
Abstract
The term cerebral small vessel disease (SVD) refers to a group of pathologic processes with various etiologies that affect small arteries, arterioles, venules, and capillaries of the brain. Magnetic resonance imaging (MRI) correlates of SVD are lacunes, recent small subcortical infarcts, white-matter hyperintensities, enlarged perivascular spaces, microbleeds, and brain atrophy. Endothelial dysfunction is thought to have a role in the mechanisms leading to SVD-related brain changes, and the study of endothelial dysfunction has been proposed as an important step for a better comprehension of cerebral SVD. Among available methods to assess endothelial function in vivo, measurement of molecules of endothelial origin in peripheral blood is currently receiving selective attention. These molecules include products of endothelial cells that change when the endothelium is activated, as well as molecules that reflect endothelial damage and repair. This review examines the main molecular factors involved in both endothelial function and dysfunction, and the evidence linking endothelial dysfunction with cerebral SVD, and gives an overview of clinical studies that have investigated the possible association between endothelial circulating biomarkers and SVD-related brain changes.
Keywords
The term cerebral small vessel disease (SVD) refers to a group of pathologic processes with various etiologies that affect the small arteries, arterioles, venules, and capillaries of the brain. 1 Age/hypertension-related SVDs and cerebral amyloid angiopathy are the most common sporadic forms of SVD. Among a few genetic forms of SVD, CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), caused by NOTCH3 gene mutations, is the most frequent one. It is a systemic arteriopathy although the clinical symptoms are those caused by brain dysfunction. The effects of both sporadic and inherited SVD on the brain parenchyma are represented by lesions mainly located in the subcortical structures, and include lacunar infarcts, ischemic white-matter lesions, and intracerebral hemorrhage. An international working group has recently published the STRIVE (STandards for ReportIng Vascular changes on nEuroimaging) to provide definitions and imaging standards for markers and consequences of SVD. 2 According to this consensus, changes currently seen on neuroimaging related to SVD include lacunes, recent small subcortical infarcts, white-matter hyperintensities (WMH), perivascular spaces (PVS), microbleeds (MB), and brain atrophy.
Over the last few decades, evidence has being accumulated regarding prevalence, clinical significance, and prognostic value of each of these changes. It is now accepted that they are strongly associated with stroke, cognitive decline, psychiatric and motor disorders, disability, and death. Overall, they are considered as a marker of poor prognosis. 1 – 5 Mechanisms linking SVD with parenchyma damage, either ischemic or hemorrhagic, are heterogeneous and not completely understood. Vessel wall changes may in fact be responsible for the rupture of the vessel, thus manifesting as hemorrhagic SVD, or for structural restriction of the vessel lumen, or for its functional dysregulation, leading to a state of chronic hypoperfusion that is responsible for incomplete infarct or acute focal necrosis (lacunar infarct). 1 Besides to and together with these mechanisms, conceptualization about the role of endothelial dysfunction, purported by some opinion leaders, leads to expect that it has a prominent role, and that the study of it may be an essential step for expanding knowledge of cerebral SVD. 6 Endothelial functioning can be assessed in vivo using instrumental tests able to reflect functional properties of normal and activated endothelium. 7 The assessment of circulating molecules of endothelial origin in blood may provide the opportunity of a wider appreciation of the various functions of the endothelium. These molecules include direct products of endothelial cells that change when the endothelium is activated, as well as molecules that reflect endothelial damage or repair. Many of these circulating biomarkers are difficult and expensive to measure, and are currently used only in the research setting.
The aim of this review is to analyze available evidence of biologic circulating markers of endothelial dysfunction in cerebral SVD. The first part of the review examines the main pathways and molecular factors involved in the physiologic function and dysfunction of the endothelium with a special focus on the peculiar situation in the central nervous system. Available evidence linking endothelial dysfunction with cerebral SVD is summarized. In the second part, we review clinical studies that have investigated potential associations between various endothelial biomarkers and the different manifestations of SVD.
Endothelial Functions and Dysfunctions
Endothelium is a monolayer of cells covering the inner surface of blood vessels, with an estimated total area in humans of about 350 m2.
8
The endothelium is a dynamic organ that serves as a functional and structural barrier between the blood and the vessel wall, and has a wide variety of critical roles in the control of vascular function and, as a consequence of its dysfunction, in many mechanisms underlying vascular disorders. Figure 1 schematically depicts the structure of vessel wall, with endothelial cells and their main functions. Endothelial cells are the main regulator of vascular homeostasis due to their interaction with both the circulating cells and those present in the vascular wall, mainly the smooth muscle cells.
9
They modulate blood flow, control permeability to plasma components, and influence adhesion and aggregation of platelets and leukocytes. The main molecules and pathways involved in the four main endothelial functions are schematically depicted in parts A, B, and C of Figure 1 (see figure legends for details).
Regulation of vascular tone which is obtained through the balanced production of vasodilators and vasoconstrictors in response to a variety of stimuli.9,10 Figure 1A shows the main mechanisms involved in vasodilatation and vasoconstriction. Regulation of fibrinolysis and coagulation pathways. The intimal surface of healthy endothelium has both anticoagulant and antithrombotic properties: endothelial cells secrete a variety of molecules important for the regulation of blood coagulation and platelet functions.
9
–
11
Vessel damage or exposure to certain cytokines or proinflammatory stimuli shift the balance toward a procoagulant/prothrombotic phenotype of the endothelial cells. Figure 1B shows the main molecules involved in this pathway.
Schematic representation of blood vessel structure and main endothelial functions. (A) EC and regulation of vascular tone. The regulation of vascular tone is obtained through the balanced production of vasodilators and vasoconstrictors in response to a variety of stimuli.9,10 The left side of (A) shows the main mechanisms involved in vasodilatation: Nitric oxide (NO) is the predominant mediator of normal vascular function and is generated from L-arginine through endothelial NO synthase (NOS). When released by the endothelium, NO diffuses in the vessel wall, to the vascular SMC, and activates guanylate cyclase causing SMC relaxation. Another endothelium-derived vasodilator is prostacyclin (PGI2), generated by cyclooxygenase (COX) and arachidonic acid metabolism. PGI2 activates adenylate cyclase with subsequent generation of cAMP and final relaxation of the underlying smooth muscle cells. NO and PGI2 have also important antiplatelet effects, as they limit aggregation; both of them are involved in the inhibition of coagulation, inflammation, and smooth muscle cells proliferation. The right side of (A) shows the main mechanisms involved in vasoconstriction. The main vasoconstrictive factor is Endothelin (ET), an endothelium-derived 21-amino-acid peptide (the ET family consists of three structurally related peptides, ET-1, ET-2, and ET-3). The product of ET transcription is prepro-ET, which is cleaved by a neutral endopeptidase to form the active precursor pro-ET. Pro-ET may be released from the non-luminal surface of the endothelial cells and converted to mature ET by the membrane-bound metalloproteinase endothelin-converting enzyme (ECE). The two ET receptors, both coupled to G proteins, are endothelin A (ETA) receptor, situated on the vascular SMC, and endothelin B (ETB) receptor, located on endothelial cells. Binding to ETA receptor stimulates phospholipase C initiating in turn events leading eventually to SMC contraction. The result of ET binding to ETB receptor on endothelial cells is the release of NO and PGI2, which opposes the vasoconstricting action of ET. ET also exert mitogenic activity on SMC (not shown in picture). Many factors stimulating ET synthesis, such as thrombin and angiotensin II, also cause the release of vasodilatator PG12 and/or NO, which oppose the vasoconstricting action of ET. (B) Endothelial cells and coagulation and fybrinolysis. The major antiplatelet agents secreted by endothelial cells are PGI2 and NO. In the quiescent state, endothelial cells maintain blood fluidity by promoting the activity of numerous anticoagulant pathways, the most important being the protein C/protein S pathway (PC/PS), which is initiated when thrombin interacts with the endothelial cell receptor thrombomodulin (TM), facilitating activation of protein C. Activated protein C (aPC) inactivates two cofactors essential for blood coagulation: factors VIIIa and Va. To be effective, protein C must form a complex with protein S, which is synthesized by endothelial cells. Complex formation between thrombin and thrombomodulin also prevents thrombin from being able to clot fibrinogen or to activate platelets. Moreover, the endothelial cell surface is rich in heparin-like glycosaminoglycans (HSPG) to which antithrombin (AT) is bound thus providing the main site for inactivation of active thrombin. Endothelial cells also synthesize tissue factor (TF) pathway inhibitor and participates in fibrinolysis by releasing tissue-type plasminogen activator (t-PA) and urokinase, allowing the transformation of plasminogen into plasmin, which degrades thrombi by digesting fibrin network. t-PA is constitutively released while urokinase is only synthesized by activated endothelial cells. The natural inhibitor of t-PA, plasminogen activator inhibitor type 1 (PAI-1) is also constitutively secreted by endothelial cells. The balance of t-PA and PAI-1, which is normally in favor of PAI-1 is also altered by (continued) Participation in inflammation (Figure 1C). Endothelial cells, together with leukocytes, mast cells, and platelets, are key cellular players of inflammatory reactions and strictly interact with the soluble factors involved in the regulation of each of the different steps of the inflammatory reaction.12,13 Left side of Figure 1C depicts the main implicated molecules. Blood vessel formation, repair and remodeling. Angiogenesis is essential for several physiologic processes, such as reproduction, development, and tissue repair, as well as in certain diseases, including inflammation and malignancies. Also on this occasion, the final outcome depends on the balance or imbalance between angiogenic mediators and inhibitors.14,15,16 Main factors involved in endothelial repair are shown in the right side of Figure 1C.
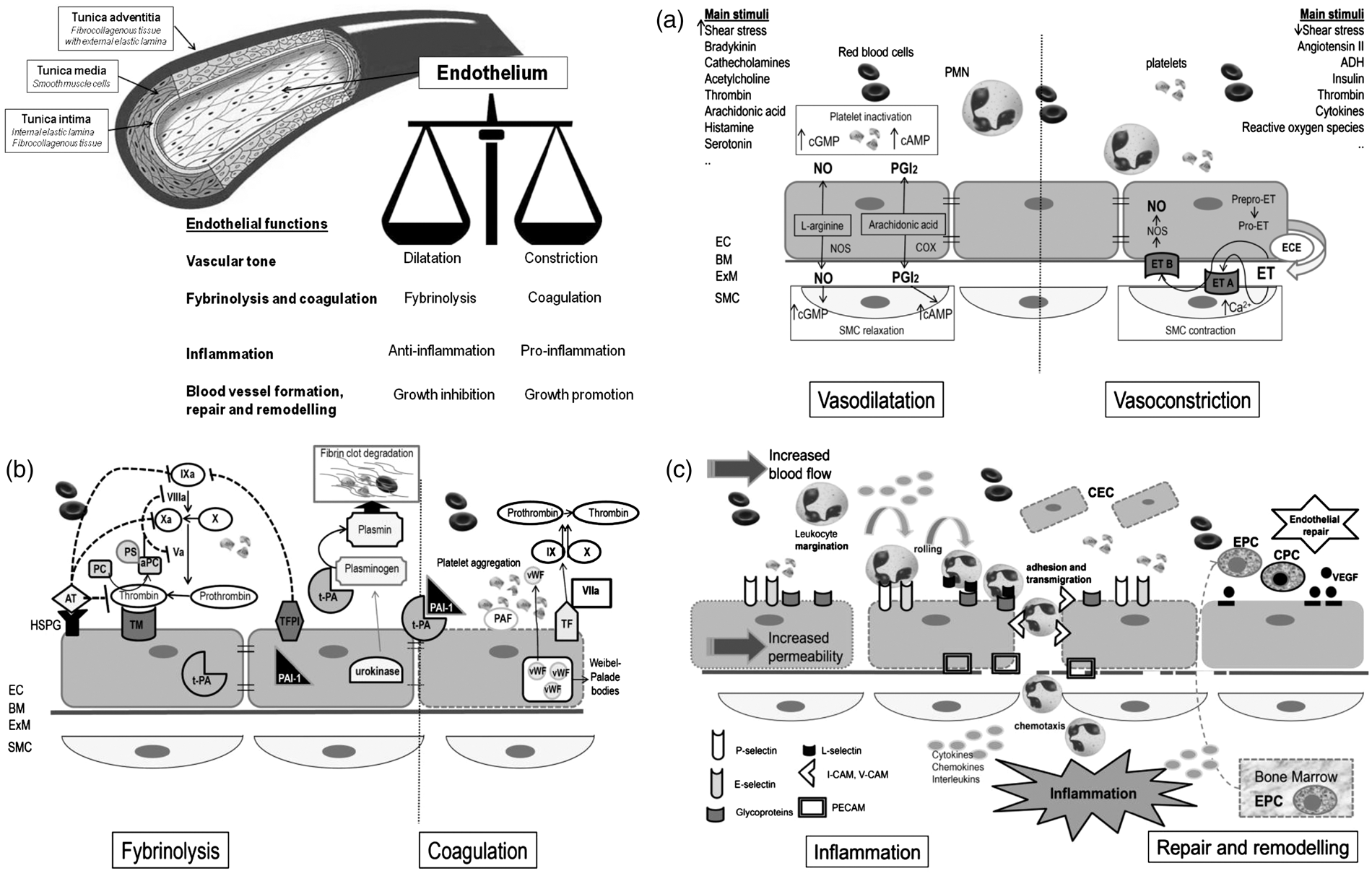
Although schematically described separately, the four different endothelial functions, with related pathways, are strictly inter-related. The term endothelial dysfunction is applied to identify the shift from a normal endothelium to a damaged one that may express with a proinflammatory, provasoconstriction, proliferative, and procoagulation phenotype. Abnormal endothelial function has been documented in different other pathologic conditions, mainly atherosclerosis, diabetes, hypertension, hyperlipidemia, and chronic kidney disease. Finally, the role of some circulating factors as important cardiovascular risk factors has been explained by their main harmful effects on endothelial cells. In this sense, homocysteine (HCY), C-reactive protein (CRP), and asymmetric dimethylarginine (ADMA) may all be considered as endothelial toxins. 17 – 19
Cerebral Endothelium: the Blood–Brain Barrier and the Neurovascular Unit
Within the vascular tree, endothelium presents a high degree of structural and functional heterogeneity. In the central nervous system, the ‘neurovascular unit (NVU)’ refers to the conceptual framework that links microvessels and neuron function and the responses of these compartments to injury.
20
The ‘NVU’ consists of microvessels [endothelial cells–basal lamina matrix–astrocyte end-feet (and pericytes)], astrocytes, neurons and their axons, and other supporting cells that are likely to modulate the function of the ‘unit’ (Figure 2A).
21
Endothelial cells in the cerebral microvessels are a part of the blood–brain barrier (BBB) (Figure 2B), where they exhibit a specialized phenotype with no fenestrations, extensive tight junctions, and sparse pinocytotic vesicular transport (Figure 2C). This barrier allows a strict control of exchange of solutes and circulating cells between the plasma and the interstitial space. Cerebral endothelial cells in most brain regions are connected by tight junctions that function as a ‘physical barrier’ preventing molecular traffic between blood and the brain.
22
Blood–brain barrier also act as a ‘transport barrier’, given to the presence of specific transport systems regulating the transcellular traffic of small hydrophilic molecules, as well as a ‘metabolic barrier’, given the presence of a combination of intracellular and extracellular enzymes.
23
Schematic representation of the cerebral endothelium as integral part of the neurovascular unit (NVU) and blood–brain barrier (BBB). (A) Schematic representation of the NVU, which consists of the monolayer of endothelial cell, connected by tight junctions and resting on the basal lamina, pericytes, smooth muscle cells, astrocytic end-feet, microglia, and neuronal terminals. Circulating blood cells, such as polymorphonuclear (PMN) cells, lymphocytes, and monocytes, are also part of the unit, given the close interaction with the luminal surface of endothelial cells and their role in immune surveillance. (B) Schematic representation of the BBB, which is an integral part of the NVU. The BBB is formed by highly specialized endothelial cells, astrocytic end-feet, and pericyte that together constitute the dynamic interface separating the brain from the circulatory system. (C) Schematic representation of tight junctions, which consist of three main groups of proteins. They are transmembrane proteins (claudins, occludin), cytoskeletal proteins (actin), and accessory proteins. Structurally, they all interact to form a continuous network of transmembrane and cytoplasmic proteins linked with the actin-based cytoskeleton, allowing the tight junction to form a barrier while remaining capable of rapid modulation and regulation.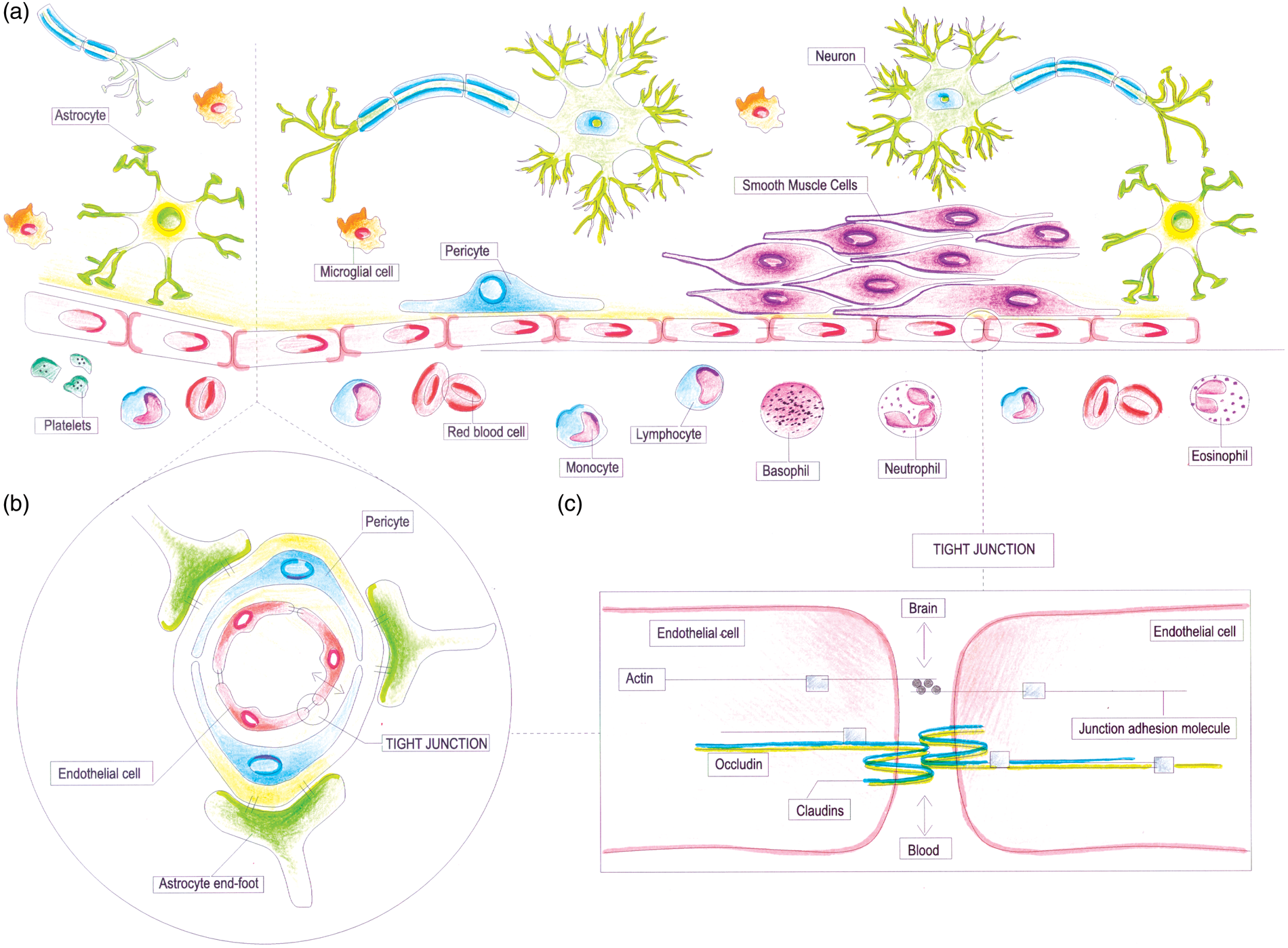
When dealing with cerebral SVD, also taking into account the different types of associated brain lesions, it is fundamental to bear in mind that all the above-mentioned components are strictly inter-related, and that presumably endothelial dysfunction is one of the major determinants of the structural and functional brain-vessel alterations. 6
Evidence of Endothelial Dysfunction/Damage in Small Vessel Disease
Endothelial dysfunction is nowadays considered as one of the pivotal mechanisms of the structural and functional brain-vessel alterations in SVD, and the underpinned evidence comes from different settings. 6
First of all, in SVD patients there is evidence suggesting the presence of reduced cerebral blood flow, and impaired cerebral blood flow autoregulation. 24 Cerebral blood flow studies using a variety of techniques, including positron emission tomography and magnetic resonance imaging (MRI), have shown hypoperfusion. Using endogenous contrast MRI perfusion, perfusion is reduced in the white matter but not in the gray matter, 25 and, within the white matter, it is reduced not only within regions of radiologic WMH but also in normal appearing white matter, although to a lesser extent. 26 Endothelium might have a role in this sense, given the fact that nitric oxide signaling is an important factor in local cerebral blood flow regulation, and that it has been used as a marker to show endothelial dysfunction and decreased vasodilation in response to external stimuli such as hypercapnia or salbutamol in patients with lacunar infarction, compared with controls.27,28
Second, it has been hypothesized that cerebral SVD may be considered as a systemic condition resulting from dysfunction of arteriolar perfusion. 29 Interestingly, there is now evidence pointing to a systemic endothelial dysfunction in patients with cerebral SVD, as indicated by several studies in which endothelial changes have been measured in other vascular beds other than the brain. This association has been documented with the kidney,30,31 with the skin, 32 and the sublingual microvasculature. 33 Impairment of endothelial function in systemic vessels, investigated by means of flow-mediated dilatation in the brachial artery, has also been associated with lacunar stroke. 34
A third body of evidence comes from pathology studies. Available pathological studies on WMH show infarct-like areas of white-matter loss, and/or area of myelin attenuation and pallor. The glial pathology includes astrogliosis, changes in astrocytes, apoptosis of oligodendrocytes and astrocytes, and axonal loss. 35 Tissue pathology changes seen postmortem are frequently interpreted as ‘ischemic’, although some of the changes also support the concept of endothelial dysfunction. An Australian study showed that reduced endothelial integrity was independently associated with increasing WMH severity, and with a significant decrease in endothelial and BBB integrity in areas of WMH compared with normal white matter. 36 In the Medical Research Council Cognitive Function and Ageing Study, a prospective population-based neuropathologic study of elderly people in the United Kingdom, postmortem MRI was used to sample lesioned and nonlesioned white matter. 35 Controls in these studies have further been divided into those from cases with lesions (controls—lesional) and those without lesions (controls—nonlesional). White-matter lesions were characterized by the expression of hypoxia-related molecules and by the expression of endothelial markers such as intracellular adhesion molecule 1 (ICAM-1), thus supporting the role of endothelial dysfunction. 37 There is evidence of BBB leakage, as identified by albumin extravasation, which is widespread in the aging brain and increased in severe white-matter lesions. 38 Together with BBB, there is evidence of the involvement of other components of the NVU. Microglial activation, identified by MHC class II upregulation, has been found to be increased in control white matter from cases with lesions compared with control white matter from individuals without white-matter lesions. 39 Very recently, it was shown that oxidative DNA damage, identified by immunohistochemistry for 8-hydroxydeoxyguanosine, is also increased not only in white matter lesions but also in control white matter from lesional cases. 40 Taken together, these data support the fact that normal-appearing white matter from cases with lesions more closely resembles the lesions themselves than it does white matter from nonlesional cases, and this suggests that lesions in the white matter exist or develop in white matter that shows a ‘field effect’ of diffuse abnormality. 35
Finally, another hypothesized mechanism by which endothelial dysfunction may contribute to brain parenchyma lesions is increased BBB permeability, with consequent leakage of plasma components into the vessel wall and surrounding parenchyma. 41 Permeability of the BBB augments with advancing age and in the presence of cerebral SVD. Increased cerebrospinal fluid/serum albumin ratio, a marker of BBB breakdown, has been documented in vascular dementia,42,43 and in patients with white-matter changes on neuroimaging. 44 More recently, contrast-enhanced MRI has been used to study BBB permeability, documenting an increased permeability in the white matter of patients with lacunar stroke compared with patients with cortical stroke. 45 Topakian et al. 46 found increased BBB permeability in normal appearing white matter in patients affected by SVD compared with controls, and white-matter lesion severity was an independent predictor of these permeability-related signal changes.
From what reported so far, endothelial dysfunction might thus be per se involved as a cause of SVD-related symptoms but also the first step on which other conditions may superimpose. This might be the case, for example, of local thrombosis with subsequent arteriolar occlusion, leading to tissue ischemia through failure of vasodilatation in response to increased neuronal activity. 47
Studies Investigating Potential Associations Between Circulating Biomarkers of Endothelial Dysfunction and Small Vessel Disease
Methods
Search Strategy
The literature search was performed using MEDLINE (Pubmed), and was restricted to the period January 2000 to September 2014 because we wanted to restrict the search to the most recent studies in which SVD-related changes had been assessed using current MRI technologies. Search strategy was as follows: (1) endothelial AND each of the following terms: lacunar, subcortical infarcts, silent brain infarcts, white-matter hyperintensities, white matter changes, leukoaraiosis, and microbleeds; (2) tPA, PAI, Fibrinogen, selectine, d-dimer, homocysteine, ICAM, VCAM, c-reactive protein, interleukin, metalloproteinase, Von Willebrand and each of the following terms: lacunar, subcortical infarcts, silent brain infarcts, white-matter hyperintensities, white-matter changes, leukoaraiosis, and microbleeds; (3) endothelial, tPA, PAI, Fibrinogen, selectine, d-dimer, homocysteine, ICAM, VCAM, c-reactive protein, interleukin, metalloproteinase, Von Willebrand, and CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy).
We included only English language written papers reporting studies that dealt with human cohorts, and considered both population- and hospital-based studies. These latter were defined as studies in which patients were recruited in hospital wards or outpatient settings. Hospital-based studies were excluded when stroke or other cardiovascular events had occurred less than 2 months before biomarkers assessment, to exclude changes consequent to the acute phase. We considered only studies regarding circulating biomarkers. Given the distinct pathophysiologic features of the arteriopathy, we report the results for sporadic SVD and CADASIL in separate tables.
Results
Sporadic Small Vessel Disease and Endothelial Biomarkers
Studies investigating the cross-sectional association between biologic endothelial markers and chronic SVD.

Abbreviations: AAA, abdominal aortic aneurysm; ADMA, asymmetric dymethil arginine; β-TG, Beta-thromboglobulin; BDNF, brain-derived neurotrophic factor; BD, Binswanger disease; BG, basal ganglia; CRP, C-reactive protein; CV, cerebrovascular; DD, D-dimer; EPC, endothelial progenitor cells; F1 + 2, prothrombin fragment 1 + 2; FA, fractional anisotropy; HCY, homocysteine; hsCRP, high sensivity C-reactive protein; HOCl, hypochlorite; ICAM-1, intercellular adhesion molecule-1; IMT, intima media thickness; L-Arg, L-arginine; iLI, isolated lacunar infarct; LI, lacunar infarcts; Lp(a), A Lipoprotein; Lp-PLA2, lipoprotein phospholipase-A2; LS, lacunar stroke; MB, microbleeds; MDA, malondialdehyde; MMPs, matrix metalloproteinases; MPO, myeloperoxidase; PAI-1, plasminogen activator inhibitor type 1; PIC, plasmin-alpha2-plasmin inhibitor complex; Pts, patients; PV, periventricular; PVS, perivascular spaces; RF, risk factor; SBI, subcortical brain infarcts; SDMA, symmetric dymethil arginine; SLI, silent lacunar infarct; SU, Stroke Unit; SVD, small vessel disease; TAT, thrombin–antithrombin complex; TF, tissue factor; TFPI, tissue factor pathways inhibitor; TIA, transient ischemic attack; TM, thrombomodulin; tPA, tissue plasminogen activator; VCAM-1, vascular adhesion molecule-1; VEGF, vascular endothelial growth factor; vWF, Von willebrand factor; WMH, white-matter hyperintensities. Studies are grouped according to types of cohort under investigation.
Longitudinal studies investigating the association between biologic endothelial markers and chronic SVD.

Abbreviations: CRP, C-reactive protein; DD, D-dimer; F1 + 2, prothrombin fragment 1 + 2; ICAM-1, intercellular adhesion molecule-1; IMT, intima media thickness; LI, lacunar infarct; pts, patients; PV, periventricular; SBI, subcortical brain infarct; TFPI, tissue factor pathway inhibitor; TM, thrombomodulin; WMH, white-matter hyperintensities.
Small vessel disease and coagulation/fibrinolysis
Ten studies evaluated markers of coagulation and fibrinolysis in relation to MRI changes consequent to SVD. Plasminogen activator inhibitor resulted independently associated with the presence of WMH and/or lacunar infarcts and reduced fractional anisotropy on diffusion tensor imaging sequences in three studies.71,74,79 Four studies investigated the association between D-dimer and markers of SVD. Gottesmann et al. 52 found an independent association between D-dimer levels and subclinical lacunar infarcts in the Atherosclerosis Risk in Communities cohort. Data from the Framingham study showed an independent association between D-dimer and total cerebral brain volume but not with WMH or silent brain infarcts. 56 In the remaining two studies, characterized by smaller sample sizes, D-dimer proved to be increased in SVD, i.e. lacunar infarcts and clinically manifest Binswanger disease (defined by the presence of WMH and evidence of subcortical vascular dysfunction such as gait disorders, incontinence, and parkinsonism), but the relation was not confirmed after multivariate analyses.73,74 Regarding tissue factor pathway inhibitor (TFPI), discordant results were found in two studies. Hassan et al. 76 found higher TFPI values in lacunar infarct patients compared with patients presenting both lacunar infarcts and moderate/severe WMH and with healthy volunteers adjusting for possible confounders. In Knottnerus et al.'s study, TFPI values were lower in patients with lacunar stroke compared with controls; the highest values were found among patients with lacunar stroke and WMH, compared with patients with only lacunar stroke. 80 These two studies examined also tissue factor (TF), which resulted independently associated with the extent of WMH only in Hassan’s et al. study. 76 Regarding thrombomodulin studies, only Hassan et al. found higher values in SVD patients in comparison with controls and a positive association with lacunar infarcts, while in the remaining two studies no association was found.52,79 Thrombin–antithrombin (TAT) was independently associated with the presence of WMH only in a small-sized study. 74 Two studies determined F1 + 2 levels in SVD, documenting an independent association with multiple subcortical lacunar infarcts.73,74 Finally, von Willebrand factors proved to be independently associated with SVD MRI markers in two studies.52,74
Small vessel disease and hyperhomocysteinemia
In the majority of the population-based cohorts, abnormally increased HCY levels were found independently associated with WMH and/or silent lacunar infarcts.53,59,62,65 In the Framingham Offspring cohort, 56 a significant association with SVD was appraised only with total brain cortical volume, while in the French cohort of the Epidemiology of Vascular Ageing study only a nonsignificant trend between HCY levels and WMH severity was found. 50 Hospital-based studies have reported less homogenous results.77,78,81
Small vessel disease and inflammation
Several studies reported on the association between inflammatory markers and SVD. One of the most widely studied biomarker was CRP. In population-based studies, results were contrasting; in three large cohorts (including overall 5,947 subjects), CRP resulted significantly associated with WMH,51,60,61 while in the remaining six cohorts, examining a total of 5,156 patients, the association was not demonstrated.49,56,63,64,65 Discrepant results were also apparent from the two longitudinal studies reporting on CRP and progression of WMH: subjects with high baseline CRP levels had significant more progression of periventricular WMH than people with low CRP levels in the Rotterdam Scan Study, while no significant association was observed between baseline CRP levels and the evolution of silent brain infarcts and WMH in the 3C Dijon study.60,61 Interestingly, the two studies that investigated the association between CRP and a more sensitive MRI marker of white-matter changes, i.e. the microstructural damage as assessed using diffusion tensor imaging, found a positive association.64,71 An independent relation between CRP and MB or CRP and periventricular spaces was reported by two different studies.49,72 Silent brain infarcts proved to be associated with higher level of CRP in three studies,60,67,69 a result that was not confirmed by Gottesmann et al.’s study. 52 Data concerning IL-6 are more consistent because an independent association between IL-6 and either WMH or silent brain infarcts was shown by the majority of population and hospital-based studies.51,60,67,69 Nevertheless, no association with the progression of MRI SVD markers was found for IL-6 in the 3C-Dijon study. 60 Only one study reported an association between IL-6, IL-18, and MB. 72
Three studies evaluated the association between fibrinogen and WMH, lacunar infarcts, PVS, and the most severe SVD-related clinical picture defined as Binswanger’s disease. In the largest of the three studies, 48 SVD patients compared with controls had higher levels of fibrinogen, a finding that was not confirmed in the two other cohorts.49,73 In all the studies in which ADMA levels were measured, an independent association with both lacunar infarcts and WMH was reported.54,55,78 Matrix metalloproteinase 9 (MMP9) was measured in a total of 234 patients, but the association with WMH was significant only at univariate analyses.69,70 The Northern Manhattan Study found an independent association of both MPO and Lp-PLA2 with WMH, but the result for Lp-PLA2 was not confirmed in the Framingham Offspring Study.66,58 Finally, one study evaluated the role of autoantibodies against oxidized-LDL finding an independent association with PVS. 83
Considering adhesion molecules, the association between ICAM-1 levels and SVD markers was investigated in three studies. Roulh et al. 68 reported an association with WMH that was confirmed after multivariate analyses in a further study cohort. Higher levels of ICAM-1 were observed in patients with lacunar infarcts and WMH compared with controls. 76 Interestingly, in the Austrian Stroke Prevention Study, ICAM-1 proved to be associated with a robust outcome measure, i.e., WMH progression at both 3 and 6 years. 85 Rouhl et al. 84 studied several endothelial biomarkers finding higher levels of E-selectine, neopterin, and VCAM-1 in patients with lacunar infarcts and/or WMH. Considering MRI markers of SVD, PVS resulted independently associated with neopterin and MB with E-selectine.
SVD and repair and remodeling
Only two studies evaluated the association between SVD and peripheral circulating cells documenting that immature EPC, and Angiogenic T cells (Tang) were significantly reduced in SVD patients. The association remained significant in multivariate analyses only for Tang.75,82
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy and Endothelial Biomarkers
Studies investigating the cross-sectional association between circulating endothelial markers and CADASIL.
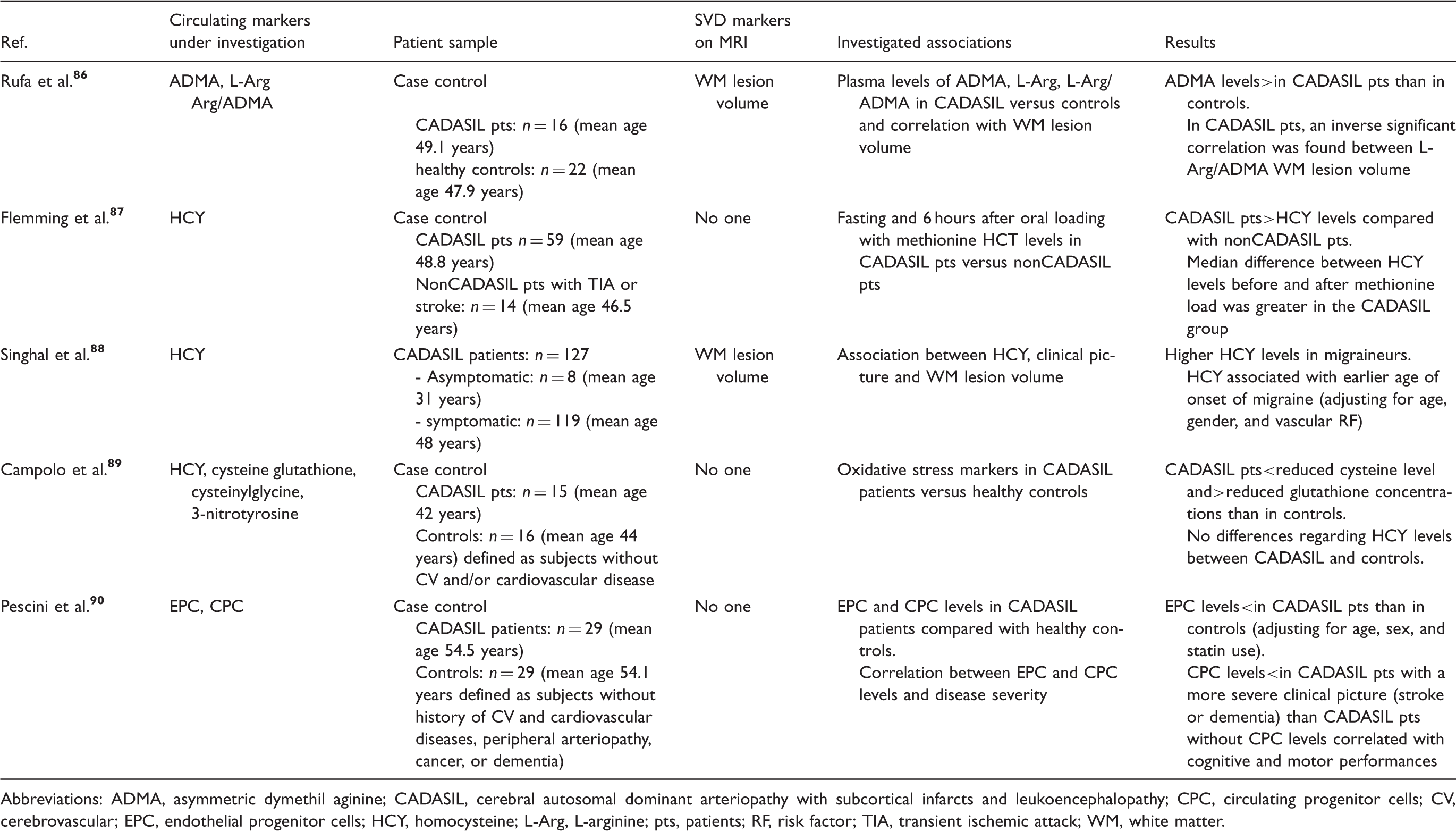
Abbreviations: ADMA, asymmetric dymethil aginine; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CPC, circulating progenitor cells; CV, cerebrovascular; EPC, endothelial progenitor cells; HCY, homocysteine; L-Arg, L-arginine; pts, patients; RF, risk factor; TIA, transient ischemic attack; WM, white matter.
Discussion
Evidence from the above considered rather large number of published studies, in which the association of several circulating endothelial biomarkers with SVD was scrutinized, looks largely heterogeneous and inconclusive concerning the identification, or even the suspicion, of a pivotal pathogenic role exerted by one or more distinct molecular factors or pathways in cerebral SVD. 40 The most consistent associations in sporadic SVD regard PAI, ICAM-1, ADMA, and IL-6. Noteworthy, these molecules are involved at different places of the endothelial dysfunction/coagulation system/inflammation cascade. Another marker of endothelial dysfunction, for which significant results seem to emerge in both sporadic SVD patients and in CADASIL cases in comparison with controls, is the level of EPC. Moreover, in CADASIL, CPC levels were associated with clinical/functional indicators of disease severity, suggesting that these cells might have a role in the determination of the final phenotypic expression of the disease. 90
Data heterogeneity may be explained by a number of differences in studies design. A first major concern relates to the different cohorts under investigation: some studies were conducted in the framework of population-based surveys of elderly individuals, while others investigated hospital cohorts without age limits. It is presumably logical to think that the brain lesion load related to SVD is lower in the population-based compared with the hospital-based cohorts, especially in stroke cohorts. Also sample sizes were largely variable, ranging from over three thousands in the largest population-based study, to less than a hundred in a number of hospital-based studies. Moreover, among the hospital-based cohorts, the clinical status of the enrolled patients was largely variable across the different studies, ranging from asymptomatic subjects, to hypertensive patients, and finally to previous symptomatic lacunar stroke patients. Concerning this latter category, it is fundamental to note that only few studies distinguished between isolated lacunar infarcts and multiple lacunar infarcts associated or not with WMH, thus recalling the concept, first introduced by Fisher, that the pathogenesis of small subcortical infarcts may be distinct: arteriolosclerosis, lipohyalinosis on one side, and (micro)atheroma on the other. 91 When this distinction has been made, results support the hypothesis of a distinct pathogenesis.76,77,79,80 Considering stroke cohorts, in some studies, infarcts cannot be defined conventionally and univocally as lacunar: in fact, while the inferior diameter was precisely defined (generally 3 mm), there was no cutoff value reported for the superior one. For this reason, even if the infarcts were subcortical, it cannot be excluded that, in some cases, they were not related to SVD.
Among the different SVD-related MRI changes selected for investigating the effect of circulating biomarkers, WMH (visual grading or volume) and lacunar infarcts were the most frequently studied, especially in the older studies. Only few recent studies considered novel MRI markers of SVD such as MB, enlarged PVS, and microstructural WM damage measured on diffusion tensor imaging. Very few studies used cellular biomarkers of endothelial dysfunction. Finally, with the exception of a few studies,52,66,74,79,84 only selective biomarkers involved in different points of the complex pathways linking together endothelial dysfunction and coagulation system/inflammation processes were analyzed. It should be underlined that the assessment of individual biomarkers cannot provide information on relative contributions by the distinct pathways. Conceptually, only the combined use of multiple circulating endothelial biomarkers may provide a comprehensive understanding of the role of selective endothelial pathways related to the variable pathologic or clinical expressions of brain SVD. In fact, the panel of biomarkers related to each single pathway was often heterogeneously studied across studies. Different methodologies can be applied to measure circulating biomarkers, thus leading to a limited reproducibility of the results obtained in different laboratories. Finally, for several circulating biomarkers investigated in many of the considered studies no definite cutoff values were reported.
Conclusion and Future Directions
The vascular endothelium, located at the interface of blood and tissue, is essential for vascular homeostasis, as it is capable of sensing changes in the hemodynamic forces and blood borne factors, and to respond by releasing different kind of substances involved in the different pathways of the above-summarized endothelial functions. The net balance between endothelial-derived factors involved in the regulation of vascular tone, of coagulation and fybrinolysis, of pro- and anti-inflammation, and factors associated with growth inhibition and promotion, is essential for the maintenance of vascular homeostasis. Any disruption of this balance may have a detrimental role in the overall pathophysiology of SVD or in the production of the final tissue changes consequent to it. Research in the field of cerebral SVD in relation to endothelial circulating biomarkers appears to be in a very preliminary stage. A major problem relates to the fact that what is measured from blood samples reflects systemic endothelial function, which does not necessarily correspond to what happens at the level of brain endothelium. As such, the study of endothelial circulating biomarkers needs to be strictly correlated with other measures specific for brain endothelial function, i.e., advanced MRI technologies with intravenous gadolinium contrast agent combined with perfusion imaging.
Finally, the issue of causality or possible reverse causality seems critical. Figure 3 is an attempt to schematically represent the possible relationship between endothelial dysfunction and brain parenchyma lesions related to SVD. We have evidence that, generally in elderly subjects, cerebral SVD-related lesions and endothelial dysfunction co-exist, but what we do not know is whether the association is causal, i.e., what happens before their coexistence. Support for a causal rather than a secondary role may come from longitudinal studies, as is the case of the three studies reported in Table 2, in which the predictive effect of the baseline circulating biomarkers levels is looked at on SVD progression.
Conceptualization of the possible relationship between endothelial dysfunction and cerebral small vessel disease (SVD).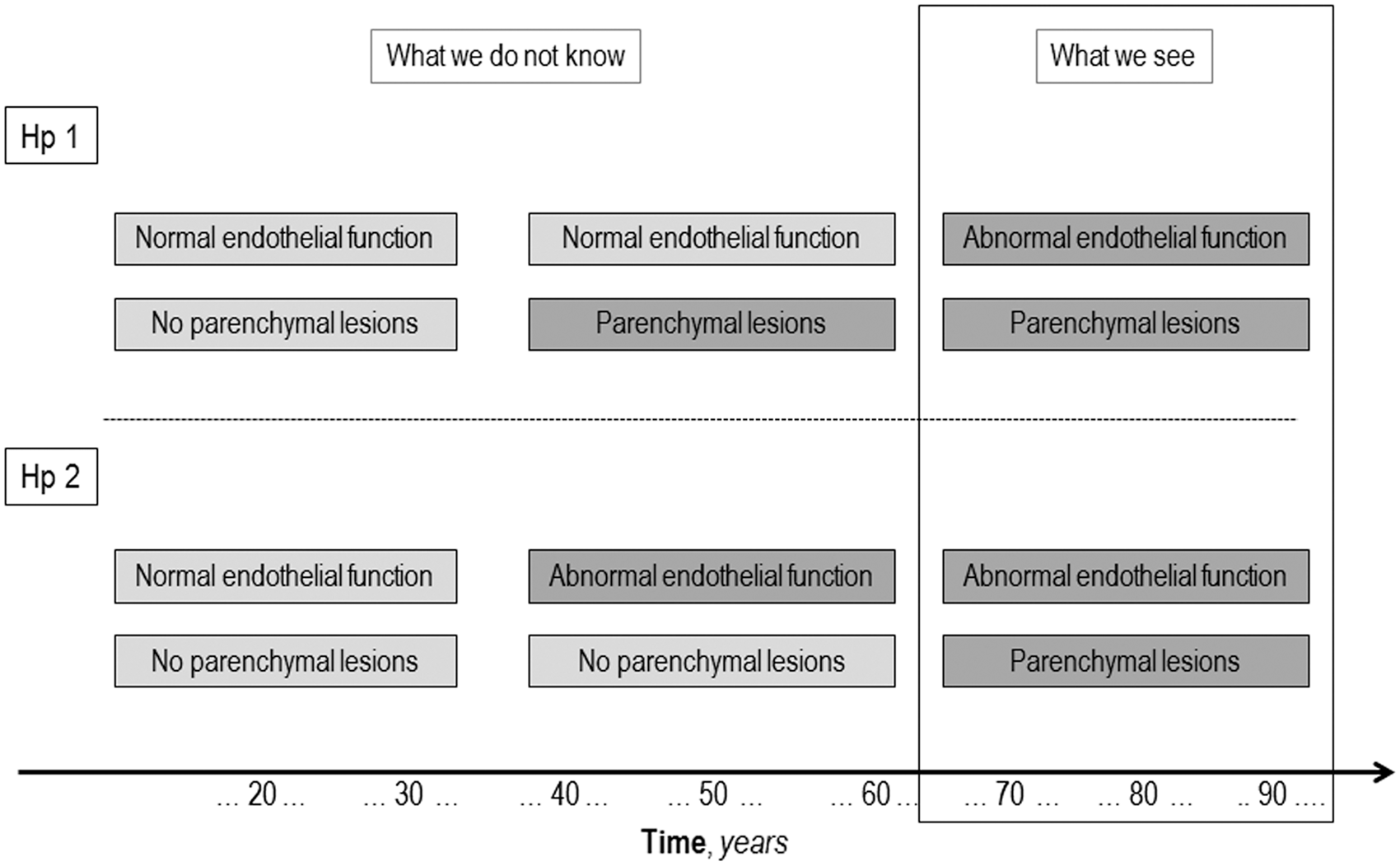
Another way of addressing the issue of causality may come from candidate gene association studies. As an example, HCY levels are known to be associated with SVD, particularly with WMH, 77 and this could either be causal or could be secondary to established disease. Examination of a genetic variant associated with increased HCY levels throughout life allows causality to be explored, an approach referred to as ‘Mendelian randomization’. 92 The methylenetetrahydrofolate reductase C677T polymorphism is associated with elevated HCY levels. A number of studies have shown no or only weak associations between a common methylenetetrahydrofolate reductase polymorphism and stroke as a whole. In contrast, an association was found in a well-phenotyped group of patients with lacunar stroke and, interestingly, this association was present only in the ischemic leukoaraiosis subtype. 64 This supports a causal role for HCY in this specific subtype.
In conclusion, considerable evidence suggests that endothelial dysfunction may have an important role in cerebral SVD, although much molecular details still need to be clarified. The use of comprehensive panels of circulating endothelial biomarkers exploring functioning of the different biologic pathways may be useful in this regard, and it might overcome limitations associated with individual assays. The final translational goal is obviously to provide a more robust pathophysiologic background for the design of experimental research finalized to reduce the burden of cerebral SVD.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The VascMCI-Tuscany study is funded by Tuscany region.
Declaration of conflicting interests
DI serves as a member in the editorial board of Stroke, and is an associate editor of Neurological Sciences Journal. He has received grants for research by Bayer Italy, and fees for conferences by Boheringer Italy and Bayer Italy. LP is a member of the editorial boards of Cerebrovascular Diseases and Acta Neurologica Scandinavica and section editor (Vascular Cognitive Impairment) of Stroke.
Authors’ contributions
AP, MP, and FP made a substantial contribution to conception, design, acquisition of data, and drafting the article. LP and DI made substantial contribution to conception, design, drafting, and revising the manuscript.