
Abstract
Pathologic conditions in the central nervous system, regardless of the underlying injury mechanism, show a certain level of blood-brain barrier (BBB) impairment. Endothelial dysfunction is the earliest event in the initiation of vascular damage caused by inflammation due to stroke, atherosclerosis, trauma, or brain infections. Recently, microRNAs (miRNAs) have emerged as a class of gene expression regulators. The relationship between neuroinflammation and miRNA expression in brain endothelium remains unexplored. Previously, we showed the BBB-protective and anti-inflammatory effects of glycogen synthase kinase (GSK) 3β inhibition in brain endothelium in in vitro and in vivo models of neuroinflammation. Using microarray screening, we identified miRNAs induced in primary human brain microvascular endothelial cells after exposure to the pro-inflammatory cytokine, tumor necrosis factor-α, with/out GSK3β inhibition. Among the highly modified miRNAs, let-7 and miR-98 were predicted to target the inflammatory molecules, CCL2 and CCL5. Overexpression of let-7 and miR-98 in vitro and in vivo resulted in reduced leukocyte adhesion to and migration across endothelium, diminished expression of pro-inflammatory cytokines, and increased BBB tightness, attenuating barrier ‘leakiness’ in neuroinflammation conditions. For the first time, we showed that miRNAs could be used as a therapeutic tool to prevent the BBB dysfunction in neuroinflammation.
INTRODUCTION
The cerebrovascular unit of the blood-brain barrier (BBB) comprises endothelial cells, pericytes, and astrocytes, which jointly maintain a tight barrier that is impermeable to ions, pathogens, and toxic agents, thus preserving homeostasis and maintaining the brain as an immunoprivileged organ. 1 Endothelial dysfunction is the earliest event in the initiation of vascular injury caused by inflammation due to ischemia, amyloid deposition, trauma, or brain infection. 2 Chronic inflammation in the brain is a consequence of immune activation of glia (microglia, macrophages, and astrocytes), oxidative stress, and BBB injury mediated by inflammatory factors produced by immune cells in the brain or blood. Due to their neuroprotective properties, inhibitors of glycogen synthase kinase (GSK) 38 were recently shown as beneficial drugs in preclinical trials in stroke, Huntington's and Parkinson's diseases, amyotrophic lateral sclerosis, spinal cord injury, and other conditions, as well as in clinical trials for Alzheimer's disease. 3 However, the potent anti-inflammatory and immunomodulatory effects of GSK3β inhibition have been less studied. Our group has shown that GSK3β inhibition: (1) increased BBB tightness under physiologic conditions, (2) decreased secretion of inflammatory factors in brain microvascular endothelial cells (BMVEC), (3) protected against BBB disruption caused by monocyte-endothelial interactions, and (4) reduced monocyte adhesion to/migration across the BBB.4,5
Recently, microRNAs (miRNAs) have emerged as gene expression regulators. Yet, the relationship between inflammation and miRNA expression is just beginning to be investigated. Various miRNAs have been shown to control angiogenesis, vascular inflammation, and atherosclerosis.6,7 MicroRNAs are post-transcriptional regulators of gene expression that function by inhibiting translation of mRNAs. They are endogenously encoded single-stranded RNAs of ~ 22 nucleotides in length that inhibit protein translation predominantly through imperfect base-pairing with sequences, which are generally located in the 3’ untranslated region (UTR) of mRNA transcripts. 8 Although the role of miRNAs in the control of proliferation, differentiation, and apoptosis has been previously recognized, the importance of these small noncoding (NC) RNAs in inflammation is just emerging. 8 For instance, miR-155 and miR-146a regulate a variety of immune reactions, including production of cytokines by T and B cells and the germinal center B-cell response. 9 miRNA profiling suggests that miR-126 is expressed mainly in endothelial cells. 10 Additional endothelial miRNA clusters have been identified: miR-17-92, miR-23-27-24, and miR-222-221 (many miRNAs are encoded by polycistronic miRNA genes). 10 The function of these miRNAs was mostly studied during angiogenesis. 10 In human umbilical vein endothelial cells, miR-126 negatively regulated tumor necrosis factor-α (TNFα)-inducible vascular cellular adhesion molecule 1, 10 consequently increasing leukocyte adhesion to the endothelium and was involved in endothelial leakiness. 11 miR-101 has been shown to regulate claudin-5 expression by targeting of VE-cadherin. 12 Despite similarity between endothelial cell types, miRNAs are clustered in miR-99b, miR-20b, and let-7b clusters that are significantly different under normal conditions in endothelial cells derived from various organs. 13 Therefore, findings from one cell type, such as human umbilical vein endothelial cells, might not be applicable to endothelial cells derived from different organs. Thus far, the association between miRNA and brain endothelial dysfunction in neuroinflammation has still not been systematically studied. We have identified miR-98 and let-7g* whose expression was regulated by GSK3β inhibition. miR-98 and let-7g* belong to highly-conserved miRNAs, the let-7 group, which consists of 12 members in mammals. Let-7 miRNAs have been implicated in the pathobiology of cancer, although their potential role in endothelial biology is still understudied. 14 In this study, we investigated in-depth the anti-inflammatory and BBB-tightening effects of these miRNAs regulated by GSK3β, both in vitro (with BMVEC) and in vivo, in an animal model of localized aseptic meningitis (induced by intracerebral (IC) injection of TNFα), utilizing intravital videomicroscopy (IVM) for surveillance of cerebral vascular changes and leukocyte-endothelial interactions.15,16
MATERIALS AND METHODS
Reagents and Cells
Recombinant human TNFα, lipopolysaccharide (LPS) and monocyte chemotactic protein-1 (MCP-1)/CCL2 were purchased from R&D Systems (Minneapolis, MN, USA). The GSK3β inhibitors were obtained as follows: lithium chloride (LiCl, Sigma/Aldrich Co., Ltd., St Louis, MO, USA), 5-iodoindirubin-3'-monoxime (I3'M, EMD Chemicals, San Diego, CA, USA), N-(4-methoxybenzyl)-N'-(5-nitro-1,3-thiazol-2-yl)urea (AR-A014418, AR, Sigma/Aldrich) and 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB 216763, SB2, Cayman Chemical Co., Ann Arbor, MI, USA).
Primary BMVEC, isolated from vessels from brain resection tissue (showing no abnormalities) of patients undergoing surgery for treatment of intractable epilepsy, were supplied by Michael Bernas and Dr Marlys Witte (University of Arizona, Tucson, AZ) and maintained as described.15,17 Brain microvascular endothelial cells were treated with GSK3β inhibitors (10 mmol/L LiCl, 1 μmol/L I3'M, 5 μmol/L SB2 or AR) for 16 hours (unless otherwise stated) and then with TNFα (50 ng/mL) for 4 hours. HEK 293 cells were obtained from Sigma/Aldrich.
Animals and Intravital Videomicroscopy
All animal experiments were approved by the Temple University Institutional Animal Care and Use Committee and conducted in accordance with the Temple University guidelines, which are based on the National Institutes of Health (NIH) guide for care and use of laboratory animals and with the ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines (study design, experimental procedures, housing and husbandry, statistical methods) (www.nc3rs.org.uk/arrive-guidelines).
Intravital videomicroscopy was performed in 8-week-old male C57BL/6 mice purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Intravital videomicroscopy for in vivo leukocyte adhesion was performed in mice that underwent craniotomy, cranial window, and cannula implantation.15,16 Six-day recovery period was allowed between implantation of the cannula and IC injections. Intravital videomicroscopy for in vivo leukocyte adhesion was performed on animals with cranial windows.15,16 Before IVM, animals were IC injected with TNFα (0.5 μg/mouse). The total maximum volume injected into the mouse brain was 5 μL, injecting 1 μL at a time with a 5-minute waiting period between injections as described. 16 The dose (0.5 μg/mouse) was established in dose-response experiments (data not shown), where consistent leukocyte infiltration was produced accompanied by mild brain edema (known markers of meningitis 18 ). No adverse effects (high fever, seizures, locomotor impairment, and/or death) were noted. 16 Two hours after injection of TNFα, leukocytes were labeled in vivo with Vybrant DiI Cell-Labeling Solution (DiI) (Life Technologies, Carlsbad, CA, USA) introduced intravenously. Leukocyte adhesion was detected in cerebral vessels through the cranial window using a Stereo Discovery V20 epifluorescence microscope (Carl Zeiss Microimaging Inc., Thornwood, NY, USA) equipped with a AxioCam MR digital camera as previously described.15,16 Transmigrated leukocytes were enumerated in an area covering a distance of 10 μm from the pial and parenchymal vessel wall by IVM and by Leica SP5 II 2-photon microscopy (Leica Microsystems Gmbh, Mannheim, Germany), respectively. The number of extravasated leukocytes was counted and normalized with respect to the immediate perivascular area surrounding the microvessel (MV) (10 μm from the endothelium × 100 μm long along the vessel). 16
In Vitro and In Vivo miRNA Transfection
Brain microvascular endothelial cells were transfected at 2×10 6 cells/mL with miRNA oligos at 50 nmol/L with the Neon Transfection System (Life Technologies) according to the manufacturer's instructions. Cells were collected at 24, 48, 72, and 96 hours after transfection and levels of transfected miRNAs were estimated by qPCR as described below. The GSK3β silencing in BMVEC with siRNA was performed as described. 5
MicroRNA (similar to siRNA) can penetrate cell membranes, but can be degraded if injected in a nonencapsulated form. Therefore, we used a liposome-based miRNA delivery system. To create miRNA-containing liposomes, 5 nmol synthetic miRNA (Dharmacon, GE Dharmacon, Lafayette, CO, USA) were mixed with Lipofectamine 2000 (Life Technologies) in RNase and DNase-free water (Life Technologies), according to the manufacturer's instructions. Lipid-oligo complexes were incubated at room temperature for at least 1 hour and, if required, were stored at 4° overnight. The miRNA-liposome complex was diluted in phosphate-buffered saline and 100 μL was injected per mouse. To verify efficiency of miRNA delivery into MVs, the mouse brains were harvested at 6, 24, or 48 hours after miRNA administration (Supplementary Materials). Microvessels were isolated and the amount of transfected miRNA was estimated by qPCR as described below. Next, mice were injected (intravenously) once with miRNA-liposomes as described19,20 or had a second injection after 24 hours before TNFα or LPS administration.
Mouse Brain Microvessel Isolation
Mouse brain MVs were isolated using a protocol based on previously published studies.21,22 For each preparation, mice (n = 4) were overdosed with 5% isoflurane and their brains were harvested and placed in 4° HBSS. The cerebellum, meninges, choroid plexus, brain stem, and large superficial blood vessels were removed. The remaining tissue was diced in HBSS (4mL/gram) and then homogenized using a Potter-Thomas homogenizer (0.25 mm clearance) (Thomas Scientific, Swedesboro, NJ). The homogenate was centrifuged (1000 × g for 10 minutes at 4°) to remove HBSS, resuspended in 17.5% dextran (Sigma/Aldrich) and centrifuged again to separate the MVs. The MV pellet was resuspended in 1% BSA in HBSS and the supernatant was centrifuged (4000xg for 10 minutes at 4 °). The MVs from each centrifugation were combined. The MV suspension was passed through a 100-μm nylon mesh filter and then through a 40-μm nylon mesh filter (Corning Life Sciences, Tewksbury, MA). The material retained on the 40-μm nylon mesh filter contained the MVs.
In Vivo Permeability Assay
Mice were injected intraperitoneally with 20 U heparin followed by an intraperitoneal injection of 200 μL of 2% sodium-fluorescein (Na-F) in saline. The amount of Na-F evaluated as described 16 was measured using a Synergy 2 plate reader (BioTek, Winooski, VT, USA). Fluorescent dye content was calculated using external standards and the data are expressed as amount of tracer per mg of tissue. 16
Monocyte Adhesion Assays
Primary human monocytes were obtained from the University of Nebraska Medical Center, Department of Pharmacology and Experimental Neuroscience, Omaha, NE from HIV-1 and hepatitis B seronegative donors by leukopheresis and were purified by countercurrent centrifugal elutriation as described. 23 Brain microvascular endothelial cells were transfected with miRNAs as described above, and grown as monolayers for 48 hours before TNFα stimulation (50 ng/mL). Treatments were removed from the BMVEC before addition of freshly isolated human calcein-AM-labeled monocytes; adhesion assays were performed as described.15–17 Fluorescence of adherent monocytes was measured using a Synergy 2 plate reader (BioTek). Results are presented as the fold difference in adhesion (mean±s.e.m.) from triplicate determinations (number of adherent monocytes for each experimental condition divided by the basal adhesion of the untreated control).
Transendothelial Migration Assays
Transendothelial migration assays were performed as described.15,17 Monocyte chemotactic protein-1/CCL2 (50 ng/mL) was used as a relevant chemokine. Brain microvascular endothelial cells were transfected with miRNA oligo or non-targeting control oligo as described above 48 hours before TNFα stimulation (50 ng/mL). Treatments were removed before monocyte addition. Chemotaxis was allowed for 2 hours. The data are presented as fold difference in migration (mean±s.e.m.) from triplicate determinations, calculated from the number of migrated monocytes for each experimental condition divided by the number of migrated monocytes in the untreated, no chemoattractant control.
Transendothelial Electrical Resistance
Brain microvascular endothelial cells transfected with miRNAs (targeting or non-targeting sequences) were plated on collagen type I-coated 96W20idf electrode arrays (Applied Biophysics, Troy, NY, USA) and were maintained for 72 hours to form a monolayer and reaching and basal levels of transendothelial electrical resistance (TEER) were 800 to 1500Ω4,5,17 which was accepted as time ‘zero’ for experiment with or without TNFα treatment. Transendothelial electrical resistance measurements were performed using the 1600R ECIS System (Applied Biophysics Inc., Troy, NY, USA) as described.15–17 The results are presented as the average percent change from baseline TEER (expressed as average±s.e.m.) from at least three independent experiments consisting of four to six replicates each.
Quantitative RT-PCR
RNA was extracted using the mirVana miRNA extraction kit (Life Technologies). Hybridization was performed on a μParaflo microfluidic chip (miRBase version 16, 1212 miRNA total, LC Sciences, Houston, TX, USA). After hybridization detection by fluorescence labeling using tag-specific Cy3 dyes, hybridization images were collected using a laser scanner and digitized using Array-Pro image analysis software (Media Cybernetics, Silver Spring, MD, USA). To validate the results and investigate the biologic meaning of differential expressed miRNAs, real-time RT-PCR was used to detect the differential expression of target genes. Real-time RT-PCR was performed using the mirVana qRT-PCR miRNA detection kit (Life Technologies) as per the manufacturer's protocol as described previously. 9 PCR primer pairs for reverse transcription and detection of mature miRs were purchased from Life Technologies (hsa-mir-98, has-let-7g*, and control U6). In general, quantitative real-time RT-PCR (qRT-PCR) on primary BMVEC was performed on three independent experiments using 25 ng of template using the StepOnePlus real-time PCR system (Life Technologies). For each sample, qRT-PCR was performed in triplicate. Amplification was analyzed using the ΔΔCt method, using a web-based data analysis tool (SABiosciences, Qiagen Inc., Valencia, CA, USA), by normalization to the corresponding values of housekeeping gene (U6) and fold-change calculated from the difference between experimental condition and untreated control.
MicroRNA Functional Analysis
The mature sequences of the miRNAs were retrieved using miRBase database: hsa-mir-98 no. MIMAT0000096: UGAGGUAGUAAGUUGUAUU GUU and has-let-7g* no. MIMAT0004584: CUGUACAGGCCACUGCCUUGC and were synthetized by Integrated DNA Technologies, Inc. (IDT, Coralville, IA, USA). The MCP-1 3'UTR and RANTES 3'UTR sequences, cloned downstream to the firefly luciferase sequence in the pMirTarget reporter vector (further pMir), were purchased from OriGene (OriGEne Technologies, Inc., Rockville, MD, USA). For the perfect match sequence, the mature sequence of hsa-miR-98 or has-let-7g* synthetic oligos was transfected together with pMir reporter plasmids, containing the corresponding 3'UTRs.
The MCP-1 3'UTR mutated in the mir-98 or let-7g* seeding sequence was generated by site directed mutagenesis (Agilent Technologies, Santa Clara, CA, USA) using the pMir/MCP-1 3'UTR as a template. The oligonucleotides for the mutagenesis were as follows: forward, 5'-agacttggggaaaAAtGAtttGGAGttgaaccacagttc (mutated bases in the mir-98 seeding sequence are capitalized and underlined) and ccctgggatgttttga gAAtGAtACGTagaatcattaa (mutated bases in the let-7g* seeding sequence are capitalized and underlined). The RANTES 3'UTR mutated in the mir-98 or let-7g* seeding sequence was generated by site directed mutagenesis (Agilent) using the pMir/Rantes 3'UTR as a template. The oligonucleotides for the mutagenesis were as follows: forward, 5'-ggtggaggcttgaggta
Luciferase Assay
For miR target validation, HEK 293 cells were plated at a concentration of 8×10 4 cells/well in a 12-well plate in DMEM with 10% FBS medium. The next day, a total amount of 0.6 μg DNA/well was transfected using Lipofectamine (Life Technologies) at a DNA to Lipofectamine ratio of 1:3. pcDNA3 was added to keep the total amount of DNA constant. Samples were harvested 48 hours after transfection and subjected to the luciferase assay system (Promega, Madison, WI) following the manufacturer's instructions using a Infinity M200PRO chemiluminometer (Tecan Group Ltd., Mannedof, Switzerland). Relative units represent the ratio between luciferase values of the sample and the nontargeting control. The experiments were performed in duplicate and repeated at least three times.
Chemokine and Cytokine ELISA
Protein expression was estimated using commercially available ELISA kits for MCP-1/CCL2 and CCL5/RANTES (R&D Systems) according to the manufacturer's instructions.
Statistical Analysis
Data are expressed as the mean±s.e.m. of experiments conducted multiple times. Multiple group comparisons were performed by one-way analysis of variance with Dunnet's posthoc tests (adhesion, migration, qPCR, ELISA, luciferase assays, animal experiments). Statistical analyses were performed utilizing Prism v6.0c software (GraphPad Software Inc., San Diego, CA, USA). Differences were considered as significant at P < 0.05.
RESULTS
Glycogen Synthase Kinase 3β Inhibition Reverses Tumor Necrosis Factor-α's Pro-Inflammatory Affects on MicroRNA Profiling of Endothelial Cells
MicroRNAs diminish target protein expression and, therefore, levels of miRNA expression are opposite to ones of their targets. Tumor necrosis factor a is one of the major inflammatory factors released by monocytes/macrophages under appropriate stimuli. 24 Using in vitro systems mimicking brain-endothelial leukocyte interactions and an animal model, we showed potent antiinflammatory and BBB protective effects of GSK3β inhibition in brain endothelium including a decrease of numerous pro-inflammatory molecules (CCL2, IL-8, IP10, CCL5, etc.) after TNFα stimulation. 4 It is possible that some of the pro-inflammatory or anti-inflammatory cytokines/chemokines are regulated through miRNAs. To date, there has been limited study on miRNA in primary human BMVEC and very little is known about the role of GSK3β in miRNA machinery.
To identify miRNAs that may potentially contribute to the involvement of GSK3β in the endothelial inflammatory response, we examined genome-wide miRNA profiles in BMVEC treated with/out the GSK3β inhibitor, LiCl (10 mmol/L), and with/out exposure to TNFα (20 ng/mL, overnight), using microarray technology (miRBase version 16, 1212 miRNAs total, LC Sciences). Differentially expressed miRNAs in response to TNFα with or without LiCl are shown in a table and Venn diagram (Figures 1A and 1B). In all, 137 miRNAs were downregulated by TNFα (< − 1fold, P < 0.0001) and 44 miRNAs were reversed by LiCl treatment (TNFα/Li) (P < 0.01) (Figure 1C). In all, 180 miRNAs were upregulated (> 1-fold, P < 0.0001) in cells treated with TNFα, and LiCl treatment was able to partially reverse 92 miRNAs (P < 0.01, Figure 1C). We ranked the miRNAs diminished by TNFα and increased by LiCl treatment to select the top differentially expressed (Figure 1C), thereby identifying a few miRNAs that were highly regulated by TNFα and reversed by GSK3β inhibition.
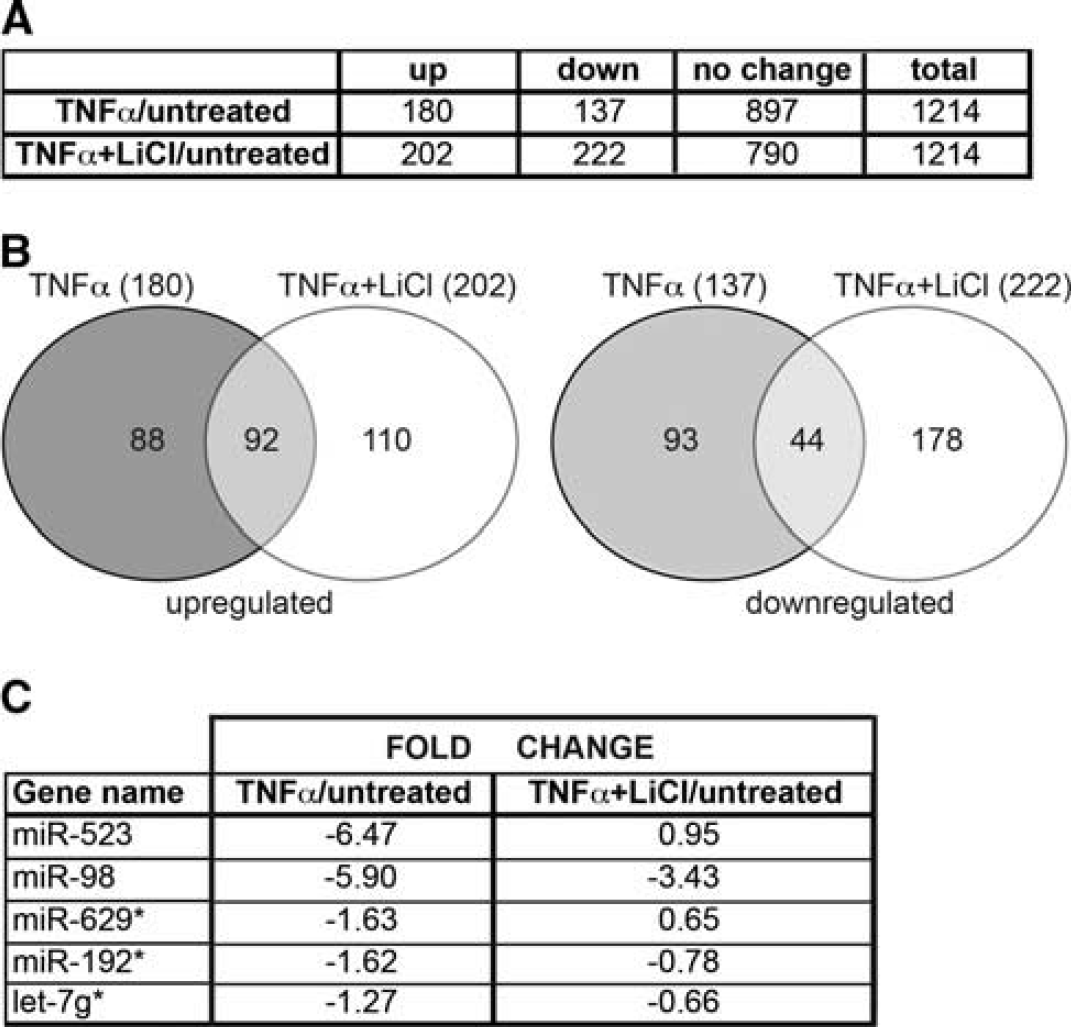
MicroRNAs (miRNAs) differentially expressed in tumor necrosis factor-α (TNFα)-treated cells compared with nontreated cells. (
To validate the microarray data, BMVEC (from four different donors) were treated in a similar manner as for the microarray, miRNAs were extracted and subjected to qPCR analysis. Expression patterns of miRNAs, such as miR-523 and miR-192, showed significant donor-to-donor variation and were not included in further study. miRNAs (mir-98 and let-7g*) were confirmed, by qPCR, to have expression patterns similar to the array results and several GSK3β inhibitors (I3'M, SB2, AR) had similar effects on these miRNAs (Figure 2) in BMVEC treated with TNFα with/out inhibitors. The TNFα stimulation decreased expression of miR-98 and let-7g* by 3.8- and 1.5-fold, respectively. GSK3β inhibitors, I3'M and AR, were able to reverse downregulation of miR-98 by 90% to 95%, and LiCl and SB2 by 70% and 80%, respectively (Figure 2A). In the case of let-7g*, LiCl and SB2 were able to increase expression to basal miR levels, while I3'M and AR increased expression by 90% and 55%, respectively (Figure 2B). The GSK3β silencing in BMVEC with siRNA 5 resulted in similar effects (data not shown).
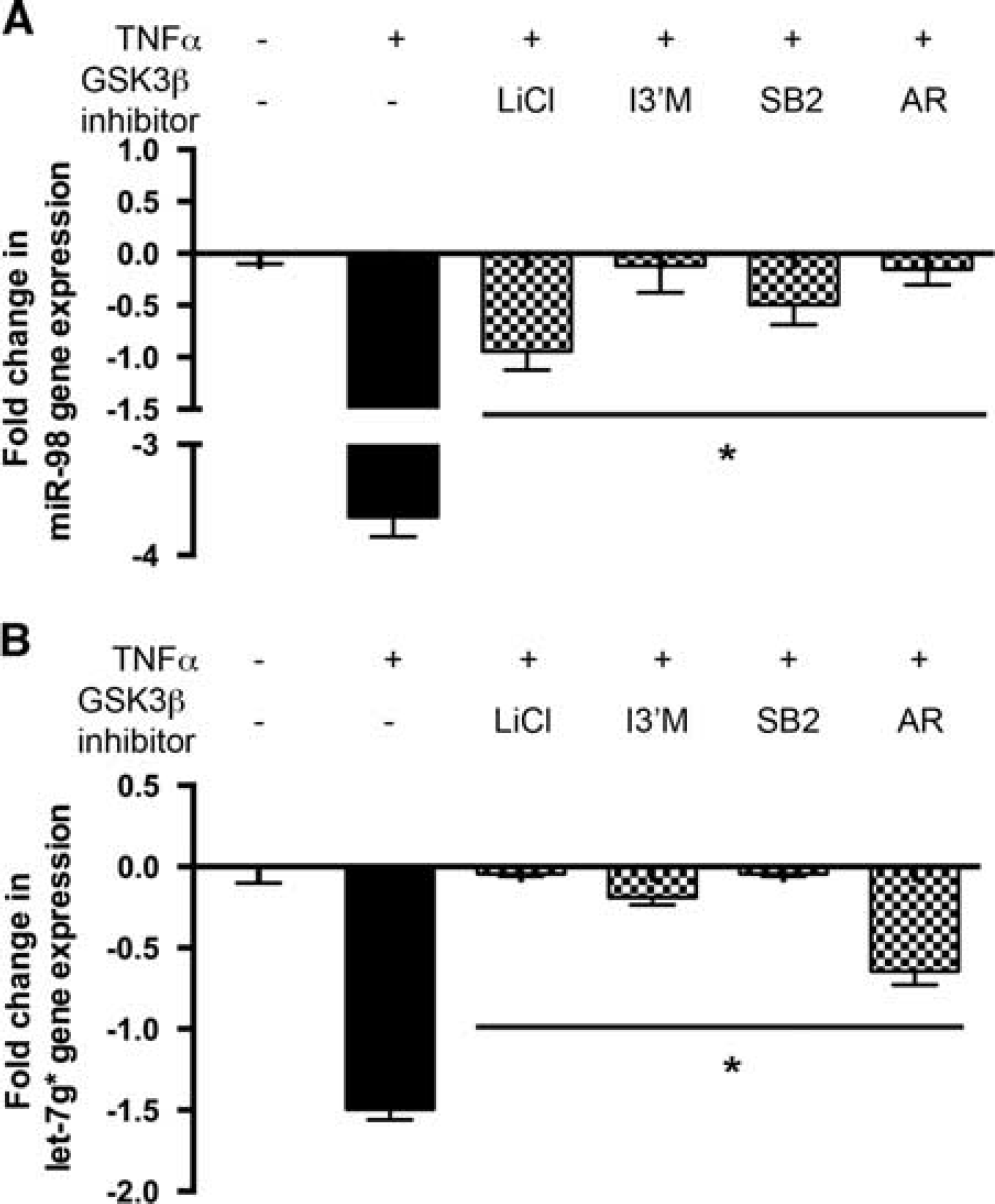
Glycogen synthase kinase 3β (GSK3β) inhibition reversed tumor necrosis factor-α (TNFα)-mediated downregulation of micro-RNA (miRNA) in human primary rain microvascular endothelial cells (BMVEC). GSK3β inhibitors were able to reverse downregulation of miR-98 (
miR-98 and let-7g* Decrease Monocyte Adhesion to and Migration Across Endothelial Monolayer
Our recent data showed that GSK3β inhibition decreased endothelial inflammation and stabilized tight junction (TJ) protein expression.4,5 Since TNFα downregulates miRNA expression and thus upregulates expression of the target proteins, we decided to reproduce the anti-inflammatory effects of GSK3β inhibition on the BBB by upregulating miRNAs that were reversed by GSK3β inhibition. To assess the role of miR-98 and let-7g* on endothelial function, we overexpressed these miRNAs in BMVEC. Brain microvascular endothelial cells were transfected with miRNA mimic oligos (GE-Dharmacon, Lafayette, CO, USA) using the Neon transfection system. With 20% efficiency of transfection, qPCR revealed 120- to 4000-fold increases in expression of selected miRNAs 72 hours after transfection into BMVEC (data not shown). The TNFα treatment of BMVEC resulted in 1.6-fold increase in monocyte adhesion to BMVEC monolayers, whereas miRNA expression reduced monocyte adhesion to BMVEC monolayers by 70% to 73% (Figure 3A). miR-98 overexpression diminished monocyte CCL-2-induced migration across BMVEC monolayers by 20% (Figure 3C), while let-7g* did not have a significant effect on migration. Simultaneous expression of both miRNAs led to a cumulative effect, with a decrease of 50% in migration of monocytes across endothelial monolayers (Figure 3C). Overexpression of NC miRNA cel-39, a nonspecific control miRNA oligo from C. elegance with no homology in mammals, did not have any effect on adhesion or migration. These data support the potential role of miR-98 and let-7g* in reducing monocyte adhesion/migration.
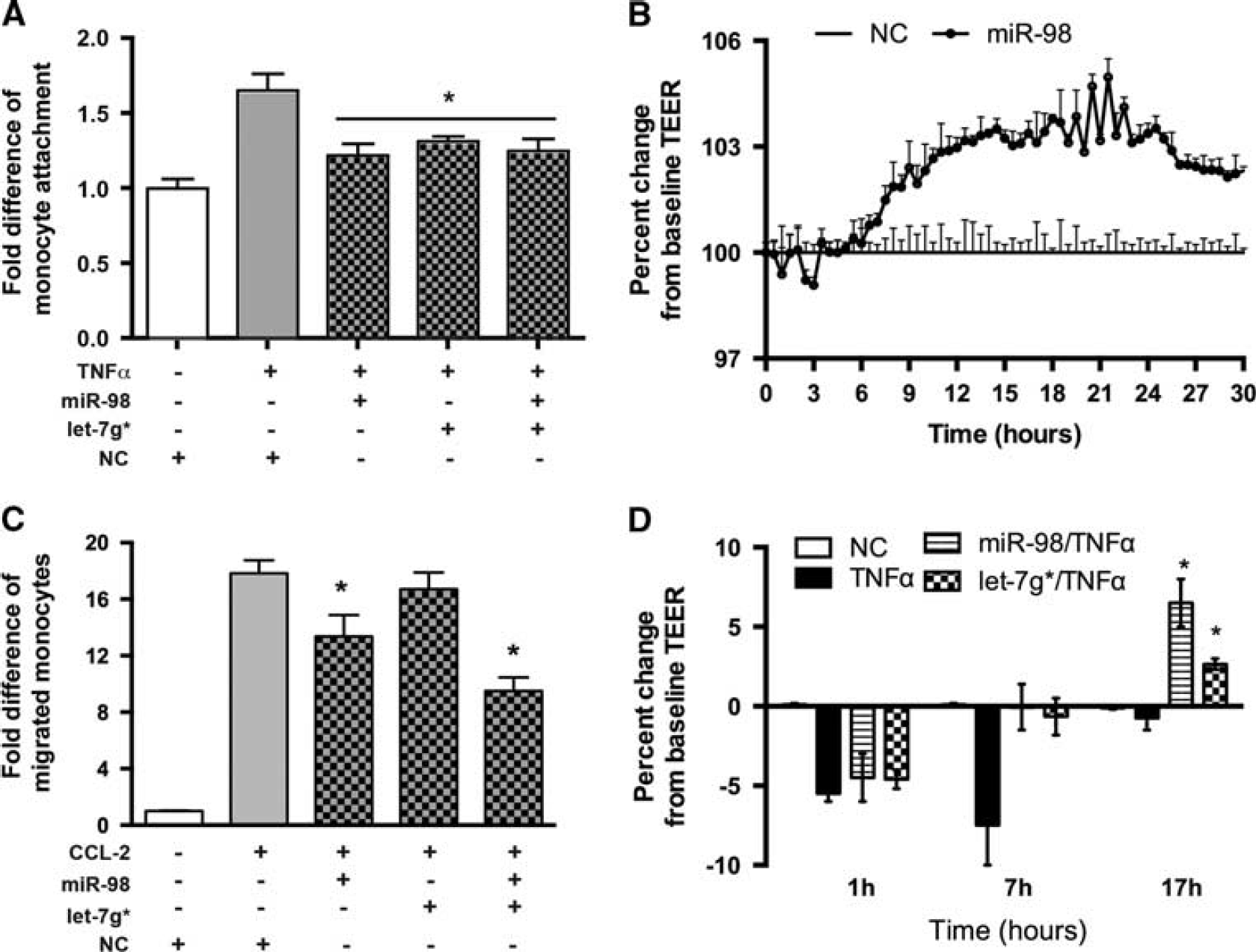
MicroRNA (miRNA) expression reduces monocyte adhesion to and migration across brain microvascular endothelial cells (BMVEC) monolayers and improves barrier function. Mimic oligos of miR-98, let-7g*, or noncoding (NC) niRNA were transfected into brain microvascular endothelial cells (BMVEC). Fold difference of adherent (
miR-98 and let-7g* Improve Blood-Brain Barrier Tightness In Vitro Next, we explored the possibility that overexpression of miRNAs in BMVEC would improve BBB tightness in vitro. Overexpression of miR-98 in primary human BMVEC led to an increase in BBB tightness 8 hours after transfection and lasted for 30 hours as measured by TEER (Figure 3B). Even though miR-98 or let7g* overexpression did not provide significant protection against TNFα insult, it did enhance recovery of BMVEC and therefore led to BBB repair (Figure 3D). Taken together, these data support the potential role of miR-98 and let-7g* in stabilization and rehabilitation of the BBB during inflammation.
miR-98 and let-7g* Decrease Leukocyte-Endothelial Interaction and Diminish Blood-Brain Barrier Permeability In Vivo
To confirm our in vitro results, we measured leukocyte adhesion to brain endothelium by utilizing IVM (Figure 4A). Using pro-inflammatory stimuli (LPS or TNFα), we have shown previously a significant increase in leukocyte adhesion to brain endothelium in vivo due to upregulation of adhesion molecules and BBB injury.4,16 Treatment with anti-inflammatory compounds (GSK3β and PARP inhibitors or cannabinoid receptor type 2 agonists) attenuated leukocyte engagement by brain endothelium, diminished expression of adhesion molecules, and decreased BBB permeability.4,5,16 First, we assessed miRNA expression in extracted MVs by qPCR in response to IC-injected TNFα (Figure 4B). Indeed, IC injection of TNFα resulted in a significant decrease in miR-98 and let-7g* expression levels in MVs (Figure 4B). Second, we tested miRNA transfection efficiency into brain endothelium in vivo before treatment with TNFα. Mice were injected intravenously with liposome-miRNA complexes once or twice (24 hours after first injection). Cel-39, a nontargeting and nonspecies-related miRNA, was used as a negative control. Microvessels were extracted from brains at different time points. Even a single injection of liposome-miRNA complexes resulted in a 22-fold increase after 2 hours; best results were obtained when mice were injected with miRNAs twice (with 24 hours between injections). The highest miRNA expression levels were obtained in MVs after 48 and 72 hours with 40- to 60-fold increases, respectively, versus injection with liposomal solution only (Figure 4C).
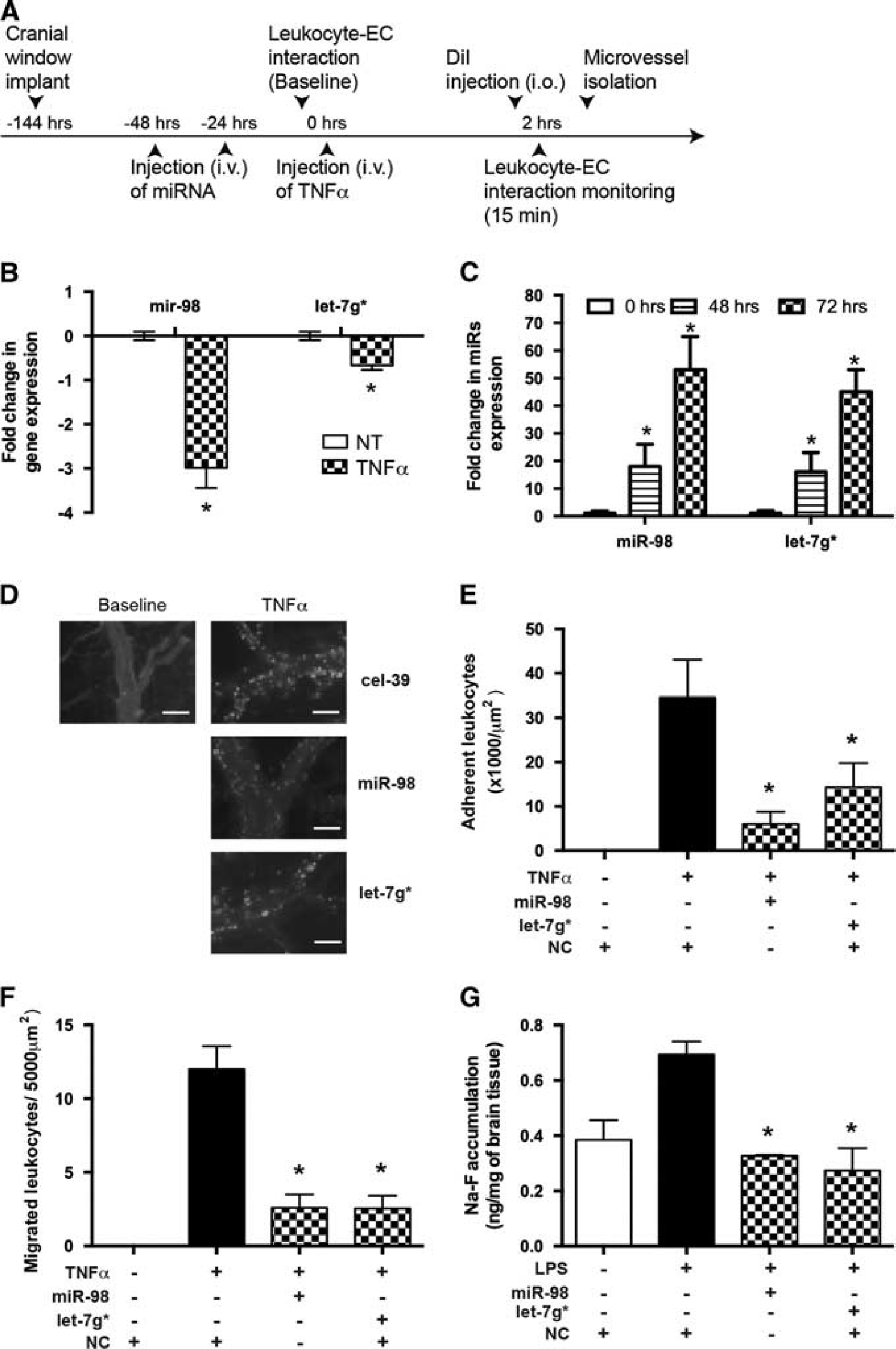
MicroRNA (miRNA) delivered into cortical microvessels (MVs) diminishes leukocyte adhesion to and migration across the cortical MVs and improves blood-brain barrier (BBB) tightness in vivo. (
Next, miR-98 or let-7g* was injected into mice before TNFα treatment and their ability to reduce leukocyte adhesion to and migration across BBB was tested by IVM (Figures 4D–4F). The TNFα injection resulted in a 32-fold increase in leukocyte adhesion to endothelium in MVs, whereas miR-98 and let-7g* diminished adhesion by 84% and 60%, respectively (Figures 4D and 4E). Overexpression of miR-98 or let-7g* led to a 4.5-fold decrease in leukocyte migration across the pial (Figure 4F) and parenchymal vessels (data not shown). Next, we assessed whether pretreatment with miRNAs would prevent BBB permeability (using FITC-labeled dextran) in a LPS-associated encephalitis model. Indeed, both let-7g* and miR-98 decreased BBB permeability to basal levels (Figure 4G). These results support a role of miRNAs in BBB stability in vivo.
miR-98 and let-7g* Lead to Reduction in Pro-Inflammatory Endothelial Responses by Targeting CCL2 and CCL5 Secretion
Our group has previously shown that pretreatment of endothelial cells with GSK3β inhibitors prevented secretion of proinflammatory mediators such as IP-10/CXCL10, MCP-1/CCL2, IL-8/CXCL8, RANTES/CCL5, and Gro a/CXCL1. 4 A bioinformatic approach revealed CCL2 and CCL5 as potential targets of miR-98 or let-7g* (Figures 5A and 6A). Since CCL2 and CCL5 are known to mediate acute inflammatory responses and are implicated in attracting leukocytes to adhere to and migrate across the BBB, 25 as well as altering BBB tightness by provoking small Rho GTPases activation and causing rearrangements in actin cytoskeleton and prompting TJ proteins, ZO-1, ZO-2, occludin, and claudin-5, redistribution, 26 we decided to check whether overexpression of miR-98 or let-7g* would result in diminished secretion of these chemokines. Brain microvascular endothelial cells were transfected with miR-98, let-7g*, or a non-targeting control, and levels of secreted chemokines were measured by ELISA in supernatants with or without TNFα treatment. Levels of secreted monocyte chemoattractant protein 1 (CCL2) were induced 3.5-fold in TNFα-treated BMVEC, while let-7g* and miR-98 overexpression reduced TNFα-induced CCL2 production by 85% to 90% (Figure 5B). Secretion of CCL5 was diminished in miR-98 or let-7g* overexpressing cells by 80% to 85% (Figure 6B).
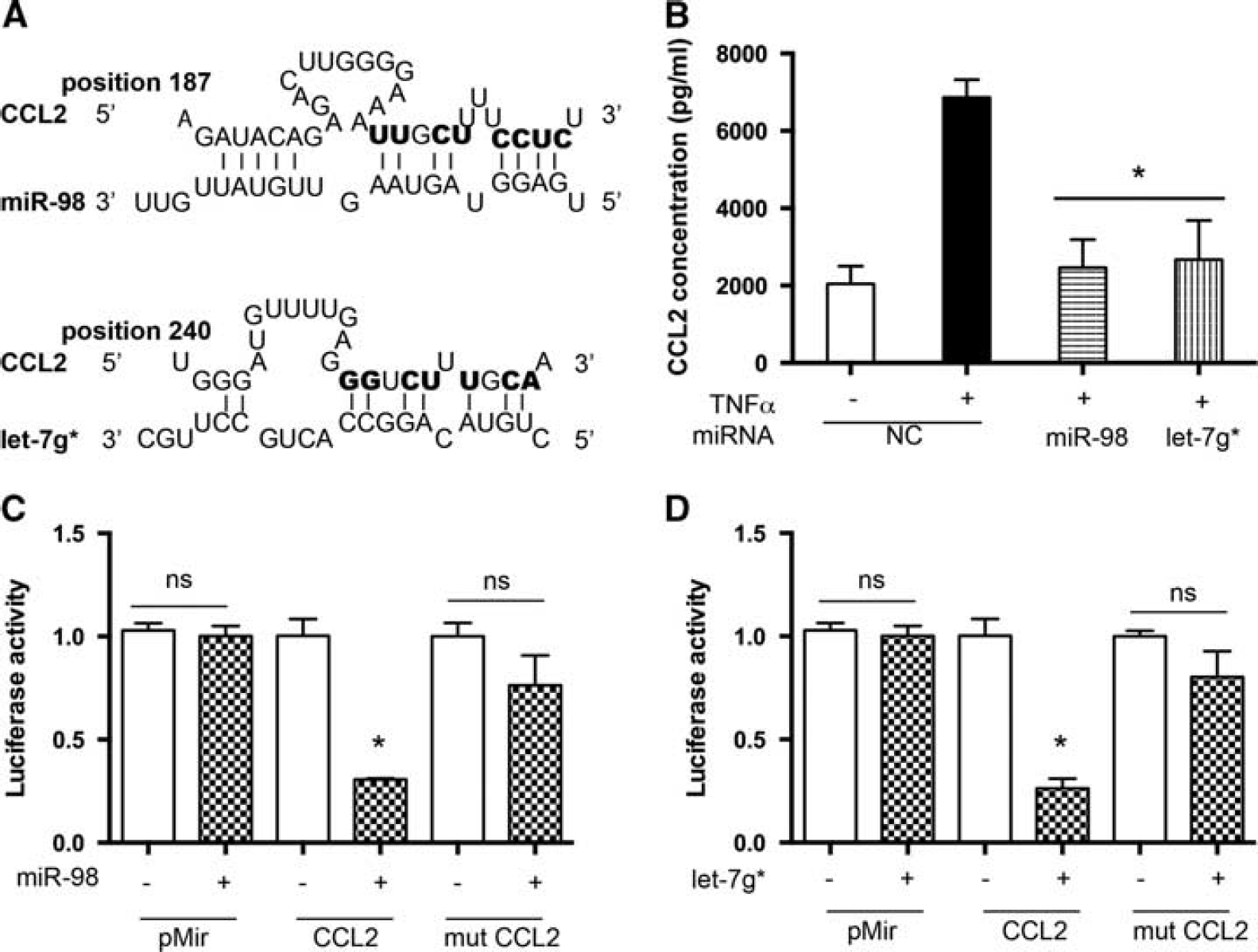
miR-98 and let-7g* directly affect CCL2 expression. (
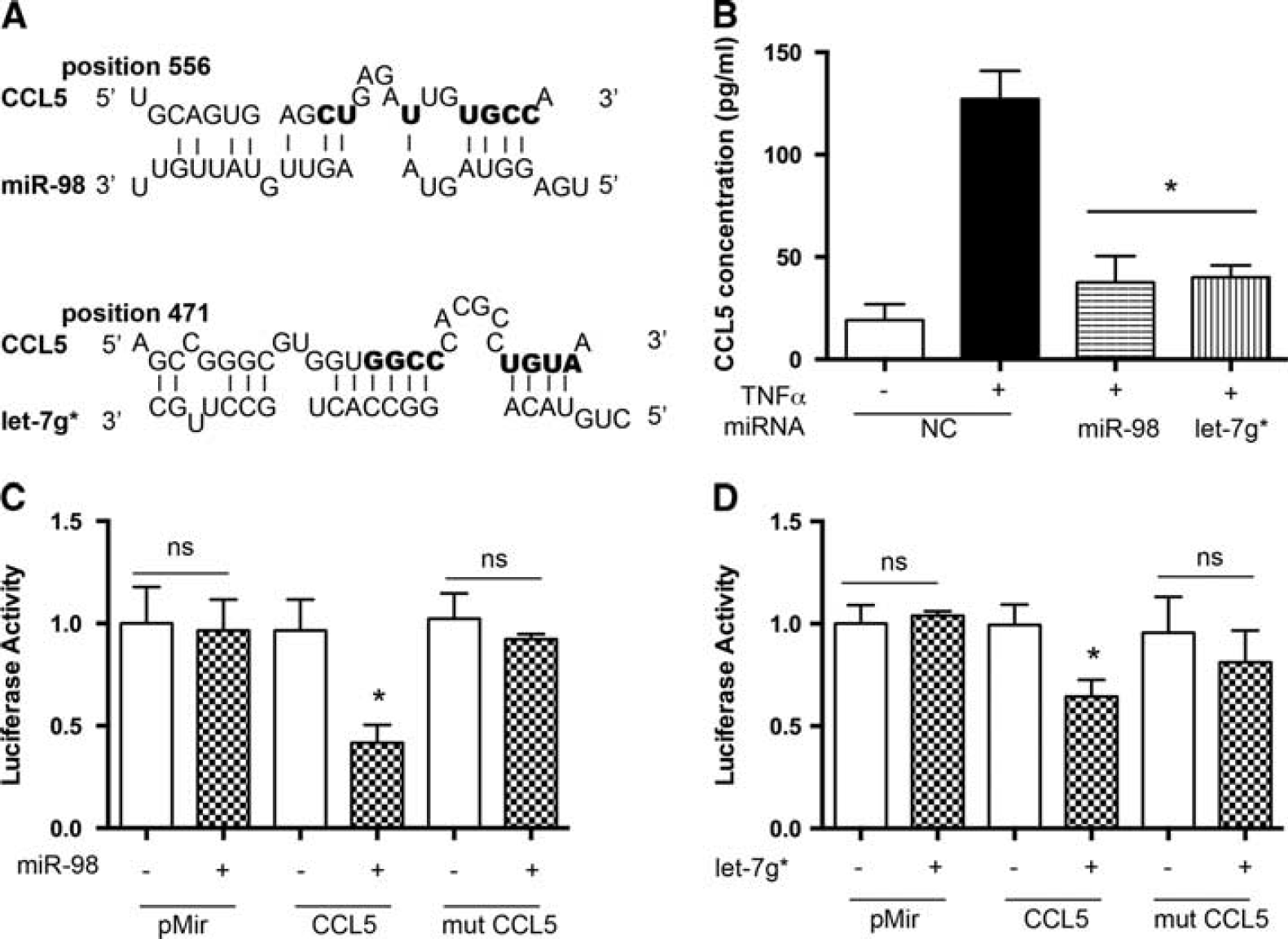
miR-98 and let-7g* directly affect CCL5 expression. (
To test the possibility that expression of CCL2 and CCL5 was post-transcriptionally regulated by miRNAs, we performed a functional assay using the luciferase reporter system. The reporter construct contained full-length CCL2 or CCL5 3'UTR fused to the luciferase gene in the pMir vector. For luciferase assays, the reporter vectors were coexpressed with miR-98 or let-7g* in HEK 293 cells and assayed 48 hours after transfection. No significant difference between the ratios was observed when empty pMir plasmid was cotransfected with either miR-98 or let-7g*. Instead, coexpression of 3'UTR CCL2 with either miR-98 or let-7g* resulted in a reproducible 70% to 75% reduction in luciferase activity (P < 0.05). However, coexpression of 3'UTR CCL5 with miR-98 or let-7g* resulted in 60% and 35% reduction of luciferase activity (P < 0.05), respectively. The control reporter plasmids carrying either CCL2 3'UTR or CCL5 3'UTR mutated in either the miR-98 or let-7g* seeding site were not affected by the addition of miRNAs, confirming the specificity of the miR-98 and let-7g* activity. Taken together, these results support a potential role for GSK3β inhibitor-dependent miRNAs, miR-98 and let-7g*, in reduction of endothelial inflammation via direct targeting of pro-inflammatory mediators, such as CCL2 and CCL5.
DISCUSSION
The BBB is a dynamic structure undergoing constant changes in physiologic and pathologic conditions. 27 Barrier dysfunction can range from mild and transient TJ opening to chronic barrier breakdown leading to increased extravasation of immune cells, poorly regulated flux of molecules and ions, impaired transport processes, and subsequent exacerbation of overall brain pathology and persistent neurologic deficits. 27 Infiltration of leukocytes across the BBB has an important role in neuroinflammatory conditions including MS, ischemia/reperfusion injury, encephalitis, and meningitis.4,5,16,17,28,29 Therefore, understanding the mechanisms that trigger and/or sustain uncontrolled inflammation might be of critical importance for the development of new therapeutic modalities to prevent neurodegeneration. Increasing evidence has accumulated that miRNAs function as key regulators in a wide variety of biologic processes, including proliferation, differentiation, apoptosis, and signal transduction, as well as organ development.30–33 Abnormal expression of miRNAs appears to be a common feature of many human diseases, ranging from cardiovascular disorders to cancer30–33 and most recently inflammatory diseases.9,34,35
Here, we present evidence showing that overexpression of the GSK3β inhibitor-dependent miRNAs, miR-98 and let-7g*, can decrease leukocyte adhesion to and migration across the BBB, in both in vitro and in vivo models. Not all miRNAs had similar abilities to improve BBB function in an in vitro model. For example, let-7g* did not affect migration, while it significantly reduced adhesion. Overall, signaling pathways activated by CCL2 and TNFα might be different, hence differences in functional assay. Even though let-7g* did not reduce migration significantly in vitro, when used together with miR-98, it had a cumulative effect. Both miRNAs improved BBB function, as shown by increased TEER values and diminished permeability. We have recently shown that GSK3β inhibition leads to increased tightness of the barrier. 5 Lithium chloride treatment resulted in 4% increase in TEER after 3 to 4 hours, which went up to 18% to 20% after 16 to 20 hours. While another GSK3β inhibitor, SB216763, showed an increase of 3% to 4% after 11 hours of treatment that went up another 7% after another 10 to 16 hours. In the current work, two GSK3β-regulated miRs, despite narrower targeting abilities than GSK3β inhibitors, were able to increase TEER values, demonstrating the remedial potential of these miRs in improving tightness. It has been shown before that overexpression of miR-125 in the human cerebral microvascular endothelial cell line, hCMEC/D3, by increasing VE-cadherin and ZO-1 proteins, enhanced BBB tightness (improved TEER values). 35 HIV-1 Tat protein-induced inflammation led to decreased levels of VE-cadherin and claudin-5 via upregulation of miR-101 in BMVEC. 12 Wu et al 36 showed an increase in miR-146a expression on MVs from MS patients. They also showed that overexpression of miR-146a in hCMEC/D3 cells diminished, whereas knockdown of miR-146a augmented, cytokine-stimulated adhesion of T cells to endothelial cells, nuclear translocation of NF-kB, and expression of adhesion molecules. Nevertheless, our study is the first to systematically study miRNA profiles in primary human BMVEC during inflammation and their inhibition of key anti-inflammatory and barrier tightening regulatory pathway (e.g., GSK-3β/Wnt). 37
Monocyte chemotactic protein-1/CCL2 and RANTES/CCL5 are mediators of acute inflammatory responses implicated in attracting leukocytes to adhere to and migrate across BBB. 25 Monocyte chemotactic protein-1 has been shown to alter BBB tightness by inducing small Rho GTPases, causing actin cytoskeleton rearrangement (stress fiber formation), and triggering redistribution of TJ proteins, ZO-1, ZO-2, occludin, and claudin-5. 26 Previously, we had shown that pretreatment of endothelial cells with GSK3β inhibitors prevented secretion of pro-inflammatory mediators such as MCP-1/CCL2 and RANTES/CCL5. 4 Reduction in expression of these pro-inflammatory cytokines would prevent their deleterious effects on TJ proteins and actin cytoskeleton and would diminish leukocyte-endothelial interactions as well. Here, we found that both chemokines are targeted by the GSK3β inhibitor-dependent miRNAs, miR-98 and let-7g*. Effects of these miRNAs on TJ protein expression and/or distribution and actin cytoskeleton remain to be studied. Other miR let-7 family members, let-7b and let-7i, have been shown to affect inflammation by targeting the pro-inflammatory cytokine IL-6 7 and Toll-like receptor 4, 38 respectively. Our study elucidates the anti-inflammatory functions of miRNAs and their potential involvement in mechanisms underlying the anti-inflammatory GSK3β inhibition functions. Since multiple miRNAs are involved in the anti-inflammatory function of GSK3β inhibition, one could argue why not just target GSK3β itself. Glycogen synthase kinase 3β is a multifunctional Ser/Thr kinase found in eukaryotes, which phosphorylates and regulates the function of more than 50 substrates and is involved in numerous essential cellular functions, such as glycogen metabolism, cell-cycle control, apoptosis, embryonic development, cell differentiation, cell motility, microtubule function, cell adhesion, and inflammation. 3 Thus, targeting GSK3β itself might lead to many adverse effects, and fine-tuning miRNA expression would be more beneficial.
We found that changes in miRNA expression seen in primary human BMVEC in vitro occur in vivo in an animal model of neuroinflammation. Overexpression of miR-98 and let-7g* in brain endothelium attenuated leukocyte adhesion/migration and diminished BBB permeability pointing to functional reproducibility of miRNA effects in vitro systems. These findings are important from two perspectives. First, we showed that miRNAs have remedial potential in neuroinflammation. Second, a combination of in vitro/in vivo approaches could be used in future testing of other miRNAs as therapeutic targets.
In summary, using a functional approach, we have identified a mechanism implicated in the negative regulation of inflammation in endothelium (Figure 7). It involves the GSK3β inhibitor-mediated increase in miR-98 and let-7g* that were downregulated during inflammation, which in turn, inhibit expression of pro-inflammatory mediators, such as CCL2 and CCL5. Our data support a role for miRNAs in modulation of inflammatory processes in endothelial cells.
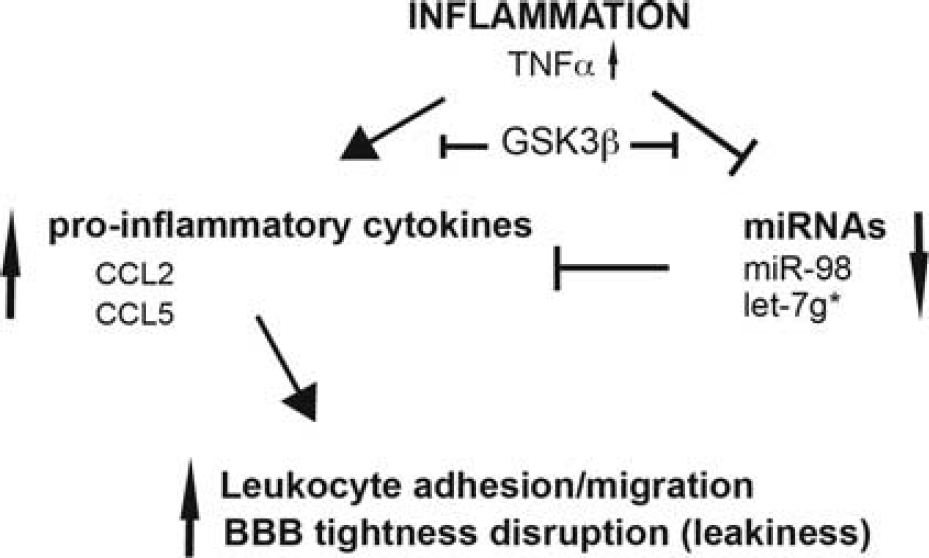
Schematic shows effects of miR-98 and let-7g* on endothelial function. BBB, blood brain barrier; GSK3β, glycogen synthase kinase 3β; miRNAs, microRNAs; TNFα, tumor necrosis factor-α.
Footnotes
SR contributed to conception and design, data acquisition, analysis and interpretation, drafting and revising article, and final approval; HD, VZ-R, and NLR contributed to data acquisition and analysis, drafting, revising and final approval of the article; YP contributed to data interpretation, drafting and revising article, and final approval.
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.