
Abstract
The effect of recombinant human tissue plasminogen activator (rtPA) on neuroinflammation after stroke remains largely unknown. Here, we tested the effect of rtPA on expression of cellular adhesion molecules, chemokines, and cytokines, and compared those with levels of inflammatory cell recruitment, brain injury, and mortality over 3 days after transient middle cerebral artery occlusion (MCAO) in mice. Mortality was dramatically increased after rtPA treatment compared with saline treatment during the first day of reperfusion. Among the animals that survived, rtPA significantly increased CCL3 expression, microglia recruitment, and cerebral infarction 6 hours after MCAO. In contrast, the extent of neutrophils and macrophages infiltration in the brain was similar in both saline- and rtPA-treated animals. Recombinant human tissue plasminogen activator induced II 1b and Tnf expression, 6 and 72 hours after MCAO, respectively, and dramatically reduced interleukin 6 (IL-6) level 24 hours after reperfusion. A dose response study confirmed the effect of rtPA on CCL3 and II1b expressions. The effect was similar at the doses of 1 and 10 mg/kg. In conclusion, we report for the first time that rtPA amplified microglia recruitment early after stroke in association with a rapid CCL3 production. This early response may take part in the higher susceptibility of rtPA-treated animals to reperfusion injury.
INTRODUCTION
Transient cerebral ischemia leads to an inflammatory response characterized by the recruitment and activation of microglial cells from within the brain and infiltration of leukocytes from the periphery. 1 A central regulator of the inflammatory response is the transcription factor, nuclear factor kappa-B. Reactive oxygen species and damage-associated molecular patterns, both being release after ischemic injury, can activate nuclear factor kappa-B resulting in the induction of proinflammatory cytokines and chemokines.2,3 Endothelial-leukocyte adhesion molecules are upregulated soon after the onset of ischemic stroke and are involved in leukocyte rolling, firm adhesion, and transmigration across the cerebral endothelium. 4 Chemokines and their receptors on leukocytes have an essential role in leukocyte trafficking during inflammation. 5 Infiltrating leukocytes release cytokines and chemokines, which amplify the brain inflammatory response further by causing more extensive activation of resident cells and infiltration of leukocytes, eventually leading to disruption of the blood–brain barrier (BBB), cerebral edema, neuronal cell death, and hemorrhagic transformation. 1 Antiinflammatory strategies in mice and rats have been shown to reduce infarct size, edema, and neurologic deficits,6–8 confirming the deleterious role of leukocytes and innate immune cells in experimental stroke.
Recombinant human tissue plasminogen activator (rtPA) is the only approved thrombolytic treatment of ischemic stroke. 9 However, a growing body of evidence indicates that rtPA has deleterious effects in the ischemic brain including cytotoxicity and vasogenic edema. 10 Additionally, endogenous tPA, which is overexpressed after stroke, may contribute to ischemic injury. 11 Endogenous tPA, initially secreted from injured neurons, may act as a cytokine to activate microglia at the site of injury. 12 These activated microglia then may secrete additional tPA, which promotes extracellular matrix degradation, neurodegeneration, and microglial selfproliferation. 12 Endogenous tPA may also regulate transendothelial migration of monocytes and reactive oxygen species-induced breakdown of occludin. 13 Treatment of photothrombotic middle cerebral artery (MCA) occlusion (MCAO) with rtPA leads to an increase in expression of intercellular adhesion molecule 1, vascular cell adhesion molecule 1, interleukin 1 beta (IL-1β), and tumor necrosis factor alpha (TNFα). 14 However, photothrombosis is known to induce weak brain cortical infarction due to the distal location of the vascular occlusion. We previously showed in a model of proximal MCAO in mice that rtPA increases vasogenic edema and mortality in a matrix metalloproteinase-dependent manner. 15 We observed a potential dissociation of rtPA-dependent mechanisms of BBB breakdown and cerebral infarction, although the neuroinflammation response was not investigated.
The role of rtPA in stroke-related inflammation should be further characterized since most of the side effects of this thrombolytic, which are described in the literature, could be due in part to reperfusion of the ischemic brain and concomitant endogenous tPA expression. 10 Moreover, to the best of our knowledge, the effects of rtPA on chemokine expression in animal models of stroke have not yet been investigated. In the current study, we analyzed cellular adhesion molecule, chemokine and cytokine expressions as well as inflammatory cell recruitment, brain injury, and mortality up to 72 hours after stroke in mice treated with rtPA or saline. We observed that rtPA amplified microglial cell recruitment early after stroke in association with a rapid CCL3 chemokine overexpression. This early response may take part in the higher susceptibility of rtPA-treated animals to reperfusion injury leading to early cerebral infarction and higher mortality.
MATERIALS AND METHODS
Middle Cerebral Artery Occlusion
Three-to four-month-old male 129/SvEV mice, weighing 25.1 ± 2.6 g, were anesthetized with 1.5% isoflurane and the left MCA was occluded for 60 minutes with an intraluminal Dafilon 6.0 nylon suture, as described previously. 15 Interruption of regional cerebral blood flow in the MCA territory was confirmed by documenting a >80% decrease in relative regional cerebral blood flow using laser Doppler flowmetry (Perimed AB, Stockholm, Sweden). A return to >50% of baseline regional cerebral blood flow within 5 minutes after suture withdrawal confirmed a reperfusion of the MCA territory. Animals not meeting both ischemic and reperfusion flow criteria were excluded from the study. In all, 6, 24, and 72 hours after reperfusion, animals were anesthetized with a lethal dose of isoflurane and perfused through the heart with 10 U/mL of heparin in 0.9% saline. Brains were harvested, frozen on dry ice for histologic assessments or in liquid nitrogen for biochemical analysis and stored at −;80°C. All procedures were approved by the Swiss Federal Veterinary Office (authorization number 1007/3714/3) and the results were reported in accordance with the ARRIVE guidelines.
Recombinant Human Tissue Plasminogen Activator Treatment
Sixty minutes after the onset of MCAO, animals were treated with 10 mg/kg of rtPA (Actilyse, Boehringer Ingelheim, Germany), injected in the left femoral vein, 10% of the dose in bolus and the remainder over 30 minutes. This relatively high dose is commonly used in rodents on the basis that specific fibrinolytic effect of tPA is at least 10 times less in rat than in human. 16 Control animals were injected with the same volume of saline (0.9% NaCl). Treatments with rtPA or saline were performed alternatively during the course of the study and the animals were chosen randomly inside cages. If an animal died, then the next animal was assigned to the same treatment.
Recombinant Human Tissue Plasminogen Activator Dose Response
For the dose response experiments, an additional set of animals was treated with saline or with 1 or 10 mg/kg of rtPA, injected in the left femoral vein, 1 hour after the onset of MCAO, and killed 6 hours after reperfusion.
Mortality Assessment
Mortality rates 6, 24, and 72 hours after MCAO were measured by counting the number of animals that died during the first 6 hours within the entire cohorts (mortality rate at 6 hours) or during the first 24 hours in the groups of animals assigned to the 24- and 72-hour time points (mortality rate at 24 hours) or during the entire course of experiments in the 72-hour groups only (mortality rate at 72 hours).
Infarct Volume and Brain Edema Measurements
Brains were cut in 20-μm thick coronal sections from the anterior to the posterior side. Every 200 μm, the brain sections were stained with a solution of 0.5% cresyl violet. Infarct size was obtained by summing infarct measurements on digital images of each section, acquired with an Epson perfection V750 scanner (Seiko Epson Corp., Amsterdam, The Netherlands), after compensation for edema as previously described. 17 Brain edema was assessed on digital images by measuring the hemispheric enlargements as described previously. 17 Infarct and hemispheric areas were manually drawn on digital images by a blinded operator using Adobe Photoshop.
Immunoglobulin Extravasation
Blood–brain barrier integrity was assessed by measuring the level of immunoglobulin G (IgG) extravasation into the brain as described previously with slight modifications. 18 Coronal brain sections (20-μm thick), spanning the entire hemispheres, were fixed in 4% formaldehyde in phosphate-buffered saline (PBS), coated with 10% normal goat serum in PBS for 1 hour at room temperature, and incubated for 4 hours with an Alexa Fluor 700 goat anti-mouse IgG (Molecular Probes, Zug, Switzerland, A-21036, dilution 1:400) in PBS containing 10% normal goat serum. After washing in water, fluorescent intensity was quantified by a blinded operator using an Odyssey infrared scanner (LI-COR, Lincoln, NE, USA) and converted into average integrated intensities.
Immune Cell Detection
Four coronal brain sections (20-μm thick), cut sequentially every 400 μm within the ischemic territory, were used for assessing macrophage and neutrophil infiltration as well as microglial cell recruitment. Brain sections were fixed in acetone (for macrophage and neutrophil detection) or in 4% formaldehyde in PBS followed by an incubation in 0.3% H2O2 in PBS for 30 minutes (for microglial cell detection). Sections were then coated at room temperature with 10% normal rabbit serum in TPBS (PBS containing 0.1% Tween-20) for 30 minutes (for macrophages and neutrophils) or 5% normal goat serum in TPBS plus 0.3% Triton X-100 for 60 minutes (for microglia), and incubated for 90 minutes at room temperature with a rat anti-mouse CD68 antibody (ABD Serotec, Oxford, UK, MCA1957GA, dilution 1:400) or a rat anti-mouse Ly-6B.2 antibody (ABD Serotec, MCA771G, dilution 1:50) or overnight at 5°C with a rabbit anti-Iba1 antibody (Wako Chemicals, Neuss, Germany, 019—19741, dilution 1:500). Immunoreactivity was revealed by a biotinylated rabbit anti-rat IgG antibody (Vector Laboratories, Peterborough, UK, BA-4001, dilution 1:100) and the VECTASTAIN ABC-Alkaline Phosphatase Kit (Vector Laboratories, AK-5000) in the presence of VECTOR RED (Vector Laboratories, SK-5100), followed by counterstaining with Mayer's hemalune (for macrophages and neutrophils), or by a biotinylated goat anti-rabbit antibody (Vector Laboratories, BA-1000, dilution 1:200) and the VECTASTAIN Elite ABC-Peroxidase Kit (Vector Laboratories, PK-6100) in the presence of 3,3′-diaminobenzidine and nickel chloride (Vector Laboratories, SK-4100), according to the manufacturer's instructions. Macrophages and neutrophils were counted by a blinded operator on the entire hemispheres and averaged for each individual brain. Microglial cells were counted on pictures acquired at a × 10 magnification within the cortex and averaged for each individual brains since striatal staining was inconsistent due to high infarction, especially in the rtPA groups.
Protein and cDNA Preparation
Right and left hemispheres were homogenized in 1 mL of cold PBS with a polytron and divided into two fractions. Half of the homogenate was mixed with 5 μL of protease inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA, P8340), snap frozen in liquid nitrogen, thawed, diluted with 500 μL of PBS, sonicated two times for 10 seconds and centrifuged at 20,000 g for 30 minutes. Protein concentration was measured in the supernatants with the Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, USA, 23227). Total RNA in the other half of the homogenate was isolated with TRI Reagent (Molecular Research Center Inc., Cincinnati, OH, USA, TR 118). Reverse transcription was performed with the ImProm-ll Reverse Transcription System (Promega Corporation, Madison, WI, USA, A3800), according to the manufacturer's instructions. Both protein and RNA sample numbers were coded before further analyses.
Real-Time Polymerase Chain Reaction
Real-time PCR was performed as described previously. 17 Specific primers and probes were used to determine mRNA expression of the adhesion molecules E-selectin (Sele), P-selectin (Selp), Icam1 and Vcam1, the chemoattractants Cd2, Cd3, Cxcl1, and Cx3d1, as well as the cytokines II1b, II6, and Tnf (Supplementary Table 1). The hypoxanthine guanine phosphoribosyl transferase gene (Hprt) was used as a housekeeping gene. Messenger RNA expressions in both hemispheres at the different time points were calculated by the 2−ΔΔCt method and compared with the contralateral expression in vehicle-treated mice killed 6 hours after reperfusion, used as a baseline value. Results were expressed as fold increase compared with baseline values.
Mortality after middle cerebral artery occlusion
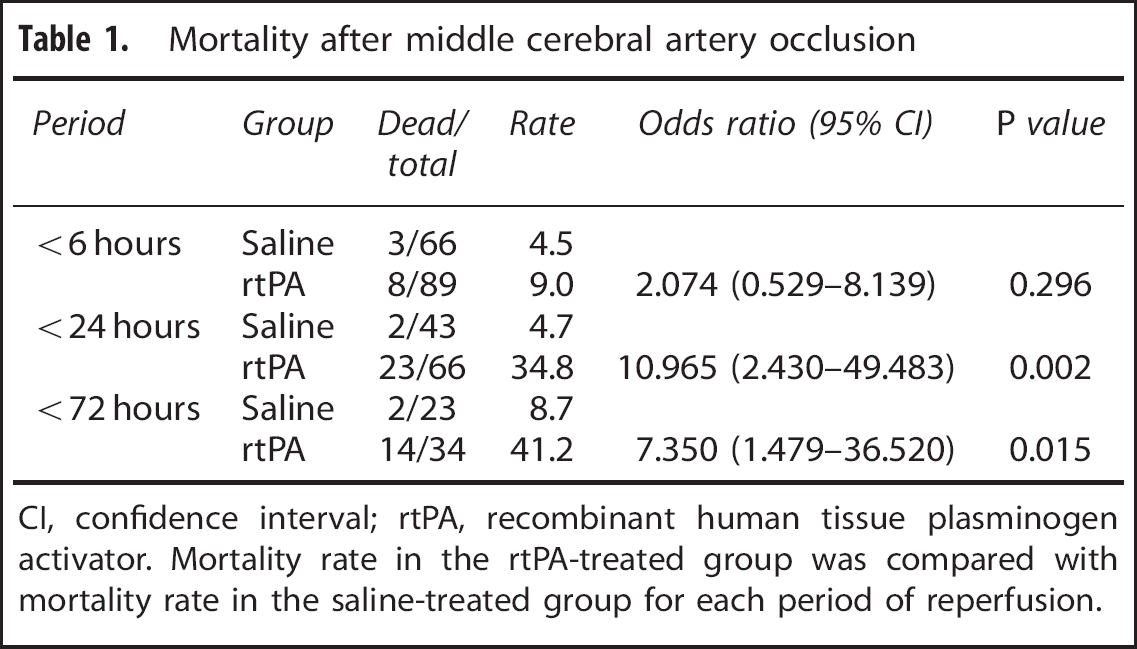
CI, confidence interval; rtPA, recombinant human tissue plasminogen activator. Mortality rate in the rtPA-treated group was compared with mortality rate in the saline-treated group for each period of reperfusion.
Enzyme-Linked Immunosorbent Assays
CCL3, IL-1β, and TNFα protein levels in brain homogenates were quantified using specific DuoSet mouse ELISAs according to the manufacturer's instructions (R&D Systems, Abingdom, UK). Detection limits were 8,19, and 25 pg/mL, respectively, and cytokine levels were expressed as pg/mg of protein.
Cytometric Bead Array
IL-6, CCL2, CCL3, and CXCL1 protein levels were measured using mouse-specific cytometric bead array flex sets (BD Biosciences, Oxford, UK) according to the manufacturer's protocol, and expressed as pg/mg of protein. Detection limits were as follows: CCL2 and CCL3—10 pg/mL; CXCL1 and IL-6—5 pg/mL. Samples were acquired on a BD FACSArray Bioanalyzer and analyzed with the FCAPArray software (BD Biosciences).
Statistical Analyses
Data were expressed as medians [interquartile ranges] or means ± standard deviations. Multiple group comparisons were performed by Kruskal-Wallis analysis of variance followed by Mann-Whitney U-tests, when appropriate. Comparisons of related samples were done by Wilcoxon Matched-Pair Signed-Rank tests. Odds ratios were determined by logistic regressions. Statistical analyses were conducted using SPSS Statistics version 20.0 (IBM Corp., Armonk NY, USA) for the Macintosh. P<0.05 was considered as statistically significant.
RESULTS
Animal Used in the Study
One hundred fifty-five animals were included in the main analyses and showed similar hemodynamic changes (Supplementary Table 2). Thirty-nine animals were excluded because of no occlusion (3 mice), early reperfusion (16 mice), subarachnoid hemorrhage (8 mice), or surgical complications leading to premature death, such as massive neck bleeding or respiratory arrest (12 mice). Twenty-nine animals were included in the dose response study and had similar hemodynamic changes (data not shown). Only 3 mice out of the 29 mice died during the 6 hours of reperfusion: two mice after 10 mg/kg rtPA treatment and one mouse after 1 mg/kg treatment. In total, surgery was performed on 223 animals.
Cell adhesion molecules and Cx3cl1 mRNA level changes during reperfusion

rtPA, recombinant human tissue plasminogen activator; Contra, contralateral hemisphere; Ipsi, ipsilateral hemisphere. Values are representative of 9 to 10 animals and are shown as relative fold increases compared with contralateral values in the saline-treated group reperfused for 6 hours. Statistically different from the corresponding contralateral level. Statistically different from the 6-hour time point level in the same treatment group. Statistically different from the 24-hour time point level in the same treatment group. Statistically different from the saline-treated group at the same time point.
Recombinant Human Tissue Plasminogen Activator Accelerates Infarct Growth and Increases Brain Edema and Mortality
The risk of mortality at 24 hours was sevenfold higher in the rtPA-treated group when compared with the saline-treated group and remained significantly higher 3 days after reperfusion (Table 1). A twofold increase in mortality was already perceived during the first 6 hours of reperfusion after rtPA treatment, although this was not statistically significant.
Cerebral infarction (Figure 1A) was significantly higher in the rtPA group 6 hours after reperfusion as compared with saline (15.1 mm3 [12.2 to 17.0] versus 7.1 mm3 [5.6 to 8.9], P = 0.002). In rtPA-treated animals, infarct volumes were almost identical between 6 and 24 hours. However during the 3 days of reperfusion, infarct volumes increased in both rtPA and saline-treated animals and no more differences could be seen between the animals by 72 hours of reperfusion (28.2 mm3 [22.0 to 32.6] and 26.5 mm3 [10.4 to 28.7], respectively).
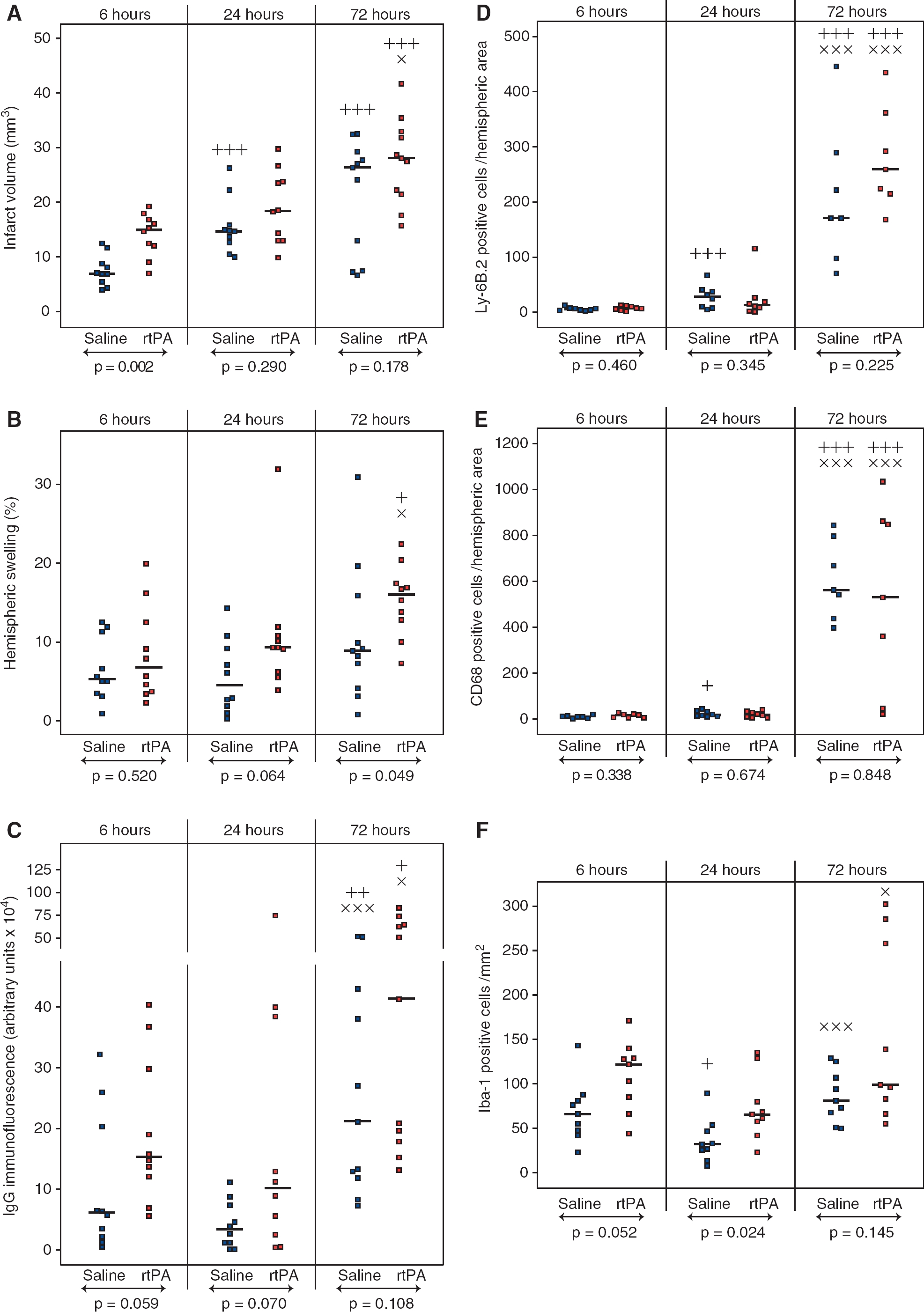
Effect of recombinant human tissue plasminogen activator (rtPA) on infarct volume, hemispheric swelling, and immunoglobulin G (IgG) extravasation, and quantification of neutrophils, macrophages, and microglia within the brain after middle cerebral artery occlusion (MCAO). Infarct volume
Hemispheric swelling (Figure 1B), a marker of vasogenic edema, was detectable as soon as 6 hours after reperfusion in both rtPA-and saline-treated animals. At 24 hours, rtPA-treated animals tended to have larger ipsilateral hemispheres than saline-treated animals. At 72 hours, vasogenic edema was dramatically increased in rtPA-treated animals compared with saline-treated animals (16.0% [12.8 to 17.4] versus 8.9% [5.7 to 12.9], P = 0.049).
Immunoglobulin G extravasation (Figure 1C), another hallmark of BBB dysfunction, was generally higher at all time points in the rtPA-treated group as compared with saline-treated animals. After 72 hours of reperfusion a significant increase in IgG extravasation, revealing a possible breakdown of the BBB, was observed in both rtPA- and saline-treated animals (41.2 × 10 4 arbitrary units [18.8 to 63.6] and 21.1 × 10 4 arbitrary units [12.4 to 40.5], respectively).
Recombinant Human Tissue Plasminogen Activator Enhances Microglia Recruitment but not Neutrophil and Macrophage Infiltration
Immune cell recruitment is a distinctive feature of cerebral inflammation after stroke. Therefore, we quantified leukocyte and microglial invasion in the ischemic territory for 3 days after MCAO and rtPA treatment.
Infiltration of neutrophils and macrophages in the ipsilateral hemispheres, as revealed by Ly-6B.2 and CD68 immunostaining (Figure 2), was low during the first 24 hours of reperfusion (Figures 1D and 1E). At 72 hours, neutrophil and macrophage counts were dramatically increased but no difference between rtPA- and saline-treated animals was found.
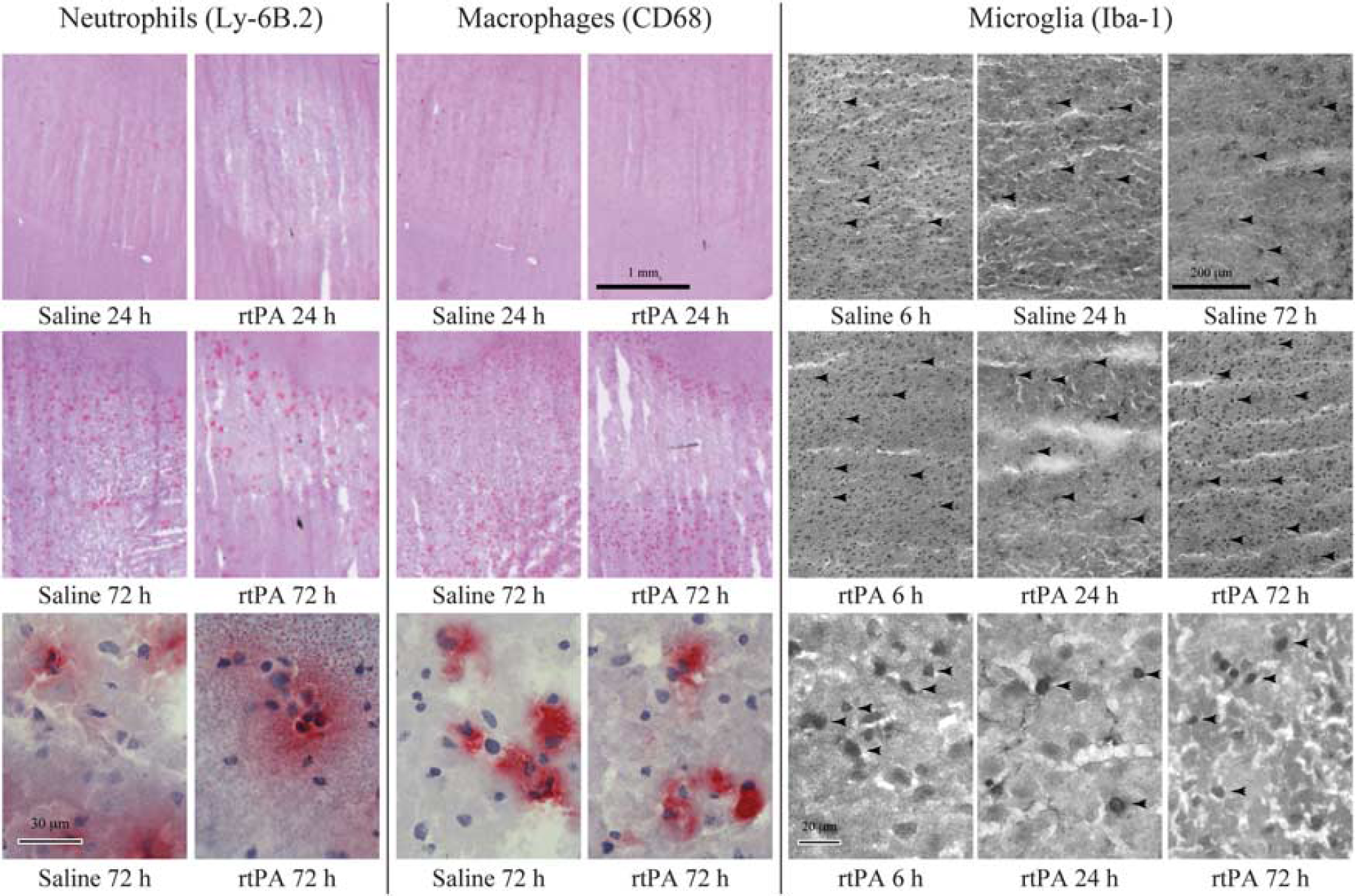
Effect of recombinant human tissue plasminogen activator (rtPA) on neutrophil and macrophage infiltration, and on microglial recruitment into the brain after middle cerebral artery occlusion (MCAO). Representative photographs of neutrophils (Ly-6B.2-positive cells), macrophages (CD68-positive cells), and microglial cells (Iba-1-positive cells) in the ischemic hemispheres of saline- and rtPA-treated mice at 6, 24, and 72 hours of reperfusion after 1 hour of MCAO. Arrows point at some positive cells. Bars are 1 mm long (neutrophils and macrophages) or 200-m long (microglia) for low magnification photographs and 30-m long (neutrophils and macrophages) or 20-m long (microglia) for high magnification photographs.
However, microglial cell (Iba-1-positive cell) recruitment was observed as early as 6 hours after reperfusion in the cortex (Figure 2) and was higher after rtPA treatment when compared with saline (122 cells/mm2 [85 to 129] versus 66 cells/mm2 [48 to 81], Figure IF). At 24 hours, the number of cells decreased in both groups but remained significantly higher in the rtPA-treated group (66 cells/mm2 [58 to 80] versus 32 cells/mm2 [26 to 47], P = 0.024). Microglial cell appearance was modified with bold and rounded body shapes, characteristic of activated cells (Figure 2). By 72 hours of reperfusion, the number of Iba-1-positive cells increased again but no differences could be detected between rtPA- and saline-treated animals (99 cells/mm2 [88 to 258] and 81 cells/mm2 [68 to 107], respectively).
Recombinant Human Tissue Plasminogen Activator does not Significantly Alter Endothelial Cell Adhesion Molecule Expression
Adhesion molecules expressed on brain microvascular endothelium cells have a crucial role in leukocyte diapedesis. So, we assessed mRNA expression of several cell adhesion molecules after cerebral ischemia and rtPA treatment. E- and P-selectin mRNA expressions were significantly increased as early as 6 hours after reperfusion within the ischemic territory of both rtPA- and saline-treated animals (Table 2). E-selectin mRNA expression peaked at 24 hours, reaching almost a 100-fold increase after rtPA treatment. P-selectin expression continued to increase up to 72 hours, reaching almost a 200-fold increase after rtPA treatment. Only a temporary difference between rtPA- and saline-treated animals could be seen at 6 hours of reperfusion with a slightly higher expression of E-selectin (28.9-fold increase [19.7 to 43.4] versus 19.7-fold increase [6.8 to 27.3], P = 0.096) and P-selectin (18.1-fold increase [13.5 to 25.8] versus 5.4-fold increase [3.5 to 14.6], P = 0.008) in the rtPA-treated group compared with saline.
A small increase in Icam1 mRNA expression (a 1.8- to 4.6-fold increase) was detected in the ipsilateral hemisphere of rtPA- and saline-treated animals during the entire course of the study, whereas Vcam1 mRNA expression remained unchanged (Table 2). Overall, no significant difference between rtPA- and saline-treated animals was observed.
Recombinant Human Tissue Plasminogen Activator Increases Ccl3 Expression but not Ccl2, Cxcl1, and Cx3cl1 Expression
Immune cell recruitment after cerebral ischemia is driven by a gradient of multiple chemoattractants. For that reason, we evaluated the expression of several chemokines after MCAO and rtPA treatment. Ccl2, Ccl3, and Cxcl1 mRNA levels were increased as early as 6 hours after reperfusion within the ischemic territory of both rtPA- and saline-treated animals (Figures 3A to 3C for ipsilateral values and Supplementary Table 3 for contralateral values). Cell and Cxcl1 expression peaked at 24 hours, with roughly a 100- to 200-fold increase for Ccl1 and a more moderate increase in 10- to 25-fold for Cxcl1. Ccl3 overexpression peaked at 6 hours and was significantly higher after rtPA treatment when compared with saline at this time point (184.6-fold increase [135.5 to 226.4] versus 139.6-fold increase [91.3 to 170.3], P = 0.049). Cx3cl1 mRNA expression remained unchanged after ischemia and rtPA or saline treatment at any of the time points tested (Table 2). CCL2 and CXCL1 proteins had a similar pattern of expression than their corresponding mRNAs with a significant increase in the ischemic territory over the 72 hours of reperfusion compared with contralateral values (Figures 3D and 3E for ipsilateral values and Supplementary Table 4 for contralateral values). Highest levels of CCL2 and CXCL1 were detected at 24 hours and saline-treated animals had significantly higher CCL2 and CXCL1 levels than rtPA-treated animals at this time point (CCL2: 772 pg/mg protein [557 to 1297] versus 493 pg/mg protein [250 to 772], P = 0.041; CXCL1: 286 pg/mg protein [202 to 475] versus 132 pg/mg protein [63 to 207], P = 0.018). CCL3 protein level was also significantly increased within the ipsilateral territory over the course of the study (Figure 3E), as it was mainly undetectable in contralateral hemispheres (Supplementary Table 4). Six hours after reperfusion, CCL3 protein concentration was twofold higher in rtPA-treated animals than in saline-treated animals (91 pg/mg protein [86 to 112] versus 44 pg/mg protein [38 to 56], P = 0.002). The highest values were observed at 24 hours in saline-treated animals (127 pg/mg protein [77 to 149]) but CCL3 concentration remained high in rtPA-treated animals at 24 hours (83 pg/mg protein [43 to 123]) and by 72 hours CCL3 concentrations were significantly reduced in both saline- and rtPA-treated animals.
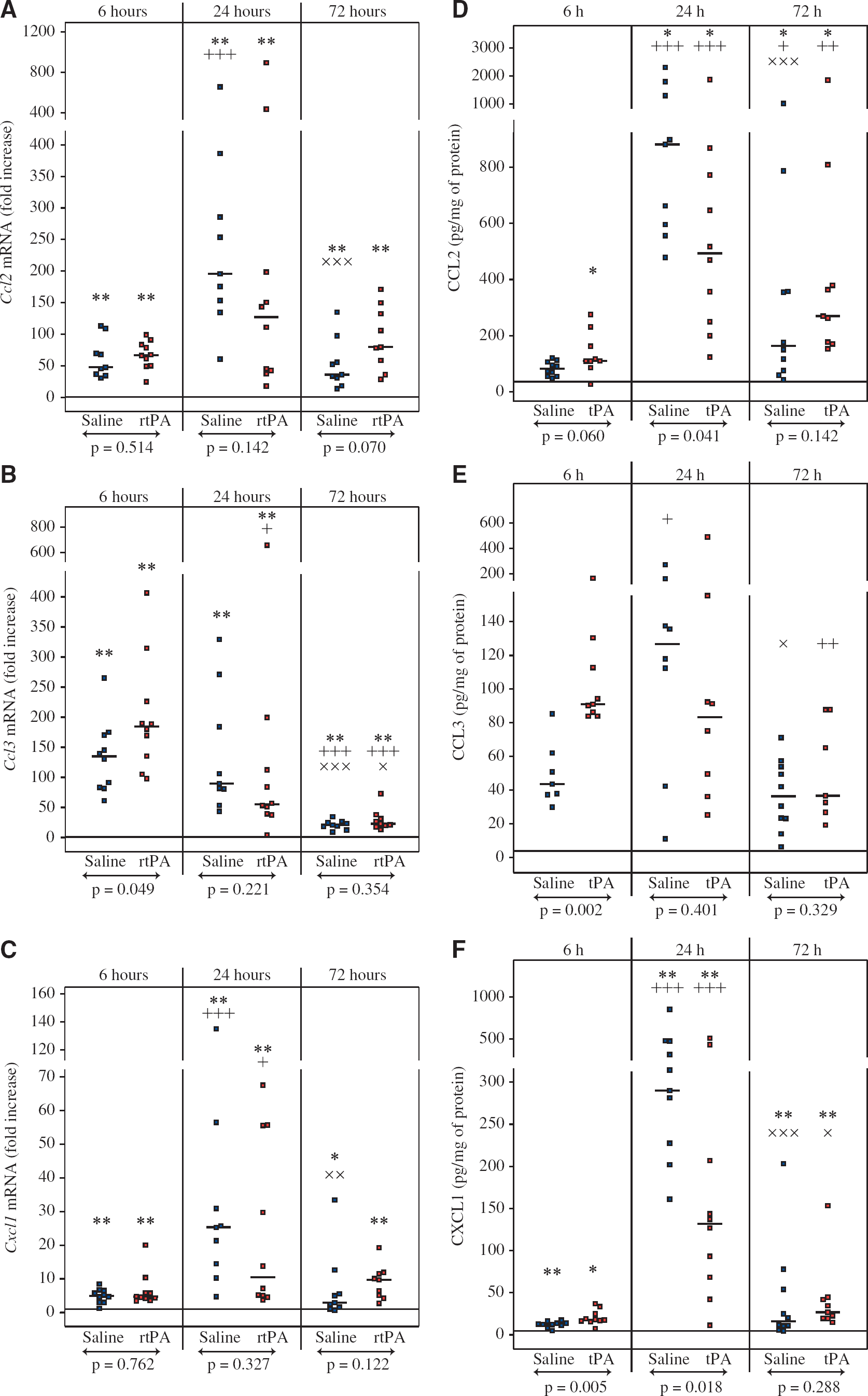
Effect of recombinant human tissue plasminogen activator (rtPA) on CCL2, CCL3, and CXCL1 expression after middle cerebral artery occlusion. Ccl2
Recombinant Human Tissue Plasminogen Activator Increases II1b and Tnf Expression but not II6 Expression
Several cytokines are involved in the initiation and perpetuation of cerebral inflammation. Consequently, we measured the expression of several representative cytokines after MCAO and rtPA treatment, II1b, II6, and Tnf mRNA levels were significantly increased within the ipsilateral territory at 6, 24, and 72 hours of reperfusion (Figures 4A to 4C for ipsilateral values and Supplementary Table 3 for contralateral values). Levels of all cytokines tested peaked at 24 hours, although a delayed increase in Tnf mRNA was observed in the rtPA group at 72 hours compared with saline (10.0-fold increase [8.0 to 16.2] versus 6.0-fold increase [4.0 to 8.2], P = 0.038). A significant higher expression of II1b was observed 6 hours after reperfusion in the rtPA-treated animals compared with saline-treated animals (17.1-fold increase [10.6 to 21.9] versus 5.8-fold increase [5.1 to 9.0], P = 0.001).
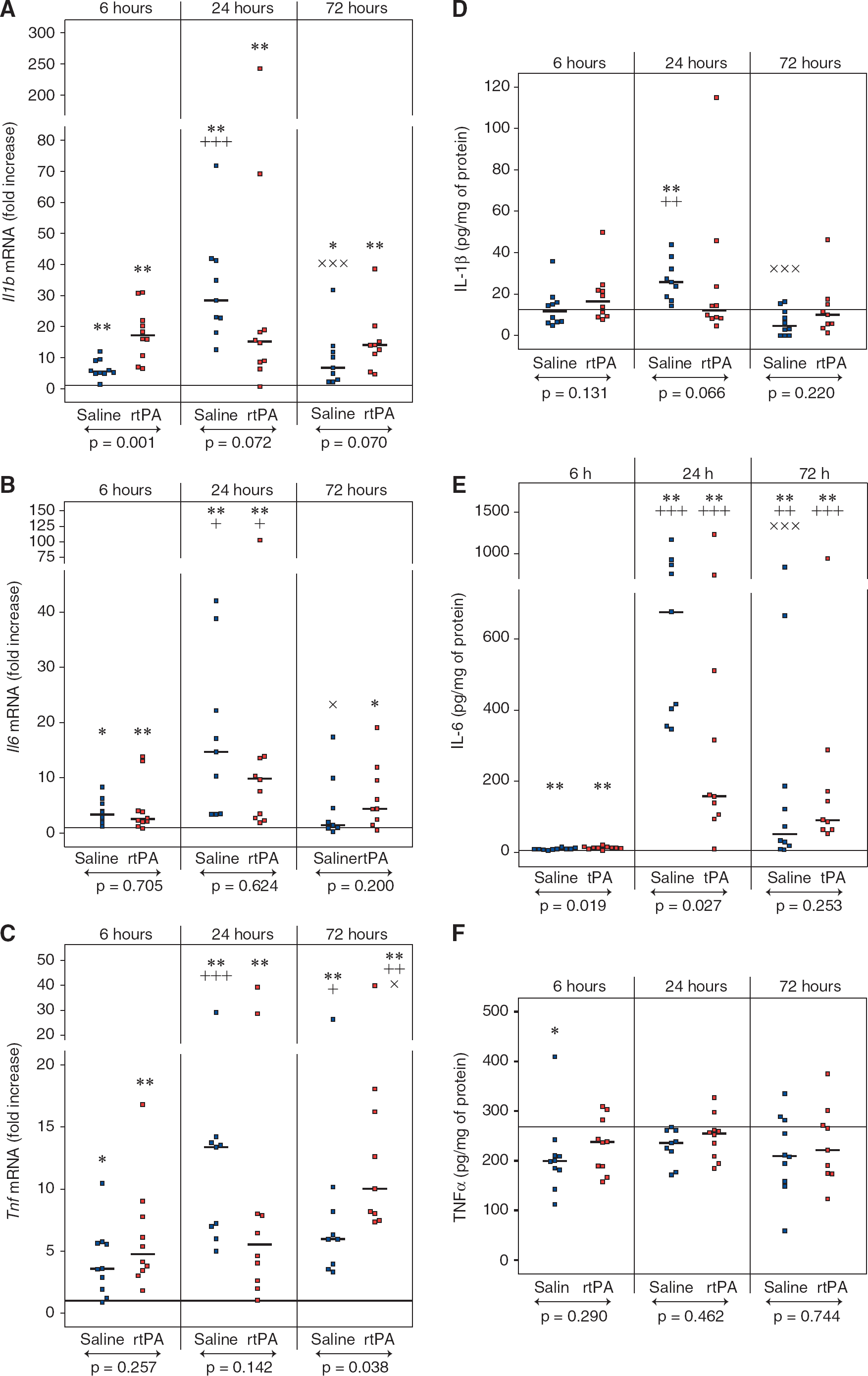
Effect of recombinant human tissue plasminogen activator (rtPA) on interleukin 1 beta (IL-1β), IL-6, and tumor necrosis factor alpha (TNFα) expression after middle cerebral artery occlusion (MCAO). II1b
Messenger RNA overexpression was barely translated into protein changes since IL-1β and TNFα protein levels in the ipsilateral territory were not different from the contralateral values at any time point, except a twofold increase in IL-1β in the saline-treated group at 24 hours of reperfusion (Figures 4D and 4F for ipsilateral values and Supplementary Table 4 for contralateral values). Only IL-6 protein levels were dramatically increased after ischemia in both saline- and rtPA-treated animals with a peak of concentration at 24 hours (Figure 4E). At this time point, IL-6 protein concentration was significantly higher in saline-treated animals compared with rtPA-treated animals (545 pg/mg protein [353 to 865] versus 158 pg/mg protein [105 to 510], P = 0.027).
A Low, Clinically Relevant Dose of Recombinant Human Tissue Plasminogen Activator Induces Rapid Priming of Inflammatory Response
The effect of a lower dose of rtPA on II1b, Ccl3, and Selp expression 6 hours after reperfusion, closer to what is used in clinic, was tested in an additional set of mice, since expression of those three genes was significantly higher in ipsilateral hemispheres compared with contralateral hemispheres after treatment with 10 mg/kg rtPA. II1b mRNA levels (Figure 5A) were significantly higher within the ipsilateral territory in the rtPA-treated animals compared with saline-treated animals after treatment with 1 and 10 mg/kg rtPA (saline: 15.9-fold increase [9.2 to 19.1]; 1 mg/kg rtPA: 30.3-fold increase [18.5 to 40.9]; 10 mg/kg rtPA: 27.8-fold increase [19.0 to 39.7]). A trend for higher expression of Ccl3 (Figure 5B) was also observed in the rtPA-treated animals compared with saline-treated animals (saline: 148.5-fold increase [99.4 to 192.1]; 1 mg/kg rtPA: 210.9-fold increase [144.3 to 304.5]; 10 mg/kg rtPA: 255.2-fold increase [187.4 to 325.0]). This was translated by higher levels of CCL3 protein (Figure 5C) after rtPA treatment than saline treatment (saline: 22 pg/mg protein [11 to 38]; 1 mg/kg rtPA: 40 pg/mg protein [28 to 84]; 10 mg/kg rtPA: 54 pg/mg protein [52 to 93]). However, reducing the dose of rtPA mitigated slightly the CCL3 response. P-selectin mRNA levels were increased to the same extent in saline- and rtPA-treated groups (saline: 7.2-fold increase [3.3 to 17.4]); 1 mg/kg rtPA: 5.4-fold increase [5.0 to 10.5]; 10 mg/kg rtPA: 10.1-fold increase [4.7 to 13.5]). No statistical differences regarding the expression of II1b, Ccl3, and Selp could be seen between animals treated with 1 or 10 mg/kg rtPA.
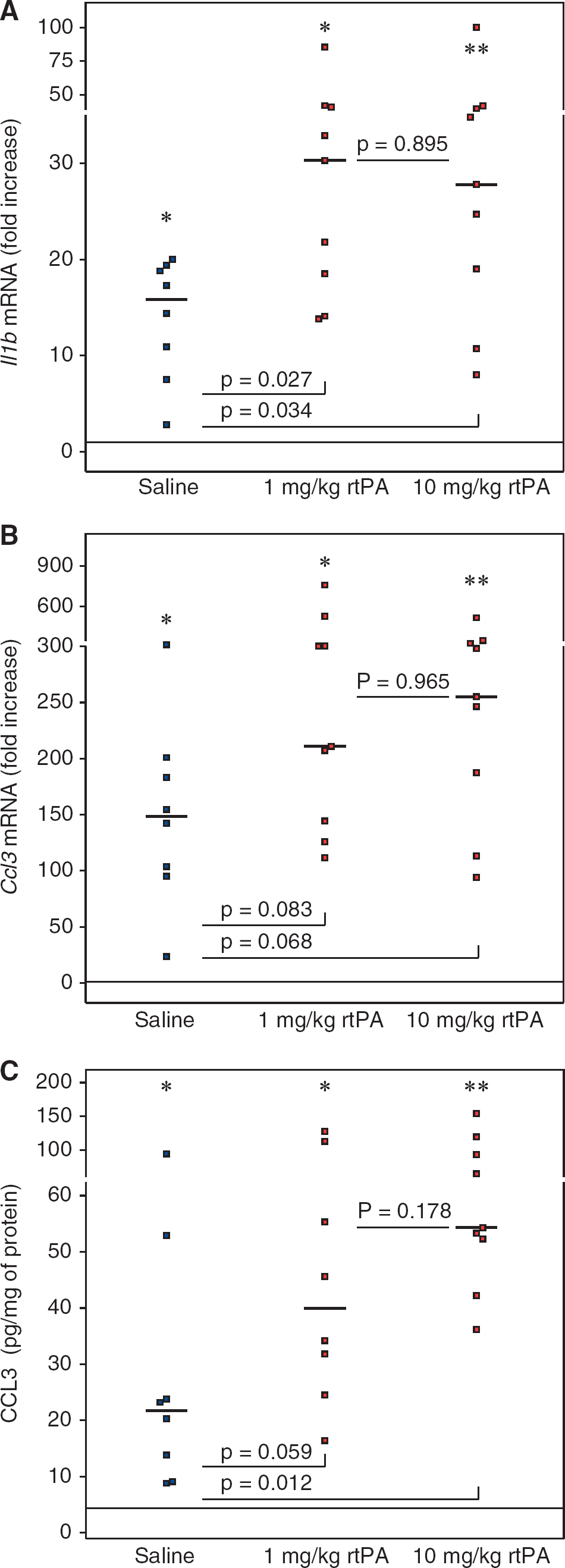
Dose response effect of recombinant human tissue plasminogen activator (rtPA) on II1b and Ccl3 mRNA levels and on CCL3 protein level after middle cerebral artery occlusion (MCAO). II1b mRNA
DISCUSSION
In this study, we confirmed the toxicity of rtPA administered at clinically relevant doses and time window after cerebral ischemia in mice. Human rtPA is the only thrombolytic treatment of ischemic stroke and leads to improvement of the clinical outcome in stroke patients despite an increase in the incidence of intracerebral hemorrhage.
9
In addition to its thrombolytic activity, rtPA could interact with different receptors in the brain, such as the low density lipoprotein receptor-related protein,
19
the N-methyl-
The role of rtPA in neuroinflammation after brain ischemia has been barely studied so far and no data on chemokine expression are available to date. In the current study, we show that, among immune cells attracted to the site of injury after cerebral ischemia, microglial cells are recruited more prominently after rtPA treatment compared with saline treatment within the first 6 hours of reperfusion in parallel to higher infarction in rtPA-treated animals. After a transient decrease in the number of microglial cells at 24 hours, concomitant to the maturation of the cells, the number of cells increased again during the following 2 days in parallel to the development of massive brain infarction in both rtPA- and saline-treated animals. Microglial cells may contribute to ischemic brain damage by producing pro-inflammatory molecules, reactive oxygen species, and matrix metalloproteinases and may release chemokines, promoting leukocyte infiltration, and leading to secondary injury. 24 It was reported that microglia could exacerbate endothelial cell death after oxygen and glucose deprivation and that treatment with minocycline, a drug known to reduce microglial activation and proliferation, 8 could attenuate this phenomenon. 25 Recombinant tPA induces a catalytic-independent activation of the extracellular signal-regulated kinase 1/2 and Jun N-terminal kinase leading to microglial activation in culture. 21 Thus, preventing rtPA-related enhancement of microglial cell recruitment and activation after stroke could possibly limit ischemic brain infarction. If our results do not permit to determine whether early microglial cell recruitment is causing or responding to early neuronal injury, then it is worth mentioning that a combination therapy with rtPA and minocycline attenuated microglial activation and decreased cerebral infarction in a model of embolic stroke in rats with diabetes mellitus. 26 The phenotype of microglial cells responding to rtPA has to be further investigated. Indeed, depending on the degree of activation characterized by the expression of specific markers on microglia and the release of different effector molecules by the cells, microglial can be in a resting state, a classic active state (Ml) or an alternative active state (M2). 24 Resting microglia constantly survey the microenvironment for potential threats, while classically activated microglia are associated with the synthesis of proinflammatory molecules, leading to an increase in neuronal death and BBB breakdown, and alternatively activated microglia are associated with termination of inflammation, tissue remodeling, and angiogenesis. 24 Interestingly, it was recently reported that microglia initially recruited at the site of injury after MCAO displayed a M2 phenotype but gradually acquired the Ml phenotype. 27 In our conditions, a rapid transition to the Ml state, if showed, could take part in higher cerebral injury after rtPA treatment.
We did not see any differences in the degree of macrophage and neutrophil infiltration between rtPA and saline treatments. Leukocyte invasion was low until the third day of reperfusion, when the BBB was dramatically damaged in both groups as shown by a significant increase in IgG extravasation. The current time course of immune cell recruitment, with microglial cells being the first cells to respond to ischemic injury, is in agreement with previous studies.1,28 Leukocyte extravasation into the brain is mediated by cell adhesion molecules that are upregulated after cerebral ischemia and promote both rolling and firm adhesion of leukocytes. 4 We did not find any marked differences neither in the expression of E- and P-selectins nor in the expression of the cellular adhesion molecules, Icam7 and Vcam1, between saline-and rtPA-treated animals during the 3 days of recovery after MCAO. However, innate immunity is orchestrated by a gradient of chemokines that are synthetized within the brain after ischemic stroke. 5 Among the different chemokines investigated in this study, we found that CCL3 was statistically higher after rtPA treatment compared with saline treatment both at the transcript level and at the protein level after 6 hours of reperfusion. CCL3 is a potent monocyte/macrophage chemoattractant, 29 and can also attract microglial cells in vitro 30 and in Wvo 31 after cerebral ischemia. There were several reports showing the induction of CCL3 after cerebral ischemia in many animal models of stroke31–33 and in stroke patients. 34 The time course of mRNA expression and protein synthesis observed in our study in the saline group is in agreement with the literature. In contrast, the role of rtPA in brain CCL3 modulation after stroke was never described, although it was reported that tPA deficiency could reduce microglial activation and neuroinflammation in a model of allergic encephalomyelitis. 35 CCL3 induction might be cell specific since rtPA had no effect on CCL3 production in cultured astrocytes. 36 CCL3 binds to the CC-chemokine receptors CCR1 and CCR5, which are expressed by all cells within the central nervous system, including microglia. 37 It was suggested that CCR5 activation on microglia might protect again microglial neurotoxicity. 38 Nevertheless, a broad-spectrum chemokine receptor antagonist, blocking in particular CCR5 and CCR1 activity, has been shown to protect the brain against focal cerebral ischemia. 6 Hence, the consequence of an rtPA-induced enhancement of CCL3 expression after stroke should be further investigated with a particular emphasis on the potential therapeutic benefit of targeting CCL3 after thrombolysis in ischemic stroke. To date, it is not known whether a twofold increase in CCL3 within the ischemic brain territory, early after reperfusion, increases microglial cell recruitment and worsen cerebral injury or whether higher microglial density, shortly after rtPA treatment, leads to larger CCL3 production at 6 hours of reperfusion. However, a study, showing that oxidative stress induced CCL3 expression in neurons, followed by microglial recruitment and delayed neuronal death in a model of transient global ischemia, 31 together with our own observations of a sustained accumulation of microglial cells during the 72 hours of reperfusion, while the protein level of CCL3 is dramatically decreased after 3 days of reperfusion, suggest that CCL3 production in transient focal ischemia is more a cause than a consequence of microglial cell recruitment.
Neuroinflammation is characterized by the induction of the proinflammatory cytokines IL-1β, TNFα, and IL-6 within the brain after transient and permanent ischemia. 39 We observed a significant increase in the three cytokines at the mRNA level during cerebral reperfusion but we observed only an increase in IL-6 at the protein level. Injection of rtPA after cerebral ischemia increased the expression of II1b compared with saline as soon as 6 hours of reperfusion and of Tnf after 3 days of reperfusion. However, rtPA did not increase the protein levels at any of the time points investigated during the 72 hours of reperfusion. Unexpectedly, we found a decrease in IL-6 at 24 hours of reperfusion in rtPA-treated animals compared with saline-treated animals and also a consistent decrease in the chemokines CCL2 and CXCL1 at the same time point. This suggests a dysregulation of the inflammatory response after rtPA treatment. The role of IL-6 in stroke remains controversial but several studies have suggested that IL-6 might be protective. 39 A significant decrease in IL-6 after rtPA treatment could therefore contribute to cerebral injury.
The dose of rtPA used in the current study was ˜10 times the dose used for the treatment of stroke patients. 9 This high dose was commonly used in rodents on the basis that specific fibrinolytic effect of rtPA is at least 10 times less in rat than in human. 16 However, to be possibly translated to the clinical field, side effects of rtPA on molecular targets potentially different from the ones belonging to the fibrinolytic cascade should be documented with doses matching those used in clinic. On a separate group of mice, we compared the effect of 1 and 10 mg/kg rtPA. We confirmed the overexpression of II1b and Ccl3 but not P-selectin after rtPA treatment. This gives more credence to the sensitivity of II1b and Ccl3 to rtPA. However, the results suggest that the previous observation of a higher expression of P-selectin 6 hours after rtPA treatment compared with saline treatment was fortuitous. More importantly, we did not see any significant differences between the effects of 1 and 10 mg/kg rtPA. Hence, the protein levels of CCL3 obtained after treatment with 1 and 10 mg/kg were higher than after saline treatment and were not statistically different. Similarly, II1b expression was amplified to the same extent with 1 and 10 mg/kg rtPA. This suggests that the maximum effect of rtPA in mouse can already be approached with a dose of 1 mg/kg.
An important limitation to our results is the high degree of mortality obtained after rtPA treatment. Only animals that survived until their designated time point were included in the final histologic or biochemical analyses. One could imagine that the data are only a weak representation of a more severe phenomenon that could have a critical role in death of the rtPA-treated animals. Indeed, many of the animals that stopped breathing after rtPA treatment showed obvious cerebral edema. Our results must also be taken with caution since other animal species, and in particular humans treated with rtPA after ischemic stroke, might respond differently. For instance, it was reported that inflammatory gene response after photothrombosis was weaker in mice than in rats 40 or that inducible nitric oxide synthase activity increased in rats after 2 days of permanent MCAO whereas it remained undetectable in mice. 41 The results might also diverge in some other models of cerebral ischemia. In our study, the effect of rtPA was investigated independently of its proteolytic effect since reperfusion was achieved by mechanical withdrawal of the thread used for proper MCAO. True thrombolysis as observed in a model of thromboembolic stroke 42 or activation of rtPA in contact with clot material 43 may increase the complexity of the pathophysiologic cascade triggered by rtPA. In addition, the duration of cerebral ischemia before rtPA treatment may also interfere with the molecular and cellular mechanisms observed. Hence, we previously reported that 52% of mice subjected to 90-minute MCAO died 24 hours after rtPA treatment 15 when, in the present study, only 35% of the animals died when subjected to 60-minute MCAO. There is clearly a need to understand the specific role of rtPA in cerebral ischemia/reperfusion and to address the deleterious effects of rtPA when used at the limit of or beyond the clinical therapeutic window, currently set at 4.5 hours after ischemia onset for selected patients. 9
In summary, intravenous injection of rtPA immediately after 1 hour of proximal MCAO in mice led to higher mortality, faster cerebral infarction, and more massive BBB dysfunction with a significant increase in II1b expression and CCL3 production within the ischemic brain territory in the first 6 hour of reperfusion and an enhancement of microglial recruitment. This is the first time that an effect of rtPA treatment after stroke on chemokine expression is shown. The use of rtPA as the only thrombolytic treatment of ischemic stroke warrants further investigations of the causal relationship between the phenomena we observed.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.