
Abstract
Cerebral small vessel disease (SVD) is a major cause of age-related cognitive impairment and dementia. The pathophysiology of SVD is not well understood and is hampered by a limited range of relevant animal models. Here, we describe gliovascular alterations and cognitive deficits in a mouse model of sustained cerebral hypoperfusion with features of SVD (microinfarcts, hemorrhage, white matter disruption) induced by bilateral common carotid stenosis. Multiple features of SVD were determined on T2-weighted and diffusion-tensor magnetic resonance imaging scans and confirmed by pathologic assessment. These features, which were absent in sham controls, included multiple T2-hyperintense infarcts and T2-hypointense hemosiderin-like regions in subcortical nuclei plus increased cerebral atrophy compared with controls. Fractional anisotropy was also significantly reduced in several white matter structures including the corpus callosum. Investigation of gliovascular changes revealed a marked increase in microvessel diameter, vascular wall disruption, fibrinoid necrosis, hemorrhage, and blood–brain barrier alterations. Widespread reactive gliosis, including displacement of the astrocytic water channel, aquaporin 4, was observed. Hypoperfused mice also demonstrated deficits in spatial working and reference memory tasks. Overall, gliovascular disruption is a prominent feature of this mouse, which could provide a useful model for early-phase testing of potential SVD treatment strategies.
Keywords
INTRODUCTION
Cerebral small vessel disease (SVD) is a major cause of age-related cognitive decline and dementia. 1 Hypertension is proposed to be a common risk factor (although it explains only a small proportion of imaging-determined SVD) and cerebral amyloid angiopathy is commonly present on pathologic examination.2,3 Clinically, SVD is associated with early impairment of attention and executive function with slowing of information processing and motor deficits. 1 These clinical manifestations result primarily from the occurrence of lesions in the cerebral white matter, multiple lacunes within subcortical structures, atrophy, and, in some cases, microbleeds in deep gray matter, white matter, or at the corticosubcortical junction, identified by neuroimaging.2,4 White matter lesions, associated with increased extracellular fluid, varying degrees of myelin and axonal pathology, and glial responses, are predominantly detected as hyperintense regions on fluid attenuation inversion recovery and T2-weighted magnetic resonance imaging (MRI). 5 Lacunes, which present as hypointense regions on fluid attenuation inversion recovery or T1-weighted MRI or hyperintense on T2-weighted MRI, are small deep cavities thought to result from small infarcts, which commonly occur in subcortical areas including the white matter and basal ganglia. 6 Cerebral microbleeds are seen as small hypointense dots on T2*- or susceptibility-weighted MRI and are thought to correspond with perivascular hemorrhages. 7 The pathologic correlates of these radiologic features include extensive vascular disruption associated with fibrinoid necrosis, arteriosclerosis, and collagen deposition. 8 Endothelial failure with altered blood-brain barrier (BBB) permeability is postulated as one pathogenetic component underlying the pathophysiology of SVD. 9 The integrity of the BBB critically depends on the cross-talk between endothelial cells, pericytes, astrocytes, and endfeet processes, which closely contact microvessels comprising the gliovascular unit. 10 Damage to selective components of the gliovascular unit may additionally contribute to the pathophysiology of SVD.
Currently available animal models mimic selective aspects of SVD (for reviews see Hachinski
Although there is no association between carotid stenosis and SVD in humans, 25 small vessel changes have been linked to histologic features that have been interpreted as representing local hypoperfusion of as yet unknown cause. 26 We therefore hypothesized that sustained hypoperfusion would produce progressive pathologic and cognitive features similar to those seen in SVD. Our study shows that, in response to sustained cerebral hypoperfusion over 6 months in mice, the model develops certain key features of SVD: namely small subcortical ischemic lesions and hemorrhages, cerebral atrophy and white matter changes, and memory impairments. Furthermore, these features were coincident with prominent disruption of the gliovasculature including the BBB, which may be potential therapeutic targets in SVD.
MATERIALS AND METHODS
Animals
Male C57Bl/6J mice (
Diffusion tensor and magnetization transfer MRI biomarker values in long-term hypoperfused (
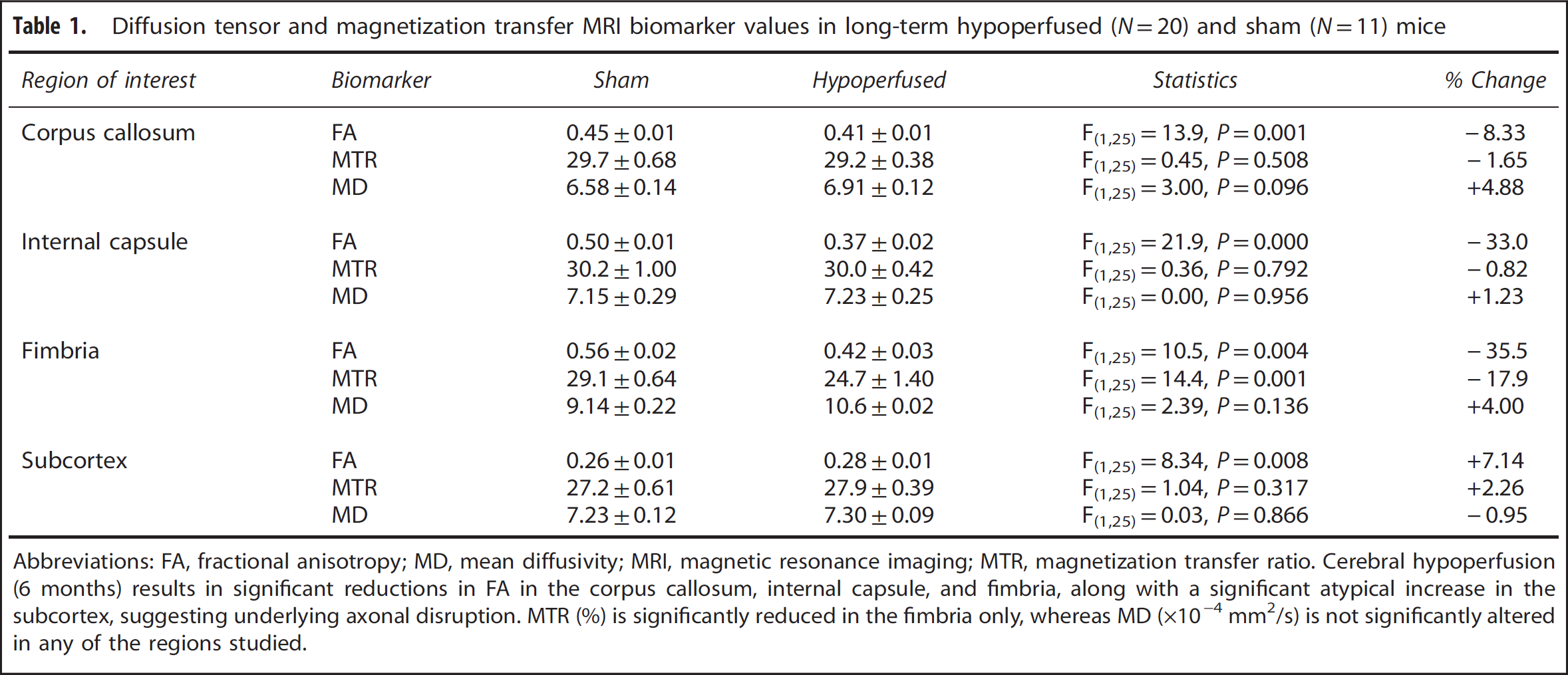
Abbreviations: FA, fractional anisotropy; MD, mean diffusivity; MRI, magnetic resonance imaging; MTR, magnetization transfer ratio.
Cerebral hypoperfusion (6 months) results in significant reductions in FA in the corpus callosum, internal capsule, and fimbria, along with a significant atypical increase in the subcortex, suggesting underlying axonal disruption. MTR (%) is significantly reduced in the fimbria only, whereas MD (× 10−4 mm2/s) is not significantly altered in any of the regions studied.
Magnetic Resonance Imaging
Structural T2-weighted, diffusion-tensor (DT), and magnetization transfer (MT) MRI data were collected using a Varian 7T preclinical scanner with a 72 mm volume coil and a four-channel phased array mouse brain coil (see Supplementary Material for detailed methods of image acquisition and protocols). Each T2-weighted MRI slice of the mouse brain was examined from anterior to posterior and recorded for (1) the presence, volume and type of cortical and/or subcortical primary ischemic lesions, classified as hyperintense signal with focal tissue loss, potentially cavitated with or without localized hypointense areas consistent with paramagnetic effects of iron/hemosiderin in the surrounding parenchyma; and (2) primary hemorrhages, their volume and anatomic location, classified as areas of rounded predominantly hypointense signal. Over consecutive T2-weighted MRI slices (1.0 to −4.6 mm Bregma), the volume of ischemic and hemorrhagic lesions were determined and then combined to give an overall lesion load; brain volume was delineated by measuring the entire volume of the brain covered by MRI (1.0 to −4.6 mm Bregma) and then data were summed to provide a regional brain volume. Mean diffusivity (MD), fractional anisotropy (FA), and magnetization transfer ratio (MTR) volumes were generated using ‘in-house’ custom software and the slice corresponding with −1.2 to −2.4 mm from the Bregma processed for region of interest analysis.
Assessment of Behavioural Alterations
In 6-month hypoperfused mice, spatial working memory was investigated using an 8-arm radial arm maze; spatial reference learning and memory was investigated using a water maze (see Supplementary Methods).
Pathologic Investigation of Small Vessel Disease
Histologic Detection of Ischemic Tissue Damage, Hemorrhage, and Fibrin Accumulation
Sections were stained with hematoxylin and eosin, Perls Prussian blue, and Martius scarlet blue, using standard protocols to determine the presence of ischemic tissue damage, hemorrhage, and vascular accumulation of fibrin. Sections from each anatomic level were screened for the presence and absence of these histologic features.
Immunohistochemical Detection of Myelin, Axonal, Glial, and Vascular Integrity
Adjacent sections to those used for histologic analysis were immunostained using standard methods (see Supplementary Methods). The primary antibodies used were as follows: anti-myelin basic protein (1:300; Millipore, Watford, UK), anti-amyloid precursor protein (1:1,000; Millipore), antiionized calcium binding adaptor molecule 1 (Iba1; 1:750; Menarini, Wokingham, UK), anti-fibrinogen (1:5,000; Abcam, Cambridge, UK); antiglial fibrillary acidic protein (GFAP; 1:100; Life Technologies, Paisley, UK), anti-aquaporin 4 (AQP4; 1:100; Millipore); and collagen IV antibody (1:2,000 (Fitzgerald, MA, USA) or 1:100 (Millipore) for double labeling with GFAP/AQP4). DAPI (4′,6-diamidino-2-phenylindole) counterstain was used to identify cell nuclei in some experiments. To assess vascular density, images of the subcortex were acquired (x400) and the percentage of area stained by collagen IV was determined using ImageJ (NIH, Bethesda, MD, USA). Total vessel width was measured in images of the thalamus (x200). Images of GFAP-positive astrocytes and astrocytic endfeet in relation to the vascualture in the thalamus were acquired with a laser scanning confocal microscope (x200; Leica SP5, Leica Microsystems, Milton Keynes, UK). GFAP-positive cell bodies with distinct DAPI-positive nuclei were counted in a grid of defined dimensions. Percentage area stained by AQP4 and collagen IV as well as their respective colocalization (Manders plugin) was determined using NIH ImageJ (Bethesda, MD, USA) software after being equally thresholded across all sections for the respective antibody. The measure of AQP4 outwith vessels was defined as the difference between percentage area stained by AQP4 and collagen IV, respectively.
Quantitation of Claudin-5 Protein
Enzyme-linked Immunosorbent assay (Cusabio) was used to quantify Claudin-5 protein in vessel-enriched fractions (see Supplementary Methods) following the manufacturer's instructions.
Statistical Analysis
For imaging studies, the brain volumes between sham and hypoperfused groups and the brain lesion volume between 1- and 6-month hypoperfused mice were compared using unpaired
RESULTS
Progression of Radiologic Features and Pathologic Correlates of Small Vessel Disease
All T2-weighted MRI scans were screened for the presence of hyperintense (Figure 1A) and hypointense (Figure 1B) lesions. These radiologic features were confirmed as areas of primary ischemic tissue damage (Figure 1C) and hemorrhagic lesions (Figure 1D) in histologic sections.
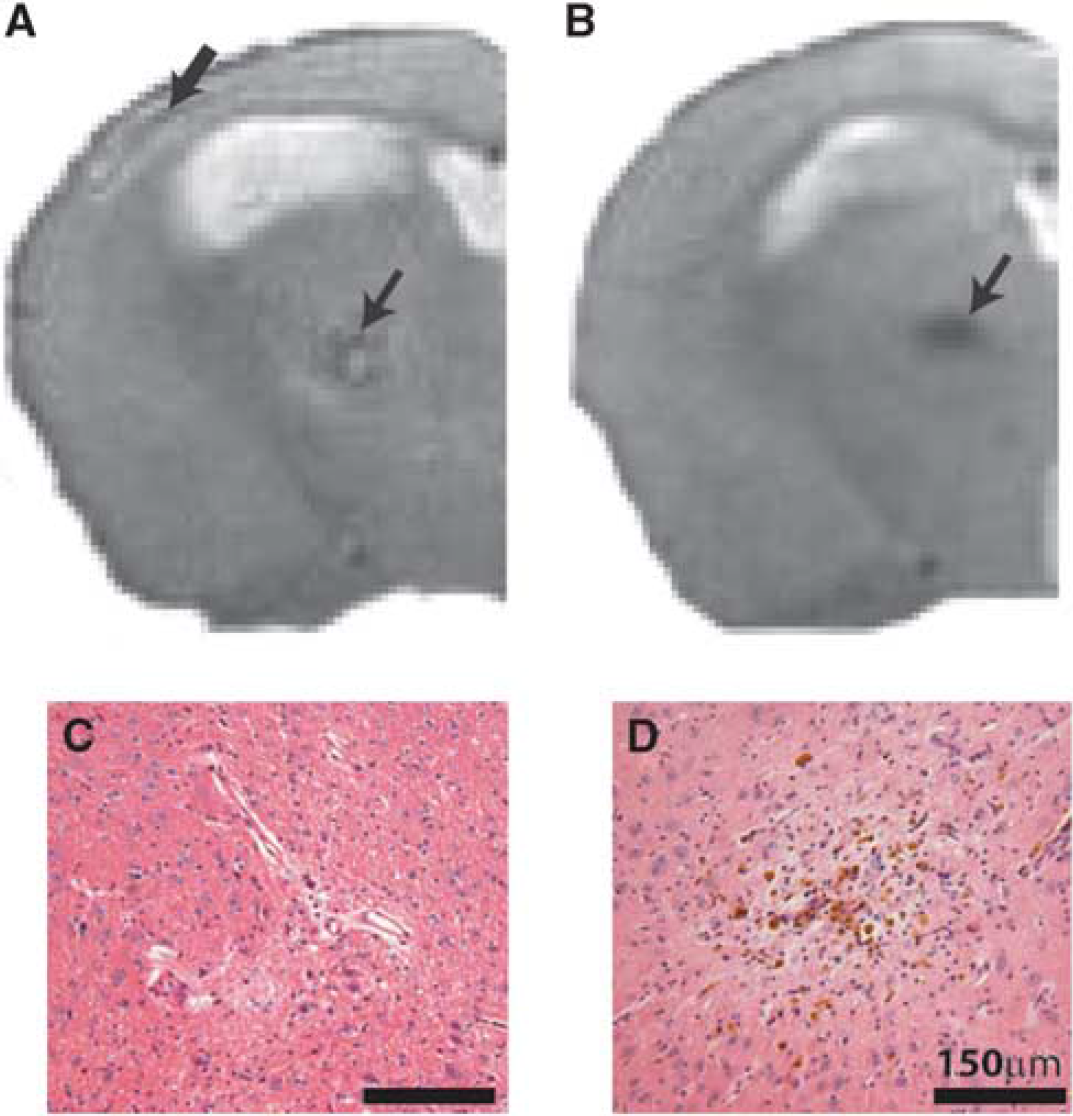
Radiologic features observed on T2-weighted structural magnetic resonance imaging (MRI). Representative images show hyperintense signal in the cortex (A; thick arrow) and subcortex (thalamus) (A; small arrow), and hypointense signal in the subcortex (B; arrow) of 6-month hypoperfused mice (slice taken −1.2 to −2.4 mm from Bregma). Parallel pathologic assessment identified ischemic-like lesions associated with hyperintense features (
Ischemic and Hemorrhagic Lesions
To examine the progression of lesion load and distribution with increasing durations of hypoperfusion, T2-weighted scans were investigated for the presence and anatomic location of primary ischemic and hemorrhagic lesions at 1 (
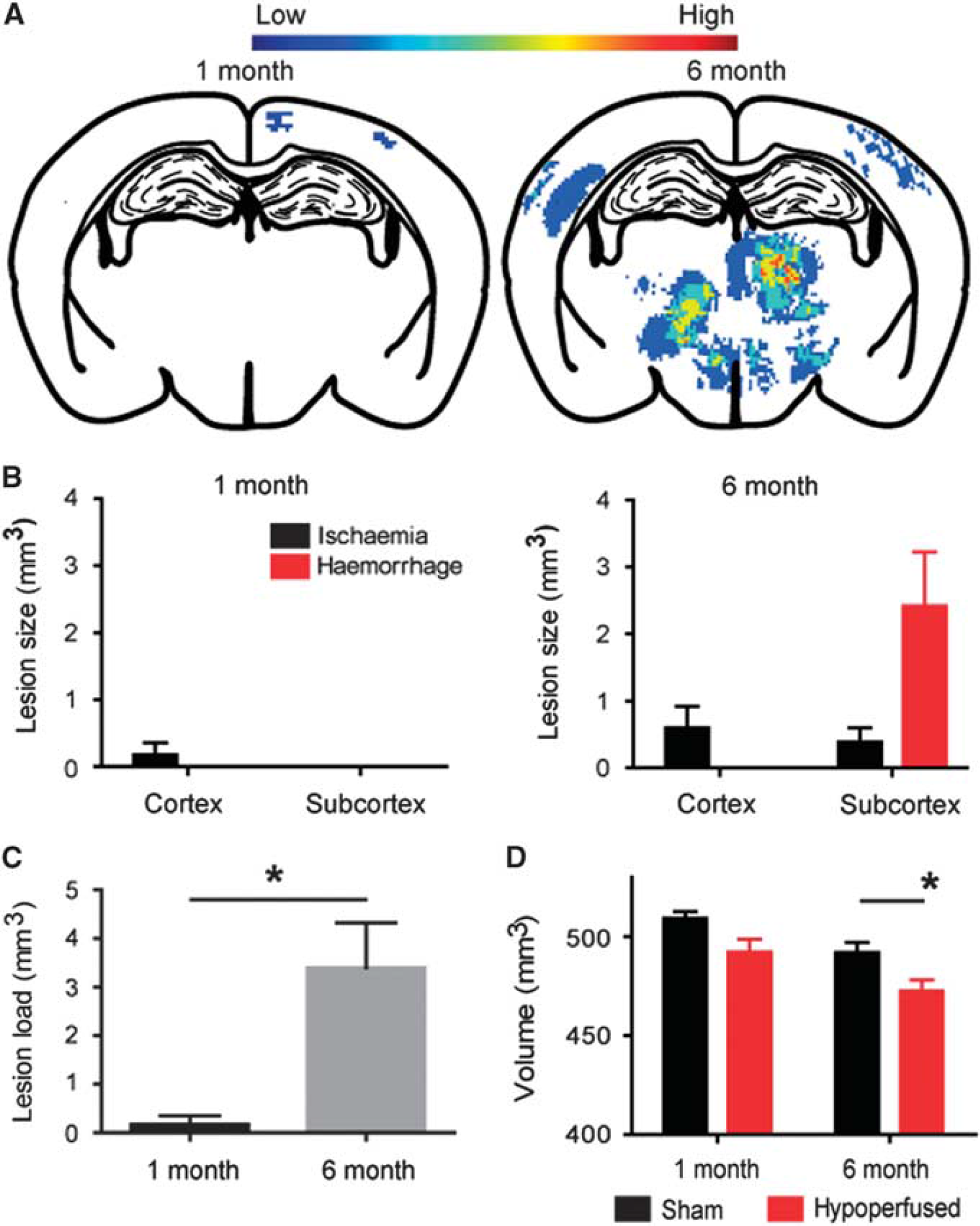
Progression of pathology in response to cerebral hypoperfusion. Heat maps depicting lesion load in the hypoperfused mice after 1 (
Brain Atrophy
To further explore progressive changes with increasing duration of hypoperfusion, the brain volume was measured on MRI. There was no significant difference in brain volume between sham and hypoperfused mice after 1-month hypoperfusion (Figure 2D; P>0.05). However, sustained hypoperfusion for 6 months resulted in a significant decrease in brain volume compared with sham mice (Figure 2D;
White Matter Disruption
Previously, we identified white matter disruption after 1 month of hypoperfusion
21
and aimed to determine the impact of 6 months of hypoperfusion on white matter integrity using MRI. We had shown previously that white matter changes primarily occur in the corpus collosum at the slice corresponding with y1.2 to −2.4 mm from Bregma. Table 1 provides a summary of white matter disruption and subcortical alterations in 6-month hypoperfused mice (
Gliovascular Alterations
Collagen IV immunostaining was used to determine whether changes in vascular basement membrane structure may occur subcortically in response to hypoperfusion at 1 month (
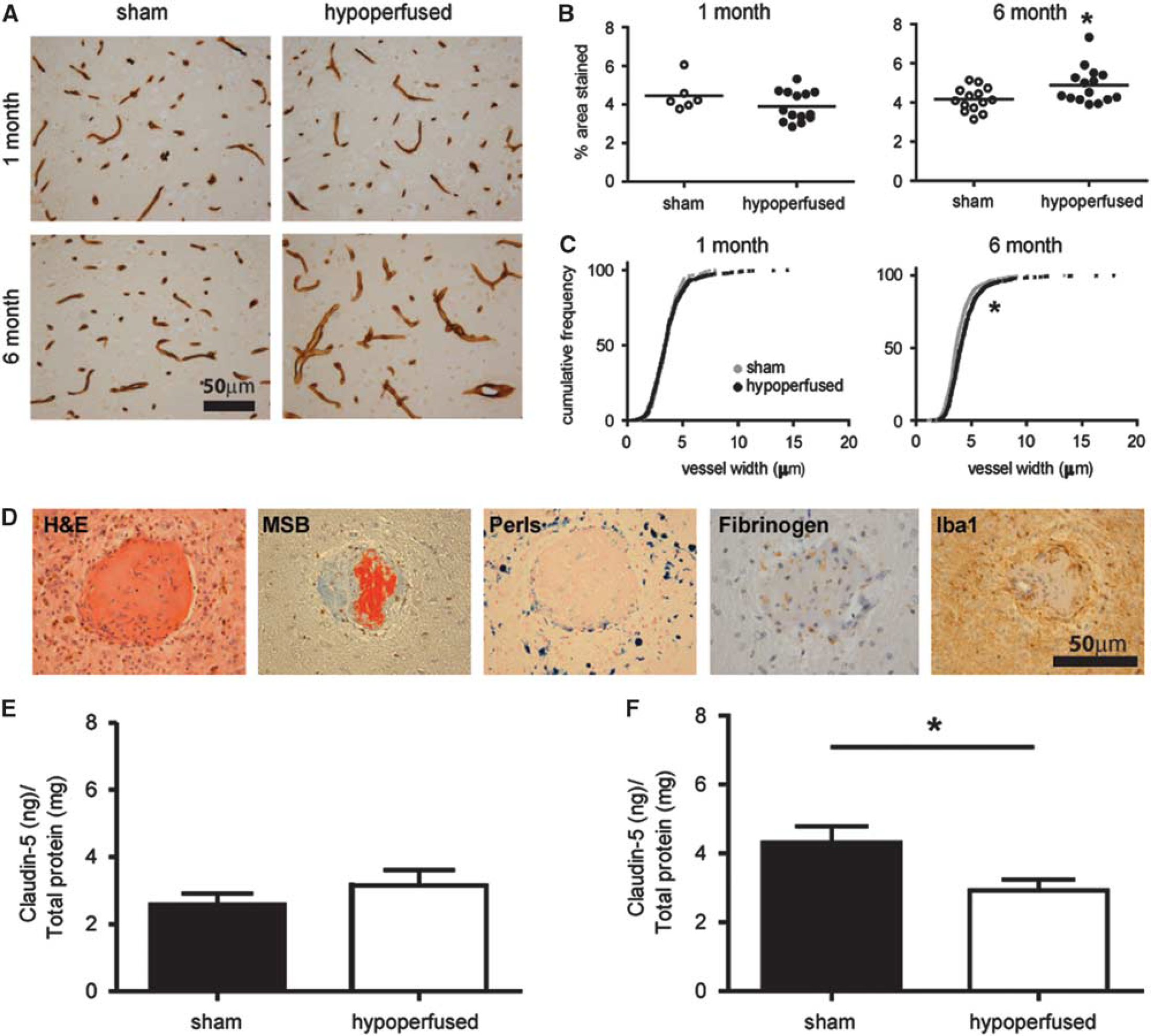
Pronounced vascular alterations and blood–brain barrier (BBB) breakdown. Collagen IV staining in sham and hypoperfused mice after 1 month (
Further investigation of the underlying mechanisms associated with the vascular pathology in 6-month hypoperfused mice was undertaken with a range of histologic markers (hematoxylin and eosin, Martius scarlet blue, Perls; Figure 3D). Notably, this revealed the presence of fibrin within the vascular wall, in subcortical areas, of 12 out of the 20 hypoperfused mice. The presence of hemosiderin-laden macrophages surrounding the vascular lesions (Figure 3D) was identified in all of the mice with evidence of fibrinoid necrosis. There was also positive Perl's staining present in an additional four hypoperfused mice. Fibrinogen accumulation within the parenchyma was also observed supportive of BBB breakdown (Figure 3D). A pronounced microglial response surrounding the vascular lesions was also detected (Figure 3D).
To further investigate whether the BBB may be progressively disrupted in the model, the levels of Claudin-5, a component of tight junctions within the BBB, were determined by enzyme-linked immunosorbent assay at 1 month (
As there is a close relationship between the vasculature, BBB, and astrocytes, we hypothesized that these prominent vascular changes would be associated with astrocytic changes. GFAP-positive astrocytes were counted within the thalamus after 1 month (
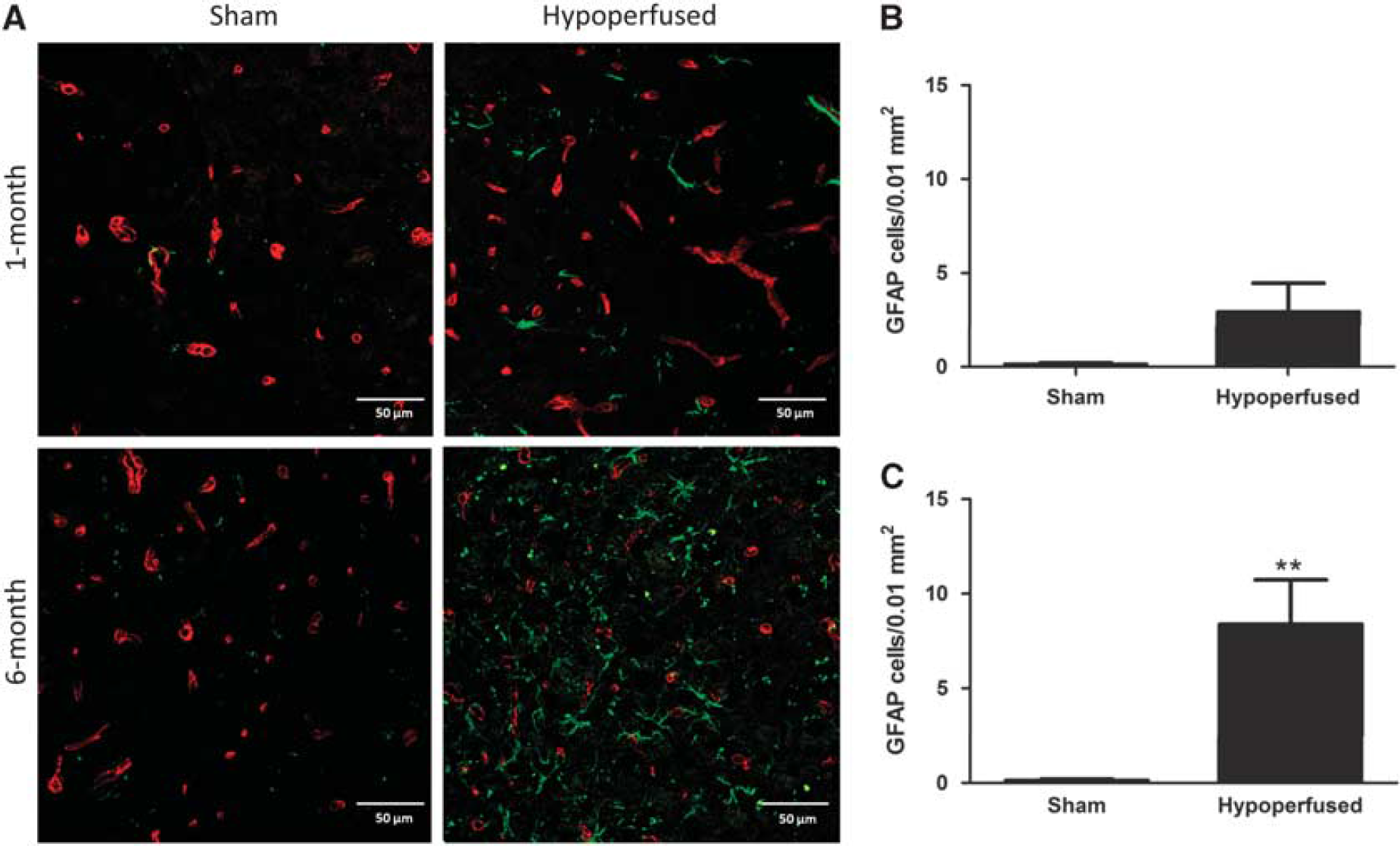
Increased astrocyte number after long-term hypoperfusion. Representative images of glial fibrillary acidic protein (GFAP) (green) and collagen IV (red) double immunostaining in the thalamus of sham and hypoperfused mice after 1 month (
To better understand potential dynamic changes in the gliovasculature, alterations in the astrocytic endfoot protein AQP4 was investigated. The mean percentage area (± s.e.m.) covered by AQP4 was not significantly different between shams or hypoperfused mice, respectively, after either 1 month (15.2±4.3 versus 15.2±2.8) or 6 months (8.4±2.9 versus 20.3±5.3) of hypoperfusion, although a trend toward an increase at 6 months was observed (
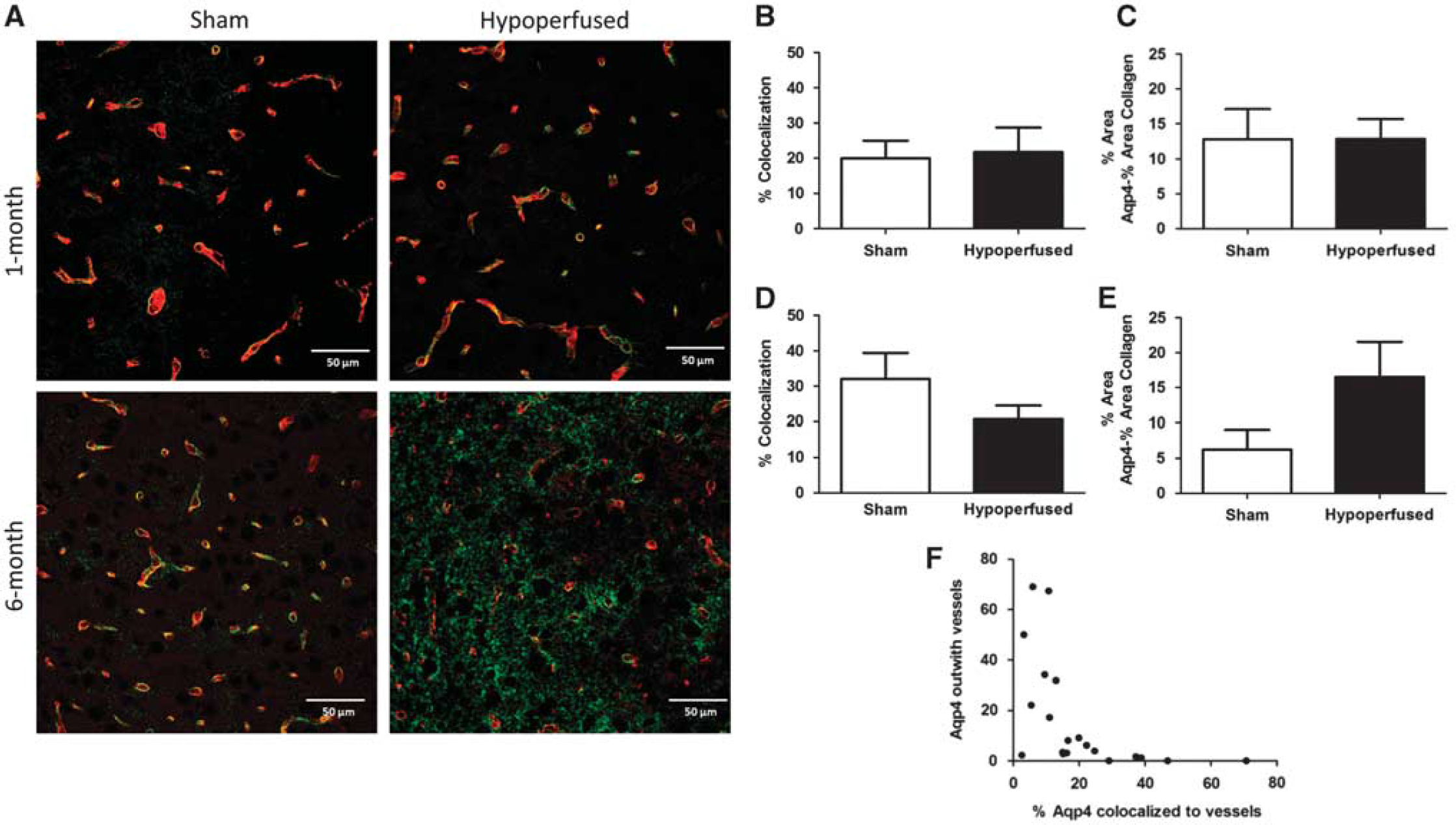
Displacement of aquaporin 4 (AQP4) after long-term hypoperfusion. Representative images of AQP4 (green) and collagen IV (red) double immunostaining staining in the thalamus of sham and hypoperfused mice after 1 month (
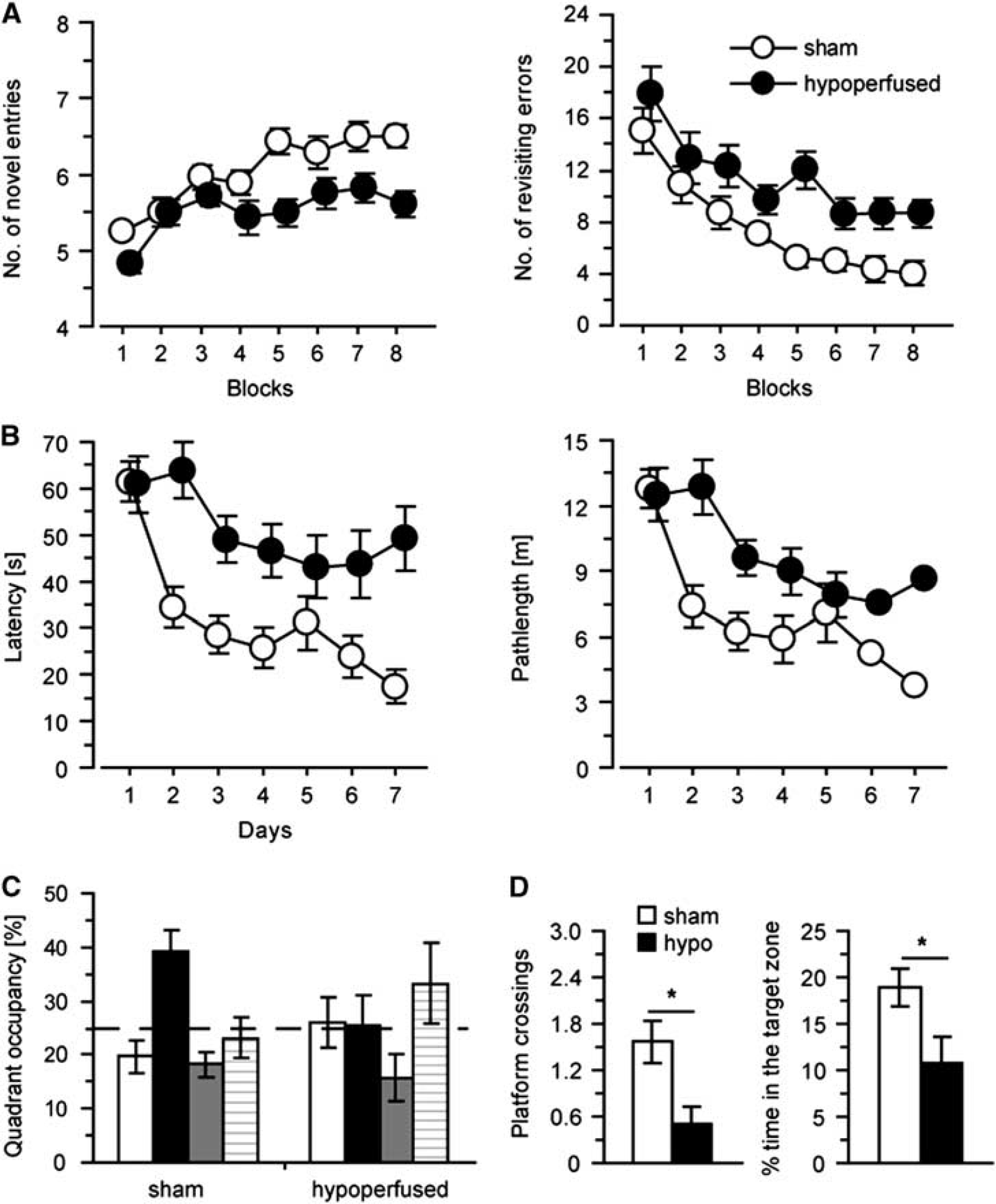
Cognitive impairments after long-term hypoperfusion (
Cognitive Assessment
DISCUSSION
Despite the increasing burden of SVD, progress towards understanding the pathophysiology and development of treatment strategies has been hampered, in part, because of the lack of relevant animal models. Here, we have evaluated a mouse model,19,20 which recapitulated some radiologic and pathologic features of SVD as well as impairment in spatial working and reference memory. Investigation of the associated pathologic correlates indicated prominent disruption of the gliovasculature including BBB breakdown.
To date, several animal models have been developed, which mimic various pathologic features of SVD.11,12 Rodent models of hypoperfusion have mainly been studied to provide mechanistic insight into the pathophysiology of white matter lesions, which are a manifestation of SVD. Previously, we demonstrated that a short, 1-month period of cerebral hypoperfusion causes subtle alterations in white matter integrity as determined by immunohistochemical approaches20,22 and DT-MRI. 21 The present study illustrates that with sustained carotid stenosis, over 6 months, there is a progressive deterioration in white matter integrity. The radiologic findings are similar to those previously reported by our group, whereby FA and MTR are altered after short-term hypoperfusion. In the present study, which examines a longer period of carotid stenosis, the magnitude of the FA/MTR changes are more pronounced, which is in agreement with our findings of significantly increased radiologic features of SVD and reduced brain volume after prolonged hypoperfusion. Consistent with our previously published data, 21 there was no change in MD detected in our model, which is somewhat unexpected as MD is a marker of white matter damage in SVD in humans. 27 A recent study has also shown that other DTI measures (AD and RD) may be sensitive to the effects of cerebral hypoperfusion, and for future studies, these additional measures would be advisable to study. 28 Notably, in the same animals in which imaging was conducted, we observed myelin and axonal disruption alongside a prominent inflammatory response.
In addition to white matter pathology, sustained hypoperfusion leads to widespread vascular disruption and progressive cognitive decline. In particular, there is a marked increase in vessel width and collagen IV density, fibrinoid necrosis, and hemorrhage, features that are all absent from short-term hypoperfused or age-matched controls. Disrupted penetrating arteries and arterioles are common in SVD, often characterized as lipohyalinosis with or without evidence of fibrinoid necrosis. The demonstration of vascular-related features, which are similar to human SVD, 29 including vessel wall disruption, collagen IV accumulation, fibrin deposition, and cavitated lacunar-type infarcts, highlights the relevance of this mouse model as a tool to explore underlying mechanisms of vascular disease. Such focal vascular disruptions were often associated with hemosiderin-laden macrophages, consistent with microbleeds. The presence of overt vascular disruption and BBB breakdown in our model is in agreement with clinical observations, suggesting that BBB disruption is a key mechanism involved in pathogenesis of human SVD. 9 To investigate the role of vascular integrity in disease progression, we measured collagen IV density and vessel width after acute and long-term hypoperfusion. Interestingly, the density of collagen IV staining, as a marker of basement membranes, was not altered after 1-month hypoperfusion, but was significantly increased at the longer time point. To further examine the progression of BBB disruption with increasing hypoperfusion, the levels of claudin-5, a transmembrane protein present at the tight junctions, was determined. Notably, 1 month of hypoperfusion had no effect in the levels of the protein, but a significant decrease was found after 6 months. Fibrinogen accumulation wihin the parenchyma was also noted at 6 months of hypoperfusion, supportive of BBB breakdown. Vascular-related changes were predominantly observed in subcortical regions, particularly the thalamus, and also in the caudate with long-term carotid stenosis. Recently, it has been shown that chronic cerebral hypoperfusion leads to vascular wall remodeling. 28 These changes may potentially lead to sheer stress in the vessels in the anterior and posterior circulation, subsequent endothelial alterations, and BBB breakdown.
Coincident with cerebrovascular changes, long-term hypoperfusion also led to marked alterations in glia. We found an increase in GFAP-positive astrocytes within the thalamus of 6-month hypoperfused mice, indicative of reactive gliosis, which is commonly observed in the brains of individuals with SVD. 8 Reactive astrocytosis was accompanied by a mislocalization of AQP4 from the vasculature to the parenchyma. AQP4 is a water channel typically confined to the astrocytic endfeet of vascular-associated astrocytes, and although the physiologic role of AQP4 is not clear, it is reported to maintain osmotic balance. Loss of AQP4 polarization to the perivascular endfeet has been reported to occur after ischemic events including the development of microinfarcts.30,31 AQP4 may act to remove edema after injury, 30 but this change in polarity can also be accompanied by disruption of the BBB. 32 In the present study, BBB alterations were coincident with reactive astrocytosis and AQP4 polarization. Whether these changes are causative of BBB disruption or a direct reaction is not entirely clear. The precise mechanism leading to these gliovascular alterations remains to be identified. Our previous work showed that early in response to hypoperfusion there is activation of inflammatory and angiogenic pathways coincident with disruption of axon–glial integrity. 22 Others have reported an involvement of the renin–angiotensin system28,33 and oxidative stress pathways. 28 It should also be noted that the microcoils themselves may cause an additional inflammatory stimuli contributing to the degenerative changes. Overall, the present study adds to these studies and highlights the detrimental effects of longer-term hypoperfusion on gliovascular alterations, identifying a potential mechanism of disruption in this model, which may allow targeted intervention studies.
In addition, sustained hypoperfusion resulted in significant impairments both in spatial working memory and in acquisition and memory deficits in a spatial reference memory task. Previously, we reported, after 1 month of hypoperfusion, that working memory was impaired while reference memory remained intact, and suggested that this was a result of the disruption of the frontal cortical circuitry.
20
The emergence of deficits in both working and reference memory with long-term hypoperfusion likely reflects the presence of white and gray matter pathology including brain atrophy. These findings are in agreement with a previous study, which examined the effects of long-term hypoperfusion using a battery of behavioral tasks and similarly reported spatial reference and working memory impairments.
24
This study of Nishio
A major strength of the current study is the demonstration of ultrastructural alterations in white matter and subcortical nuclei using quantitative MRI biomarkers. Human data have indicated that such measures may more accurately model disease burden and reflect degree of cognitive decline compared with overt radiologic features. 34 In agreement with this, we observed multiple subtle pathologic alterations in hypoperfused mice, which individually were not sufficient to be visible on T2-weighted MRI screening, although global brain atrophy was very evident, suggesting that while critical for SVD diagnosis, especially in humans where acute pathologic confirmation is often impossible, the need for more advanced imaging methods is required to detect the full spectrum of disease presentation.
As yet, the mechanisms causing predisposition to SVD are not known. Vascular risk factors including diabetes and hypertension were reported to have a key role in the epidemiology of SVD, with hypertension thought to be the most important modifiable risk factor. However, it has since been shown that all vascular risk factors combined account for < 2% of the variance in white matter hyperintensities. 2 Furthermore, despite major advances in blood pressure control, the incidence of SVD remains high, and this has failed to prevent the progression of white matter hyperintensities or lacunar stroke, 35 suggesting that other risk factors may be crucial to disease progression. Recent evidence from animal models also suggest that hypertension is not the key driver of white matter changes.36,37 Our results show that some radiologic, pathologic, and cognitive features of human SVD can be recapitulated in a mouse model by common carotid stenosis. Our findings also provide evidence that hypoperfusion may precipitate SVD-like disease culminating in a progressive brain and gliovascular pathology. The model may provide opportunities for testing novel treatment strategies for this highly prevalent disabling condition.
DISCLOSURE/CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.