglial activation; in contrast, these improved outcomes were observed with anti-S100B IgG treatment, suggesting that the non-specific IgG component of the neutralizing S100B antibodies contributes to only a small part of the neuroprotection provided by anti-S100B IgG treatment. Future studies will provide insight into the non-specific IgG effect and will highlight the specific inhibitory effect of S100B.
Our data strongly implicate a role for S100B protein in the pathophysiology of TBI and demonstrate that inhibition of S100B limits microglial activation and reduces the extent of trauma-induced neurologic damage and associated functional deficits. Previous studies using primary microglia from newborn rat cortex or BV-2 microglial cell lines have demonstrated that micromolar to nanomolar concentrations of S100B cause IFNγ-induced expression of iNOS as well as NO secretion.10,36 In contrast, our studies using BV2 and primary microglial cultures stimulated by LPS and/or IFNγ showed no effects of S100B on key markers of microglial activation, such as NO release or ROS production. Furthermore, it has been proposed that S100B may act through AGER and subsequently Toll-like receptors to induce inflammatory responses.7,36 Although our proximity ligation assays do not support a direct interaction in the brain between S100B and AGER at early time points after TBI in our model, the relationships between S100B and AGER are complex. The AGER-transducing activity may not have a significant role in S100B-stimulated NO production by microglia, but the AGER extracellular domain may be important for concentrating S100B on the BV-2 cell surface. 36
Studies have demonstrated that S100B-stimulated NO release from the microglia may be dependent on the density of AGER molecules on microglial surface and activation of pathways such as p38 MAPK (mitogen-activated protein kinase) that result in production of ROS. 36 Extracellular S100B can promote AGER-dependent AP1 and nuclear factor (NF)-kB-dependent gene transcription of cytokines, chemokines, and iNOS, as well as AGER-independent NO production in microglia. 27 In astrocytes, extracellular S100B can activate AGER/Rac-1/Cdc42, AGER/Erk-Akt, and AGER/NF-KB pathways. 37 Extracellular S100B can also facilitate neurodegeneration via AGER/MAPK/cyclinD1-CDK4 activation and cell cycle re-entry 38 as well as AGER/NF-KB activation. 39 Furthermore, S100B has been shown to stimulate the production of TNF-α in the microglia through concurrent activation of Rac1-NF-KB module, Rac1-JNK-AP-1 module, and potential involvement of ERK1/1-NF-KB and p38MAPK-NF-KB modules.7,40 TNF-α has an important role in S100B release from the astrocytes, suggesting that TNF-α might elevate the extracellular concentration of S100B favoring activating effects of S100B on microglia.7,40 Collectively, our data and previously demonstrated mechanisms indicate that S100B contributes to the activation and progression of neuroinflammatory processes following TBI. However, S100B inhibition may directly or indirectly attenuate neuroinflammation after TBI, and future studies are needed to determine the mechanisms of S100B-mediated neuroinflammatory effects.
In conclusion, our study indicates that S100B has a role in TBI-induced microglial activation and associated neurologic dysfunction. Furthermore, our data support the potential of S100B as a therapeutic target for treatment of TBI. The mechanisms by which S100B contributes to the pathobiology of TBI do not appear to involve direct S100B-GER interactions. Further studies are needed to elucidate the specific pathways underlying S100B-mediated neuroinflammatory and related neurodegenerative actions after TBI.
Footnotes
DBZ and AIF conceived the study. SVK designed the in vivo experiments and performed surgeries on S100B knockout as well as C57bl/6 mice followed by behavioral assessments, histology, and statistical analysis. BAS and DJL designed and performed the in vitro experiments on primary rat and BV-2 microglial cultures. LA and KBD generated the S100B knockout mice, formulated the antibodies, and performed the proximity ligation assays. SVK, BAS, DJL, and AIF drafted and finalized the manuscript. All authors have read and approved the final manuscript.
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Dr. David Weber for his invaluable intellectual contribution, Dr. Alexander Marks (University of Toronto) for the generous gift of S100B knockout mouse line, and Bethyl Laboratories (Montgomery, TX, USA) for the S100B neutralizing antibody. The authors also like to thank Dr. Hegang Chen, a senior biostatistician from the Department of Epidemiology and Public Health at the University of Maryland School of Medicine, for his expert consultation with the statistical analysis and Katherine Cardiff and Juliane Faden for expert technical assistance.