
Abstract
Transient suppression of peripheral immunity is a major source of complication for patients suffering from ischemic stroke. The release of Arginase I (ArgI) from activated neutrophils has recently been associated with T-cell dysfunction in a number of pathologies. However, this pathway has not been previously explored in ischemic stroke. Using the murine model of transient middle cerebral artery occlusion, we explored effects of stroke on peripheral T-cell function and evaluated the role of neutrophils and ArgI. Stimulation of splenic T cells from post-stroke animals with anti-CD3/CD28 resulted in decreased proliferation and interferon-γ production when compared with sham-surgery controls. Flow cytometric analysis of intrasplenic leukocytes exposed the presence of a transient population of activated neutrophils that correlated quantitatively with elevated ArgI levels in culture media.
INTRODUCTION
Despite intense research, interventions that improve outcome following ischemic stroke remain elusive. Intensive care remains a major ongoing source of cost and stroke-associated morbidity for affected patients, who often require several weeks of aggressive post-stroke medical management. During this time, infection is a major source of secondary complication. 1 Data has suggested incidence of post-stroke infection in 25% to 65% of patients, a phenomenon that notably appears to be highly correlated with severity of stroke.1–3 Aside from prolonging intensive care unit stays, secondary infection may worsen overall outcome.4,5 Therefore, delineation and targeting of stroke-associated factors that induce suppression of immunity may be critical for improving outcomes.
Although it has been generally accepted that some degree of stroke-associated immunosuppression exists, specific mechanisms leading to this effect remain to be determined. The most commonly reported immunological deficits associated with ischemic stroke are seen within the cellular immune compartment, which include lymphopenia and decreased T-cell function in response to mitogenic stimulation.6–9 We have noted that stroke-induced abnormalities of cellular immunity mirror those seen in patients with malignant brain tumors, in whom we recently identified a novel axis of peripheral immunosuppression involving the release of the enzyme Arginase I (ArgI) from circulating activated neutrophils. 10 ArgI is well known to metabolize L-arginine to L-ornithine and urea and has recently been observed to be a key regulator of cellular immunity in both mouse and man.11–13 In mice, ArgI expression has been described in a range of myeloid cells, most notably myeloid-derived suppressor cells (MDSCs). 14 In humans, ArgI appears to be constitutively expressed in neutrophil granules (from which it is liberated during inflammation) and can be induced in a range of monocyte-lineage cells. 15 In contrast, ArgI expression in murine MDSC has been primarily described to be restricted to the intracellular compartment. 16 ArgI-mediated depletion of L-arginine suppresses T-cell function and has been proposed as a central pathway of immunosuppression in a range of pathological conditions. Mechanistically, L-arginine depletion results in an inability of activated T cells to express the CD3ζ subunit of the T-cell receptor, a critical component of signal transduction and proliferation following antigenic activation.11–13
Based on these observations, we hypothesized that neutrophil activation and subsequent peripheral release of ArgI may have a role in stroke-related suppression of cellular immunity. To test this hypothesis, we used an established model of murine stroke to explore phenotypic and functional changes in cellular immunity potentially associated with ArgI activity.
MATERIALS AND METHODS
Mouse Model of Stroke
All experimental protocols were approved by the Institutional Animal Care and Use Committee and conformed to the National Institutes of Health guidelines for the care and use of animals in research. Experiments were written in accordance with the ARRIVE guidelines. Following isoflourane anesthesia, male C57Bl/6 mice at 8 to 10 weeks of age were subjected to 60 minutes middle cerebral artery occlusion (MCAO) via the intraluminal suture method as we have previously described.17,18 Adequacy of MCAO was confirmed by laser Doppler flowmetry measured (> 70% drop required for inclusion) over the ipsilateral parietal cortex in all mice. Groups of randomized and masked animals were killed at 4 and 10 days after MCAO and tissues were harvested for analysis. Successful occlusion was confirmed by slicing each brain into five 2-mm-thick coronal sections and placing each slice in a 1.2% solution of 2,3,5-triphenyltetrazolium chloride (Sigma, St Louis, MO, USA) for 30 minutes at 37°C to identify infarcted tissue. Sham surgeries were performed by exposing the external carotid artery without insertion of the filament. Tissues from sham mice were collected 4 days post surgery. All experiments were performed at least in triplicate with 4 to 6 animals per group. A total of 25 sham and 68 MCAO surgeries were performed, resulting in a survival rate of 75% (51/68) in the MCAO groups. When possible, the experimenter performing immunology experiments was masked to group.
Isolation of Splenic Leukocytes and Purification of Low-Density Cells
Spleens collected from MCAO or sham mice were placed in unsupplemented RPMI (Invitrogen, Grand Island, NY, USA), mechanically disrupted into single-cell suspensions, and filtered through a 70 μm strainer. Splenic cells were resuspended into phosphate-buffered saline (Invitrogen) and poured over an equal volume of Ficoll Histopaque 1077 (Sigma). The low-density cell layer (which typically includes only mononuclear cells) was collected after centrifugation at 1,650 r.p.m. for 30 minutes. The resulting splenic mononuclear cell (SMC) layer was subsequently used for cell marker analysis by flow cytometry and T-cell functional assays.
Flow Cytometry of Immune Cell Populations
Splenic mononuclear cells were stained for myeloid lineage and T-cell markers, along with the appropriate isotype controls, to identify immune cell populations. Murine myeloid cell-specific antibodies included FITC-anti-Ly6C, PE-anti-Ly6G, and PerCP-Cy5.5-anti-CD11b from BD Biosciences (San Diego, CA, USA) and PE-Cy5-anti-MHCII, and APC-anti-F4/80 from eBioscience (San Diego, CA, USA). Myeloid cell populations were delineated using the following phenotypes: CD11b+Ly6GloLy6Chi for monocytes, CD11b+Ly6GhiLy6Clo for granulocytes, and MHCII+F4/80+ for macrophages. T-cell markers included FITC-anti-CD8, PE-anti-CD3, and PE-Cy5-anti-CD4 from BD Biosciences. The fluorescently labeled antibodies were added to 2.5 × 10 5 cells in Stain Buffer (BD Biosciences) and incubated on ice at 4°C for 30 minutes before washing and performing an analysis on a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Intracellular staining for CD3ζ was performed by resuspending SMC into 100 μL of Stain Buffer, staining with FITC-anti-CD3 (BD Biosciences) for 20 minutes, transferring to 100 μL of Cytofix/Cytoperm buffer (BD Biosciences) for 25 minutes, and staining with PE-anti-CD3ζ(CD247) (eBioscience) in 100 μL of Stain Buffer for 20 minutes before flow cytometry.
Flow data was analyzed using the FlowJo software program (Treestar, Ashland OR, USA). Positivity for the various markers was determined based on comparison with isotype controls.
T-Cell Functional Assays
Splenic mononuclear cell were plated at a concentration of 1 × 10 5 cells per well in 96-well round-bottom plates with 200 μL of RPMI supplemented with 10% fetal bovine serum (Invitrogen) and 1% Penicillin–streptomycin (Invitrogen). T cells were stimulated using 1 × 10 5 anti-CD3/CD28 microbeads (Dynabeads, Invitrogen). Media were collected at 48 and 72 hours and tested for interferon-γ (IFN-γ) concentration by enzyme-linked immunosorbent assay (ELISA; ThermoScientific, Rockford, IL, USA). To evaluate mitogen-induced T-cell proliferation, BrdU (BD Biosciences) was added to mitogen-stimulated SMC cultures at a concentration of 10 μmol/L. Following 24 hours of stimulation in the presence of BrdU, SMC were harvested, washed, and stained with PE-anti-CD3 for 20 minutes, fixed and permeabilized for 25 minutes with Cytofix/Cytoperm buffer, treated with DNAase (Sigma) for 1 hour to expose incorporated BrdU, and stained with APC-anti-BrdU (BD Biosciences) for 20 minutes before analysis by flow cytometry. The T-cell population was initially identified by positive expression of CD3 (Figures 1B and 1C, left panels) and the percent of BrdU+ cells within that gated population (Figures 1B and 1C, right panels) was recorded.
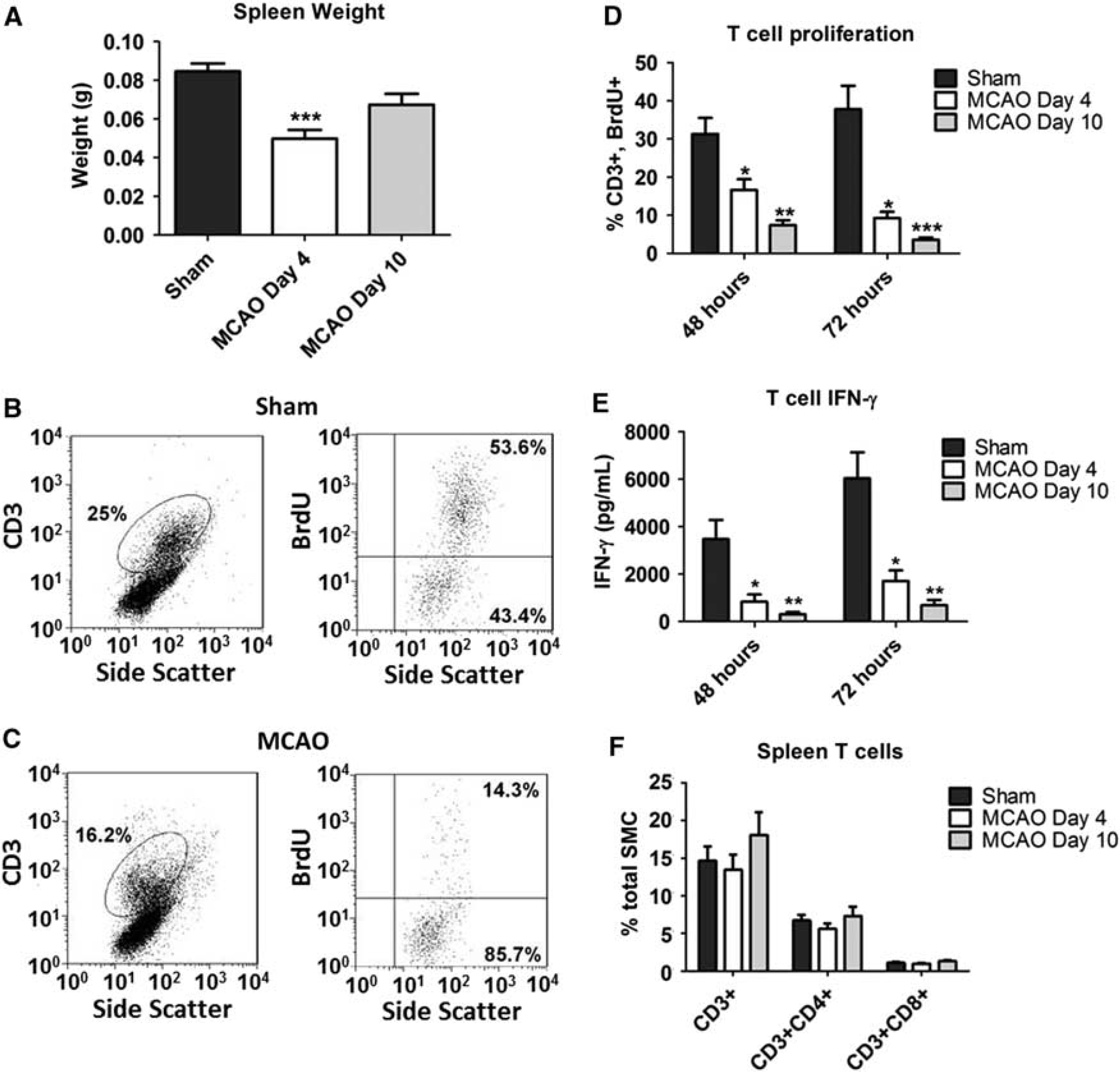
MCAO is associated with splenic atrophy and T-cell hypofunction. (
Mouse Neutrophil Degranulation and Measurement of ArgI Release
To confirm the potential for murine neutrophils to shift to the low-density layer of a Ficoll density gradient following activation and degranulation, single-cell splenocyte preps were generated (as above) from unmanipulated animals. These preps were mixed with 1 μmol/L N-formyl-methionyl-leucyl-phenylalanine (fMLP, Sigma) or left untreated for 20 minutes. Cells were then resuspended in phosphate-buffered saline and poured over an equal volume of Ficoll Histopaque 1077. Splenic mononuclear cell were collected and analyzed by flow cytometry as above to detect an increased frequency of neutrophils within the low-density fraction following
To explore the potential that ArgI could be released from preformed neutrophilic granules in the murine system, neutrophils were isolated from bone marrow of unmanipulated animals as described by Siemsen
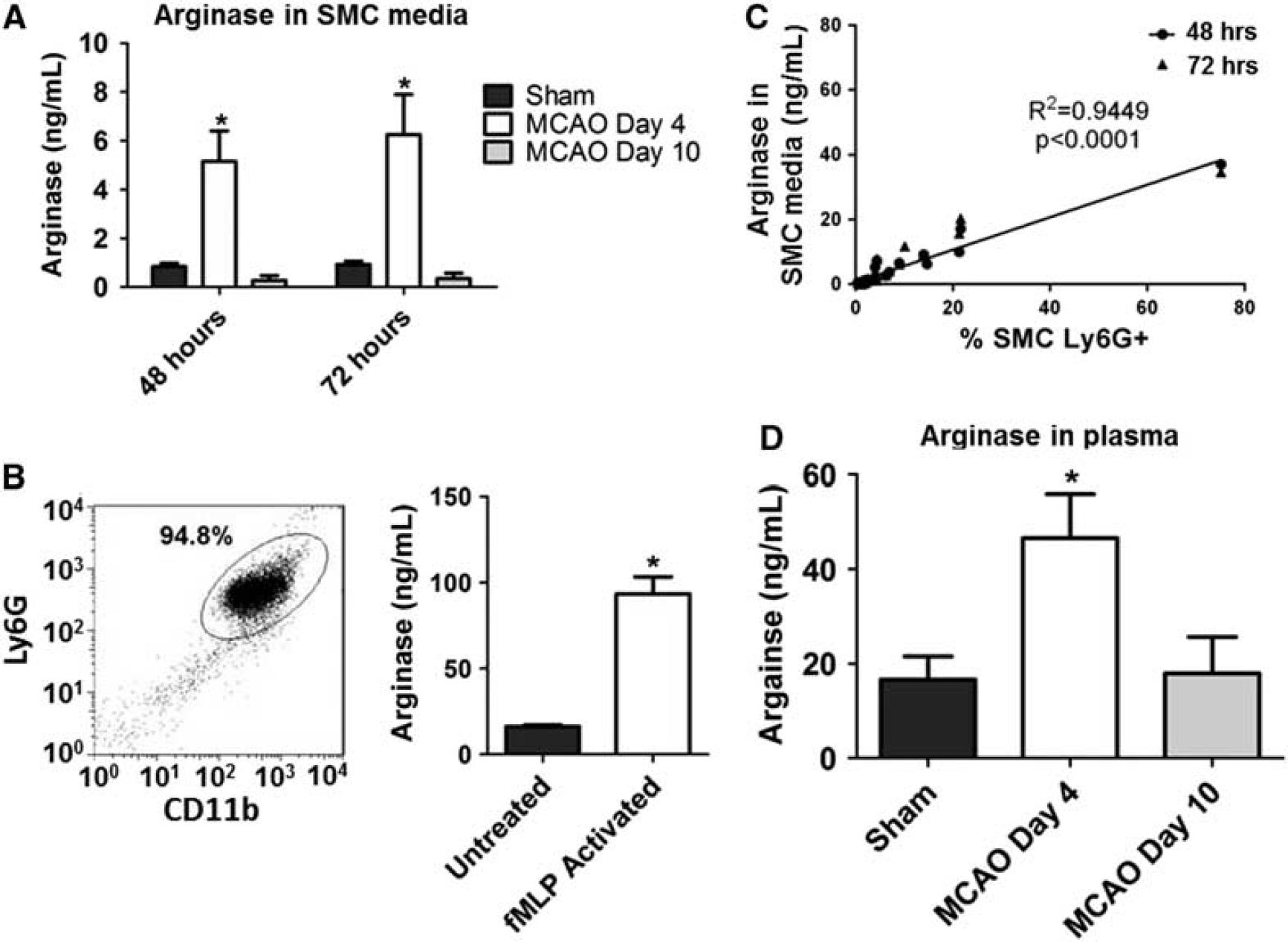
Release of ArgI and correlation with the frequency of activated neutrophils within spleens following MCAO. (
Arginase I was measured in media from T-cell functional cultures, SMC fractions, or purified bone marrow neutrophil cultures by ELISA (Uscn Life Science, Wuhan, China). Arginase I was also measured in plasma collected from sham or MCAO animals by ELISA. To collect plasma, whole blood was collected into sodium citrate anticoagulant and centrifuged for 10 minutes at 1,200 r.p.m. Plasma was diluted 1:10 before analysis by ELISA.
L-Arginine Supplementation in vitro
Splenic mononuclear cell preps from MCAO-treated animals were used for T-cell functional analysis as described above. Culture media was supplemented with 4 μmol/L L-arginine. Cultures were incubated for 48 or 72 hours before cells were harvested for BrdU proliferation analysis and media was collected for IFN-γ measurement as outlined above.
Statistical Analysis
All data are presented as mean ± s.e.m. Multi-group analyses were conducted using non-parametric one-way analysis of variance (Kruskal–Wallis) followed by Dunn's
RESULTS
Experimental Stroke Induces Splenic Atrophy and T-Cell Dysfunction
As has been previously reported,6,8,20 we noted a significant decrease in the splenic mass of animals after MCAO (Figure 1A). Splenic atrophy appeared to be temporally associated with the onset of stroke, as we observed partial reconstitution of weight by day 10 (Figure 1A).
T-cell function in animals after experimental stroke was quantitatively evaluated through
Ischemic Stroke Induces Accumulation of Activated Neutrophils in the Spleen
To test our hypothesis that experimental stroke results in the activation of peripheral neutrophils, we purified SMC as described above and analyzed the frequency of various myeloid-lineage populations using flow cytometry. We observed a significant relative expansion of CD11b+ cells within atrophic spleens by 4 days after MCAO (Figures 2A and 2B, left panels). Critically, further investigation of the expression of myeloid-lineage markers demonstrated that the CD11b+ population was predominantly Ly6GhiLy6Clo (Figures 2A and 2B, right panels), confirming a neutrophilic phenotype. The expansion of this population appeared to be temporally correlated with the induction of MCAO, as the frequency of intrasplenic neutrophils approached baseline levels by 10 days after stroke (Figure 2C). In contrast to the expansion of intrasplenic neutrophils, we observed no change in the frequency of other myeloid-lineage cells that have previously been associated with immunosuppression (Figure 2C).
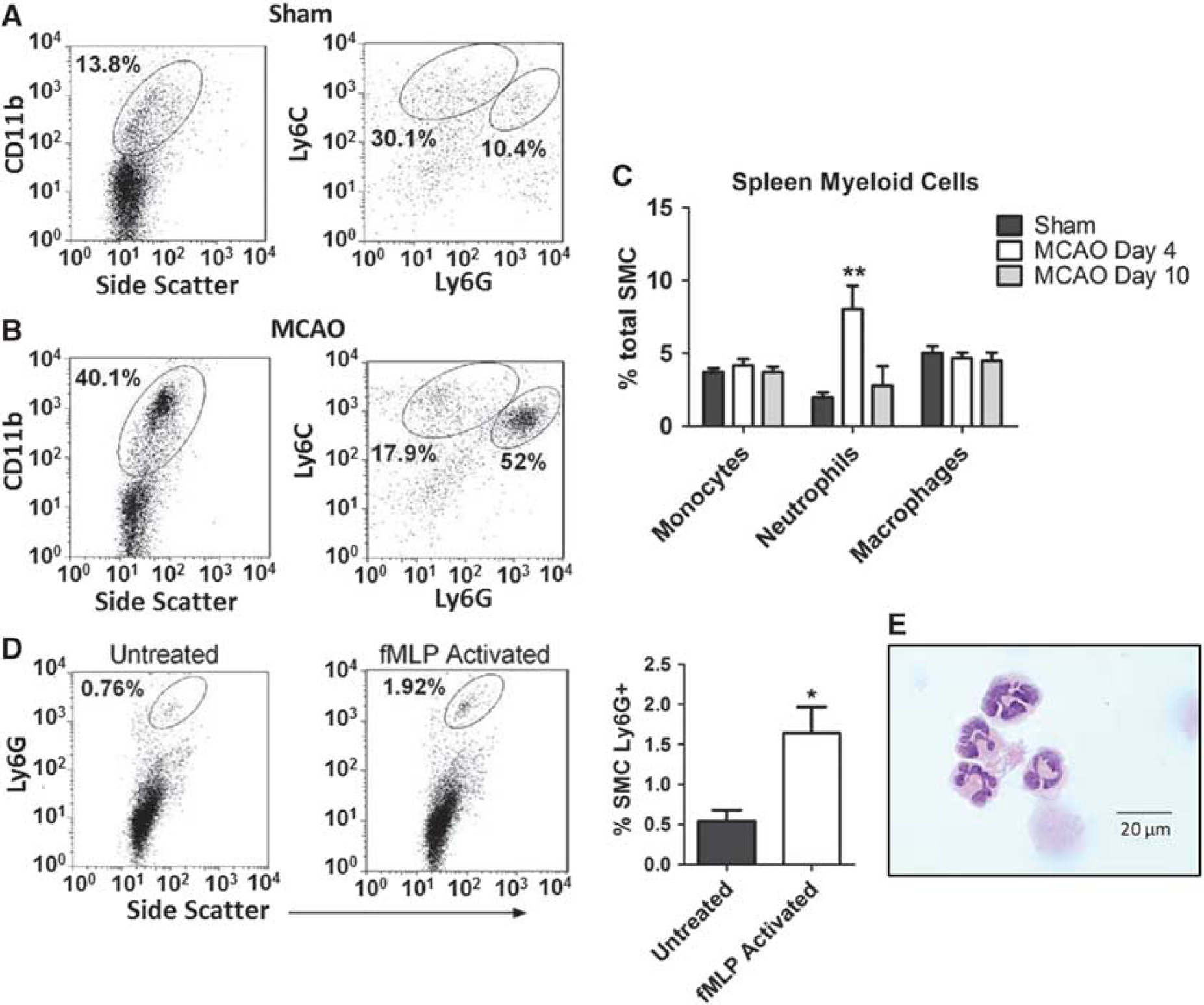
Spleens from MCAO mice harbor increased numbers of activated neutrophils. (
It is generally expected that Ficoll purification of leukocytes should separate high-density neutrophils from lower-density mononuclear cell populations. We, and others, have previously reported that human neutrophils can ‘shift’ to the lower-density population after activation and degranulation.
10
However, this functional change has not been previously demonstrated within the murine system. To explore this possibility, neutrophils within splenic single-cell suspensions from unmanipulated animals were activated
Together, these data suggest that the increased frequency of neutrophils in the SMC fraction from animals following MCAO is because of
Activated Murine Neutrophils Release ArgI from Preformed Granules; Increased Levels of ArgI are Associated with Experimental Stroke and Reversible T-Cell Hypofunction
ArgI release from preformed granules following neutrophil activation has been widely described in humans.21,22 However, to our knowledge, a similar phenomenon has not been previously described in the murine system. To explore this possibility, murine neutrophils were isolated from bone marrow of unmanipulated mice (Figure 3B) and stimulated
To further support the hypothesis that activated neutrophils in post-MCAO animals release ArgI, we measured levels of the relevant protein in the media of SMC cultures from study animals. Figure 3A illustrates our seminal finding that experimental stroke is associated with a significant increase in the level of ArgI release from splenic leukocytes at day 4 following MCAO. Critically, we noted a return to baseline levels of ArgI by day 10, providing a temporal relationship to the onset of stroke, the extent of splenic atrophy, and most notably the relative accumulation of activated neutrophils within post-stroke spleens. The association between ArgI and neutrophil activation following experimental stroke was further strengthened through quantitative correlation of ArgI protein levels with the frequency of Ly6G+ cells within SMC from animals at day 4 following MCAO. As predicted, we noted an extremely strong correlation between ArgI protein concentration and the frequency of activated neutrophils (Figure 3C). In addition, to confirm that the increase in ArgI also occurs
To provide indirect mechanistic confirmation for the immunosuppressive effects of ArgI in stroke, we evaluated levels of CD3ζ expression on direct
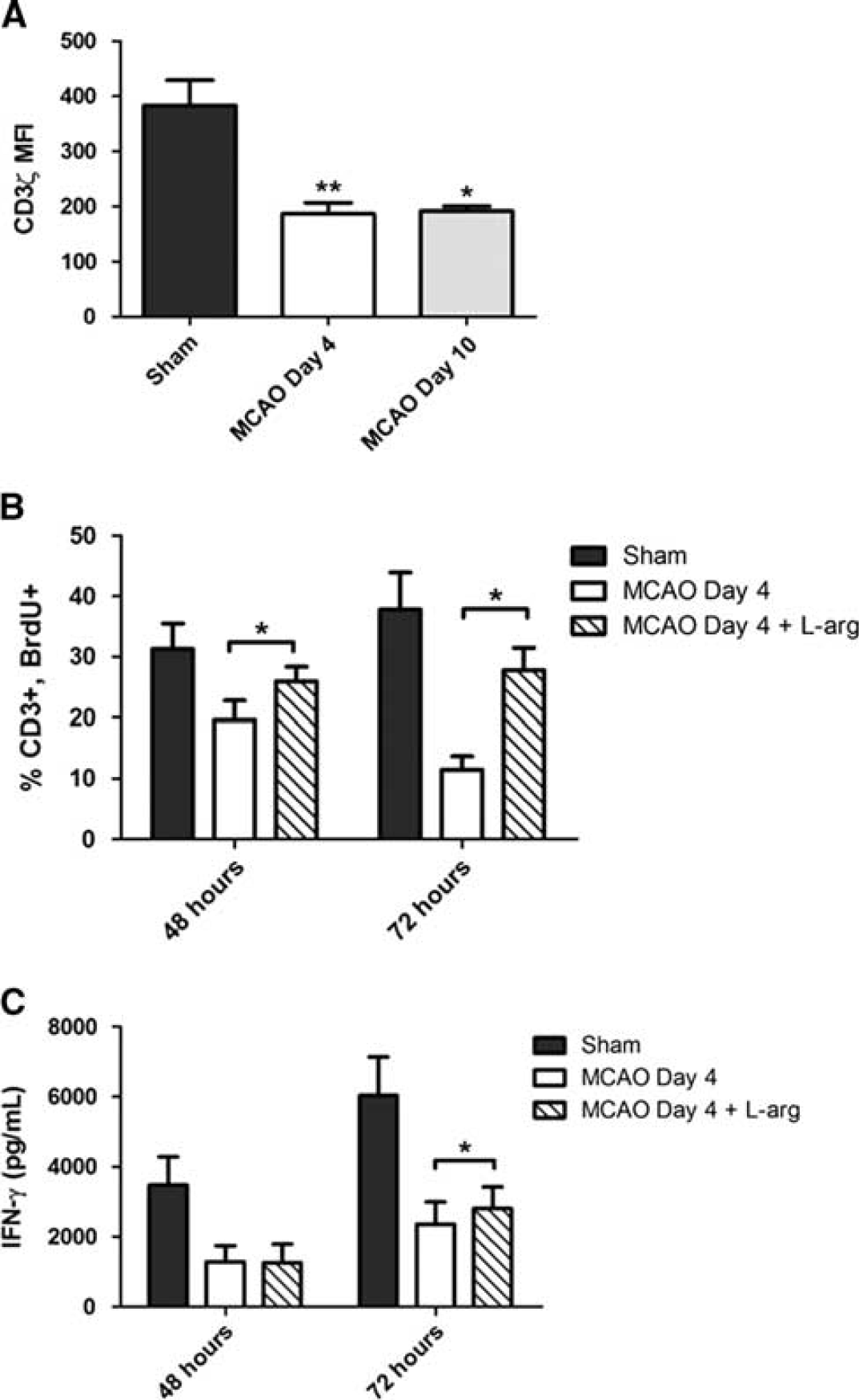
Splenic T-cell dysfunction following ischemic stroke correlates with decreased CD3ζ expression and can be rescued through L-arg supplementation. (
DISCUSSION
The number of stroke-related deaths in the US has decreased over the past decade, an improvement that can be primarily attributed to early medical intervention, dedicated stroke units, and more aggressive approaches for thrombolysis.23,24 However, intensive care of surviving patients remains a significant source of healthcare cost. As systemic infection in stroke patients is an independent risk factor for poor clinical outcome and may contribute to increased mortality rates,4,5 efforts to improve immune function in the days to weeks following stroke may provide significant gains in both time to discharge as well as overall outcome.
Here, we describe a novel pathway of stroke-associated immunosuppression, through which splenic atrophy and T-cell hypofunction are associated with the accumulation of activated neutrophils and ArgI release. The potential role for neutrophils in the suppression of cellular immunity has not been widely explored in mice, although these cells could be included under broad definitions of the MDSC. Although prior studies of murine MDSC have widely focused on ArgI as a mediator of cellular immunosuppression, the enzyme has generally been described to be active within the intracellular compartment rather than released into the extracellular environment.
14
To our knowledge, we are the first to describe the capacity for murine neutrophils to release ArgI from preformed neutrophilic granules following activation. This observation may have important implications for modeling the role of neutrophils in other pathologies where suppression of T-cell function occurs. Critically, we confirmed that post-stroke T-cell proliferation can be restored
In addition to restoration of T-cell proliferation, we also observed a significant increase in IFN-γ release at 72 hours post stimulation with L-arg supplementation. Though increased, IFN-γ levels were not restored to the level observed in T-cell cultures from sham animals. It has previously been shown that T-cell IFN-γ production is critical for decreasing susceptibility to infection in a mice following ischemic stroke.
8
Prass
There are certainly several caveats to be taken into consideration when evaluating these findings. As has been consistently demonstrated in many systems, immunosuppression is generally a multifactorial process. ArgI-mediated immunosuppression likely represents only one arm of a more global effect. Therefore, isolated therapeutic targeting of this pathway may offer only partial benefit, mandating continued exploration of additional sources of stroke-related immunosuppression. In addition, it remains unclear whether L-arg inhibition and/or the presence of excess L-arg will exert systemically beneficial or harmful effects outside of the immune system. Notably, L-arg serves as the substrate for various isoforms of nitric oxide synthase in the biosynthesis of nitric oxide, which has been widely explored in the pathophysiology of brain ischemia and related injury.
25
Therefore, it would be important to consider the acute effects of excessive nitric oxide in stroke outcome. Our data indicates that Arginase levels are elevated for at least 4 days after stroke, with a concurrent decrease in T-cell function which can be partially rescued
For these reasons, greater knowledge of the peak influence and subsequent resolution of the ArgI-specific effect on immunity, as well as preclinical modeling of targeting this pathway in the setting of illness will be essential for driving clinical decision making. Ultimately, confirmation of these early experimental results within the human system may lead to the development of new therapeutic options for augmenting immunity and improving outcomes for patients with ischemic stroke.
Footnotes
Experimental stroke surgeries (MCAO) were performed by TS and FS; and analysis of cell function following MCAO was performed by TRS. TRS, RJT, PSH, and AW were involved in the experimental design, data analysis, and in writing of the manuscript.
The authors declare no conflict of interest.