
Abstract
Glycogen is present in the brain, where it has been found mainly in glial cells but not in neurons. Therefore, all physiologic roles of brain glycogen have been attributed exclusively to astrocytic glycogen. Working with primary cultured neurons, as well as with genetically modified mice and flies, here we report that—against general belief—neurons contain a low but measurable amount of glycogen. Moreover, we also show that these cells express the brain isoform of glycogen Phosphorylase, allowing glycogen to be fully metabolized. Most importantly, we show an active neuronal glycogen metabolism that protects cultured neurons from hypoxia-induced death and flies from hypoxia-induced stupor. Our findings change the current view of the role of glycogen in the brain and reveal that endogenous neuronal glycogen metabolism participates in the neuronal tolerance to hypoxic stress.
INTRODUCTION
Glycogen, the storage form of glucose in animals, is found in the brain. Its presence has been restricted mainly to astrocytes, while only a few studies have located it in neurons, particularly in motoneurons.1–6 Therefore, in brain function, all the physiologic roles of glycogen as an energy reserve in hypoglycemia and ischemia 7 or contributing to the consolidation of memory8,9 have been essentially attributed to astrocytic glycogen.
Glycogen synthase (GS, for a full list of abbreviations see Supplementary Information) is the enzyme responsible for the formation of glycogen. It is regulated by phosphorylation at multiple serine residues, and the phosphorylation of these sites induces the inactivation of the enzyme. 10 It is also allosterically activated by glucose-6-phosphate (Glc-6-P). 11 In mammals there are two GS isoforms, namely liver glycogen synthase, whose expression is specific to this organ, and muscle glycogen synthase (MGS), which is expressed elsewhere, including the brain. 12 The degradation of glycogen is mediated by glycogen Phosphorylase (GP), which releases glucose-1-phosphate, which in turn is rapidly converted into Glc-6-P. Glycogen Phosphorylase is activated by phosphorylation 13 and allosterically by AMP. 14 Three isoforms of GP have been described: liver, muscle, and brain. In the brain, both the brain and the muscle isoforms of GP are expressed. 15
Strikingly, while it is accepted that neurons do not contain glycogen, these cells do express MGS.16,17 Moreover, although the presence of GP in neurons was described many years ago,18,19 more recent studies have reported that they do not express GP15,17 and thus lack the capacity to degrade glycogen. Only in several pathologies do polymers of glucose, normally referred to as polyglucosan bodies, build up in neuronal tissue. 20 In addition, we previously showed that neuron-specific hyperactivation of GS and the subsequent accumulation of glycogen severely compromise neuronal function and survival.17,21 All together, these data raise the question as to why neurons express MGS if they do not accumulate the polysaccharide under normal conditions and would be unable to degrade it.
Working with pure neuronal cultures and genetically engineered flies and mice, here we show that, against general belief, neurons do accumulate glycogen, although in small amounts, and do express GP, which confers them full capacity to degrade it. We also show that an active endogenous glycogen metabolism proves to be protective for neurons during oxygen deprivation. Our observations modify the traditionally accepted view of glycogen metabolism in the brain and reveal that neuronal glycogen metabolism has a conserved role in tolerance to hypoxia.
MATERIALS AND METHODS
Primary Cultures of Neurons and Hypoxia Exposure
Neuronal cultures were obtained from the neocortex and the hippocampus of mouse embryos at embryonic day 16 (OF1 mice; Charles River Laboratories, Chatillon-sur-Chalaronne, France). Mouse brains were dissected in phosphate-buffered saline (PBS) containing 0.6% glucose (w/v; Sigma, St Gallen, Switzerland). After removal of the meninges, the telencephalon was mechanically and enzymatically dissociated with trypsin (0.25%; Invitrogen, Alcobendas, Spain). Dissociated cells were then seeded on plates (Nunc) pretreated with poly-L-lysine (10 μ/mL; Sigma) and maintained in serum-free, 25mmol/L D-(+)-glucose neurobasal medium (Invitrogen) supplemented with L-glutamine (2mmol/L; Invitrogen), extra D-(+)-glucose (until a final concentration of 55mmol/L), sodium bicarbonate (5 mmol/L; Invitrogen), penicillin/streptomycin (100U/mL and 100mg/mL; Invitrogen) and B27 supplement (1/50; Invitrogen). After 1 day in culture, the cells were treated with Uridine/5-fluoro-2′-deoxyuridine (50 μg/mL and 20 μ/mL; Sigma) to eliminate astrocytes. The purity of the cultures was determined by immunocytochemistry with neuron- (anti-β-III-tubulin (TUJ1, BABCO, Richmond, CA, USA)) and astrocyte- (anti-glial fibrillar acidic protein, GFAP (Dako, Hamburg, Germany)) specific antibodies. Cells were maintained 5 days in vitro (DIV5) with the above-mentioned neurobasal medium. All experiments were performed at DIV5. For hypoxia treatments, cell medium at DIV5 was changed with fresh neurobasal medium (unless otherwise stated), and neurons were incubated for the indicated times at 37°C in an oxygen-regulated incubator (IN VIVO2 200; Ruskin, Bridgend, UK) set at 1% O2, 5% C02, 94% N2. For GS knockout (KO) cultures, neurons were isolated from brain-specific MGS KO mice, 9 following the same protocol.
Glycogen Synthesis
Glycogen synthesis was measured at DIV5 after incubation of primary cultured neurons with 15 μCi/mL [U- 14 C] glucose (PerkinElmer Life Sciences, Tres Cantos, Spain) in neurobasal medium containing 25 mmol/L glucose. On termination of the incubations, neurons were rapidly washed three times with ice-cold 150 mmol/L NaCl to remove the remaining labeled glucose from the medium and then extracted in 0.1 mol/L NaOH. To ensure full precipitation of the radioactive glycogen in the following step, extracts were deproteinized with trichloroacetic acid (10%, w/v) containing glycogen carrier. 22 Glycogen was then isolated by selective precipitation in ice-cold 66% ethanol. 23 This precipitation step also removed the traces of labeled glucose, which is soluble in ethanol. The radioactivity incorporated into the polysaccharide was measured.
Metabolite Determinations
Glycogen content was determined using a modification of a previously described method, 23 which increased the sensitivity to measure the small amounts of glycogen present in neurons. Briefly, after incubation, medium was removed, and neurons were rapidly frozen in liquid nitrogen and kept at −80°C until the analyses. Cells were scraped into 30% KOH and the extract was heated for 15 minutes at 100°C. Glycogen was selectively precipitated with cold 66% ethanol overnight, and the pellet was digested with α-amyloglucosidase. In contrast to the method traditionally used, glucose converted into Glc-6-P was measured with the fluorometric assay described in Zhu et al. 24 This increased the sensitivity of the measurement more than 50-fold. The intracellular concentration of Glc-6-P and uridine diphosphate glucose (UDP glucose) was measured in perchloric acid-treated cells by means of the same fluorometric assay using Glc-6-P dehydrogenase and UDP glucose dehydrogenase, respectively.
Enzyme Activity Assays
To measure GS and GP activity, cell cultures were scraped using 100 μL of ice-cold homogenization buffer (Supplementary Information). Activity of GS was measured in the presence or absence of 6.6 mmol/L Glc-6-P. 25 The GS activity measured in the presence of Glc-6-P ((+)Glc-6-P) corresponds to the total amount of enzyme, whereas measurement in its absence ((—)Glc-6-P) is an indication of the active (unphosphorylated) GS form. The (—)Glc-6-P/(+)Glc-6-P activity ratio (GS activity ratio) is an estimation of the activation state of the enzyme, and it does not depend on the total levels of GS. Thus, the GS activity ratio can be directly compared between tissues with different GS expression levels. Activity of GP was determined by measuring the incorporation of [U- 14 C] glucose-1-phosphate into glycogen in the absence or presence of 5 mmol/L AMP, as previously described. 26 The ratio (+)caffeine (which is accepted as the measurement of the phosphorylated active form of GP)/(+)AMP (which, in the case of Brain GP, is a measure of the total amount of the enzyme) is an estimation of the activation state of the brain isoform of GP. Enzyme activities are expressed as milliunits (mU) per milligram of protein, where 1 mU is the amount of enzyme converting 1 nmol substrate per minute, while enzyme activity ratios are adimensional.
Immunocytochemistry
Cells were fixed for 30 minutes in PBS containing 4% (w/v) paraformaldehyde. For glycogen detection, cells were washed with PBS, followed by three 10-minute sequential incubations with PBS containing the following: (1) NaBH4 (1 mg/mL), (2) 0.2% (v/v), Triton X-100, and (3) 3% bovine serum albumin with 0.2% Triton X-100. Incubation with the primary and secondary antibodies was performed as previously described. 27 Nuclei were stained with Hoechst (1 mg/mL) parallel to secondary antibody incubation. The primary monoclonal antibody raised against glycogen was a gift from O. Baba (Tokyo Medical and Dental University).
For the detection of GS and GP, an amplification protocol was applied based on the Tyramide Signal Amplification (TSA) technology (PerkinElmer Life Sciences) (Supplementary Information).
Immunohistochemistry
Forty-week-old mice were anesthetized and perfused transcardially with PBS containing 4% (w/v) paraformaldehyde. Brains were removed, postfixed in the same solution overnight, cryoprotected with 30% (w/v) sucrose, and sectioned at coronal planes (30 μmol/L thick). For immunodetection of antigens, sections were washed in PBS and PBS-0.1% Triton X-100, and blocked for 2 hours at room temperature with PBS containing 10% normal goat serum and 0.2% gelatin. Primary antibodies (brain GP, produced by Eurogentec, Köln, Germany; Parvalbumin, Swant and GFAP, DAKO) were incubated overnight at 4°C with PBS-5% normal goat serum. Sections were incubated with dye-labeled secondary antibodies and Hoechst 33342 for 2 hours at room temperature in PBS-5% normal goat serum and mounted in Mowiol. Confocal images were taken with a Leica SPE microscope (Mannheim, Germany). 3D and surface reconstructions of GP and Parvalbumin-positive neurons were made using Imaris 7.6.1 × 64 (Bitplane, Inc., Zürich, Switzerland) Image Processing Software.
Cell Viability Assay
Neurons were seeded in poly-L-lysine-coated coverslips and incubated in normoxia and hypoxia for the indicated times. After treatments, analysis was performed using the Live Dead Cell Staining kit (Abeam, Cambridge, UK). This assay is based on membrane permeabilization discrimination of two dyes, which differentiate between live and dead cells. Neurons were examined and randomly photographed under a fluorescence microscope. The average number of dead cells was estimated by cell counts in 10 fields from 5 to 8 coverslips of three independent experiments.
Adenoviral Production and Neuronal Infection
The adenoviral vector (Supplementary Information) was transfected into HEK293A cells. Cells were kept in culture for 7 days until the adenovirus was generated. The medium and the cells were then collected, frozen and thawed three times, and centrifuged. Neurons at 3 days in vitro (DIV3) were infected with the adenovirus (at a multiplicity of infection of 30) for 12 hours, with an infection efficiency of around 60%, and protein was overexpressed until DIV5, when cells were exposed to hypoxia.
Hypoxia Exposure of Flies
The hypoxia experiments were based on previous reports. 28 Briefly, groups of 16 to 20 flies, aged 7 days, were placed into nylon-sealed tubes, which allow air exchange, and put into an oxygen-regulated incubator (IN VIVO2 200; Ruskin) set at 0.6% O2 and 99.4% N2 for 1 hour before recovery in room air (21% O2). Flies became uncoordinated within 30 seconds of hypoxic exposure and fell to the bottom of the tubes, where they remained motionless for the rest of the insult. Recovery time was measured as the period of latency between the end of the insult and the time at which flies crossed an arbitrary line drawn 2 cm from the bottom of the tube.
Mouse Studies
All procedures were approved by the Barcelona Science Park's Animal Experimentation Committee and were performed in accordance with the European Community Council Directive and National Institutes of Health guidelines for the care and use of laboratory animals. Mice were allowed free access to a standard chow diet and water and were maintained on a 12:12-hour light-dark cycle under specific pathogen-free conditions in the Animal Research Centre at the Barcelona Science Park. MGS-9APcp2 mice (males) were generated as previously reported. 21 Eight-week-old control and MGS-9APcp2 mice were anesthetized (sodium thiopental, 0.25 mg/g of body weight) and perfused transcardially with 150 mmol/L NaCl (saline) to induce glycogen degradation. After 5 minutes, the cerebellum was removed and frozen for later glycogen determination. Nonperfused mice (Control and MGS-9APcp2) were anesthetized and immediately decapitated, and the cerebella were rapidly dissected and frozen in liquid nitrogen. Due to the high turnover of glycogen postmortem, part of the glycogen might have been degraded during the isolation process, and thus the levels of glycogen of all groups may be underestimated.
Statistical Analysis
Results are presented as mean±s.e.m. of n = 4 to 8 independent experiments. Unless otherwise stated, significance between two variables was analyzed by the Student's t test using the GraphPad Prism software (La Jolla, CA, USA). P<0.05 was considered to be statistically significant. *P<0.05, **P<0.01, and ***P<0.001. For the statistical analysis of fly recovery time, the Cox Proportional Hazards likelihood ratio test was used. 29 P values were obtained using the R function coxph from the package survival with the different trials as an adjustment variable.
RESULTS
Neurons Synthesize Glycogen Under Basal Conditions
To study glycogen metabolism exclusively in neurons, we worked with pure neuronal cultures. Neurons were isolated from embryonic brains and cultured for 5 days with antimitotics. At DIV5, after the antimitotic treatment, the neuronal purity of the cultures was above 99.9%, as shown by neuronal- (TUJ1) and astroglial- (GFAP) specific immunolabeling (Figure 1A).
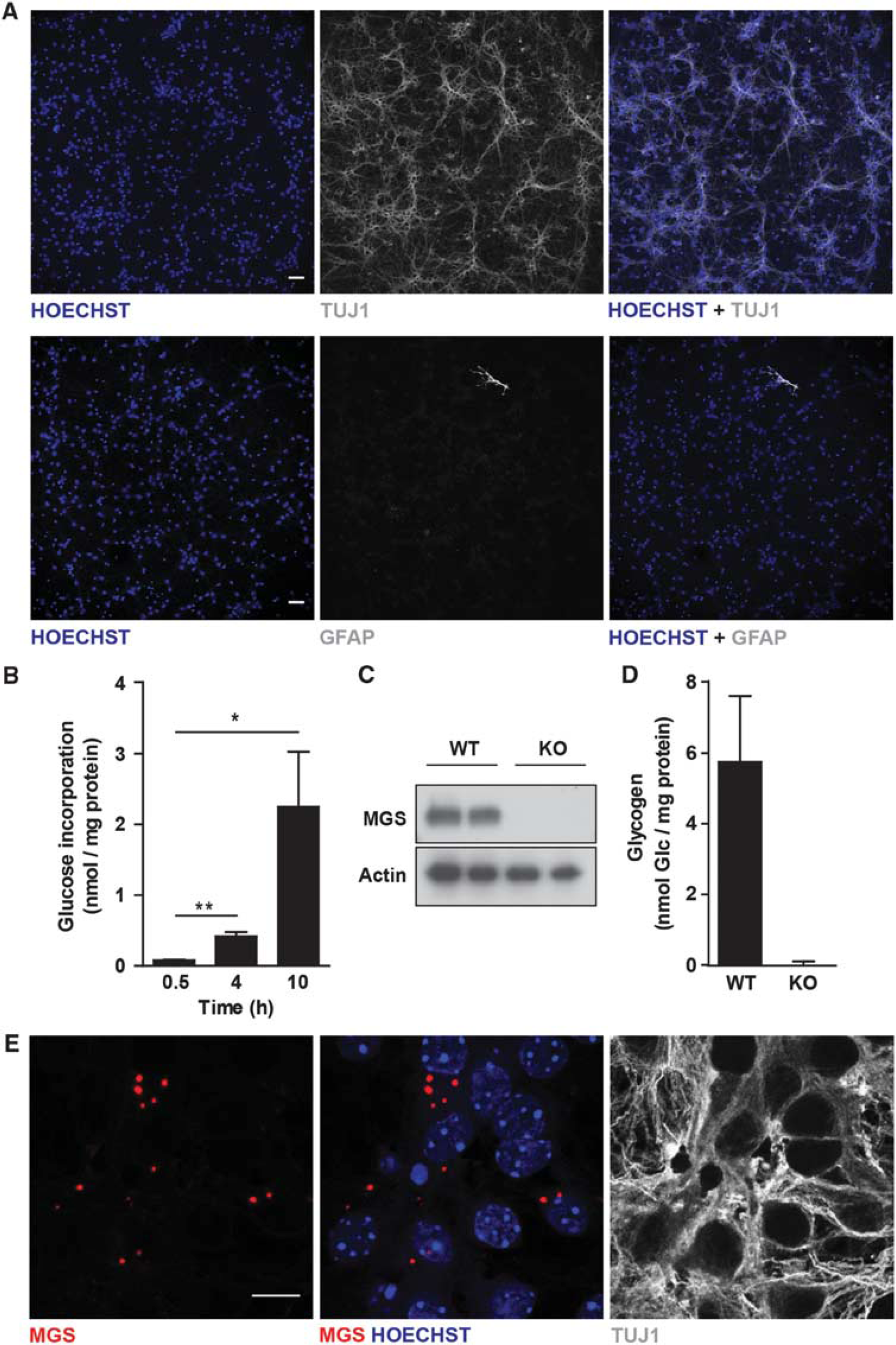
Neurons synthesize glycogen under basal conditions. (
At DIV5, glucose concentration in the medium had practically not decreased. To determine whether neurons synthesize glycogen under basal conditions, they were incubated with medium containing [ 14 C]-Glucose for increasing periods, and the amount of radioactivity incorporated into glycogen was measured. Increasing amounts of glucose were incorporated with time (Figure 1B), thus pointing to active glycogen synthesis. Given the lability of glycogen, partial degradation of the polysaccharide may have occurred during the extraction procedure, thus leading to an underestimation of the values. To determine the total amount of glycogen normally present in neurons, we developed a new, ultrasensitive method to measure the concentration of the polysaccharide. This method was based on fluorescence detection, instead of absorbance, thus enabling us to detect up to 50 times lower glycogen concentrations. To validate the result, we compared glycogen content in MGS wild-type (WT) neurons with that in neurons isolated from a brain-specific GS KO mouse model 9 (Figure 1C). Detectable amounts of glycogen were found in WT neurons, but not in the KO cells (Figure 1D). To assess the effect of glucose availability on neuronal glycogen content, neurons were incubated in media containing increasing concentrations of glucose (ranging from 5 to 55 mmol/L), and glycogen was determined. Interestingly, in contrast to what happens in astrocytes, glycogen content did not vary with increasing glucose concentrations, thereby suggesting a complex regulation of this molecule in neurons (Supplementary Figure S1). We next determined the subcellular distribution of endogenous MGS in neurons. To overcome the sensitivity problems associated with the low concentration of this enzyme, we worked with an amplified immunofluorescence protocol (Figure 1E). Muscle glycogen synthase accumulated in aggregates in the cytoplasm of the neurons, which is the typical location for GS under conditions of active glycogen synthesis. 30 This observation thus reinforces the notion that neurons indeed accumulated glycogen.
Neurons Have the Machinery Necessary to Degrade Glycogen
To analyze whether neurons have the machinery necessary to mobilize glycogen, we analyzed the mobilization of this polysaccharide under a glycogenolytic condition. For this purpose, cells were incubated under diminished oxygen (Hypoxia), a situation in which brain glycogen is degraded. 31 Due to the energy-demanding nature of this condition, neuronal glycogen might be of great relevance. After exposing neurons to 1% O2 (Hypoxia) for 4 hours, glycogen content was reduced (Figure 2A), in comparison with neurons incubated at 21% O2 (Normoxia); however, the concentration of glucose in the medium did not significantly change after hypoxic exposure (data not shown). After the hypoxic period, the intracellular distribution of MGS no longer showed the punctate pattern (Figure 2B). This observation is consistent with glycogen degradation. To further characterize the capacity of glycogen degradation of neurons, these cells were forced to accumulate large amounts of glycogen by transducing an active form of MGS using a recombinant adenovirus (Ad MGS-9A). 27 Neurons were infected with Ad MGS-9A at DIV3. At DIV5, when neurons had accumulated a high amount of the polysaccharide (but still reaching levels below those proven to be toxic for these cells—in the range of 400 nmol Glc/mg protein), they were exposed to 4 hours of hypoxia. Remarkably, glycogen was diminished after hypoxia, thus pointing to a degradation capacity that was activated during incubation under low oxygen availability (Figure 2C). This reduction in glycogen was confirmed by immunofluorescence analysis with a glycogen antibody (Figure 2D). Accordingly, in hypoxia, the subcellular localization of MGS-9A changed, being found in the nucleus, where it is detected only in glycogen-depleted cells. 30
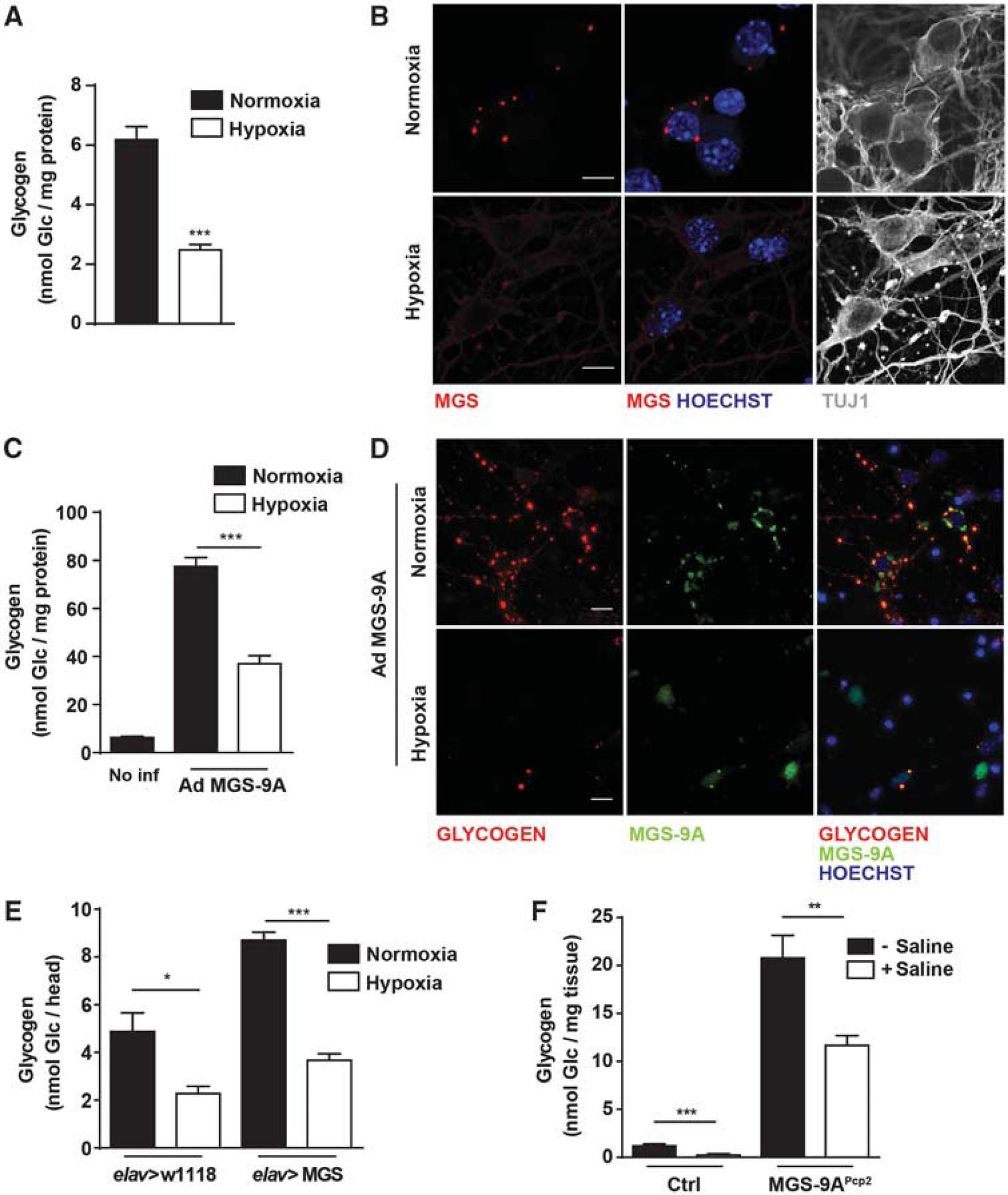
Neurons have the machinery necessary to degrade glycogen. Neurons were cultured for 5 days in Neurobasal with antimitotics. At 5 days in vitro (DIV5) medium was replaced with fresh neurobasal. (
To study the capacity of neurons to degrade glycogen in vivo, we first used the fly model Drosophila melanogaster. We used a previously reported transgenic line 21 that expresses MGS and accumulates glycogen specifically in neurons (elav>MGS). Both the control (elav>w1118) and the MGS-overexpressing lines were exposed to hypoxia (1 hour at 0.6% O2), and glycogen from whole heads was measured (Figure 2E). The control line showed a reduction in glycogen content. Such a reduction could take place in any kind of brain cell. However, the net decrease in glycogen content after hypoxia was higher in the elav>MGS line (Figure 2E). Given the neuronal specificity of the transgene, this difference could be attributed, at least in part, to the degradation of glycogen in neurons that overexpressed MGS.
This result was reinforced using a genetically engineered mouse that we previously described. 21 This line expresses the active form of GS, MGS-9A, specifically in Purkinje neurons (MGS-9APcp2). To mimic an extreme aglycemic anoxic condition, cerebella of WT and MGS-9APcp2 mice were isolated either immediately after anesthesia or 5 minutes after perfusion with saline, and total glycogen content was determined (Figure 2F). The glycogen content of WT mice, which was in accordance with the values of brain glycogen reported by other groups,32,33 dropped to almost zero after the saline perfusion. MGS-9APcp2 mice, which had a 15-fold increase in glycogen accumulation, also showed a marked reduction in glycogen levels after saline administration. This finding is explained only by the degradation of glycogen in Purkinje neurons. Taken together, our results show that neurons have the capacity to degrade glycogen in vitro and in vivo.
Neurons Degrade Glycogen Using Brain Glycogen Phosphorylase
We next characterized the molecular mechanism through which glycogen is mobilized. For this purpose, neurons infected with the adenovirus containing MGS-9A were exposed to hypoxia and incubated in the presence of inhibitors of GP (1,4-dideoxy-1,4-imino-D-arabinitol, DAB), autophagy (chloroquine) or lysosomal acid alpha-1,4–glucosidase (acarbose). Only by selectively blocking GP was glycogen degradation prevented (Figure 3A). In addition, under normoxic conditions, glycogen content almost doubled when GP was inhibited, thus indicating that GP was the enzyme responsible for the glycogen degradation. These observations were further confirmed by detecting glycogen using a specific antibody in neurons infected with the adenovirus and exposed to the same experimental conditions (Figure 3B). To confirm that neurons were indeed expressing GP, we analyzed its presence by western blot (WB) using isoform-specific antibodies. We introduced technical improvements that increased the sensitivity over that of our previous study. 17 The analysis of WT neurons revealed the presence of the brain but not the muscle isoform of GP (Figure 3C, quantification of the WB can be found in Supplementary Figure S2). In addition, no changes in GP total protein levels were observed after exposing WT neurons to 4 hours of hypoxia. Analysis of the subcellular distribution of GP in WT neurons revealed GP both in cell soma and in neuntes, in a punctate pattern, similar to that found for MGS (Figure 3D). However, after hypoxia GP activation state (expressed as the ratio (+)caffeine/(+)AMP) was increased (Figure 3E), although (+)AMP GP levels were unchanged (Figure 3F). The protein showed a diffuse pattern throughout the soma, indicating that degradation of glycogen had taken place (Figure 3D). To test whether GP was also expressed in adult neurons, brain GP was analyzed in parvalbumin-positive neurons in hippocampal sections (CA2) from adult mouse brains. Parvalbumin-positive neurons are a subtype of interneuron that express high levels of MGS 34 and are therefore good candidates for having glycogen degradation machinery. When these neurons were stained for brain GP, neuronal soma showed a positive signal (Figure 3G), thus indicating that these cells express the enzyme. Brain GP detection in astrocytes, as assessed by colocalization of this isoform and GFAP, was used as a positive control of the study. The 3D reconstruction of the images shown in Figure 3G confirmed the neuronal presence of GP in vivo (Figure 3H). Taken together, these findings show that neurons have the capacity to degrade glycogen—through the action of brain GP—and that this degradation is greatly enhanced under conditions of oxygen depletion.
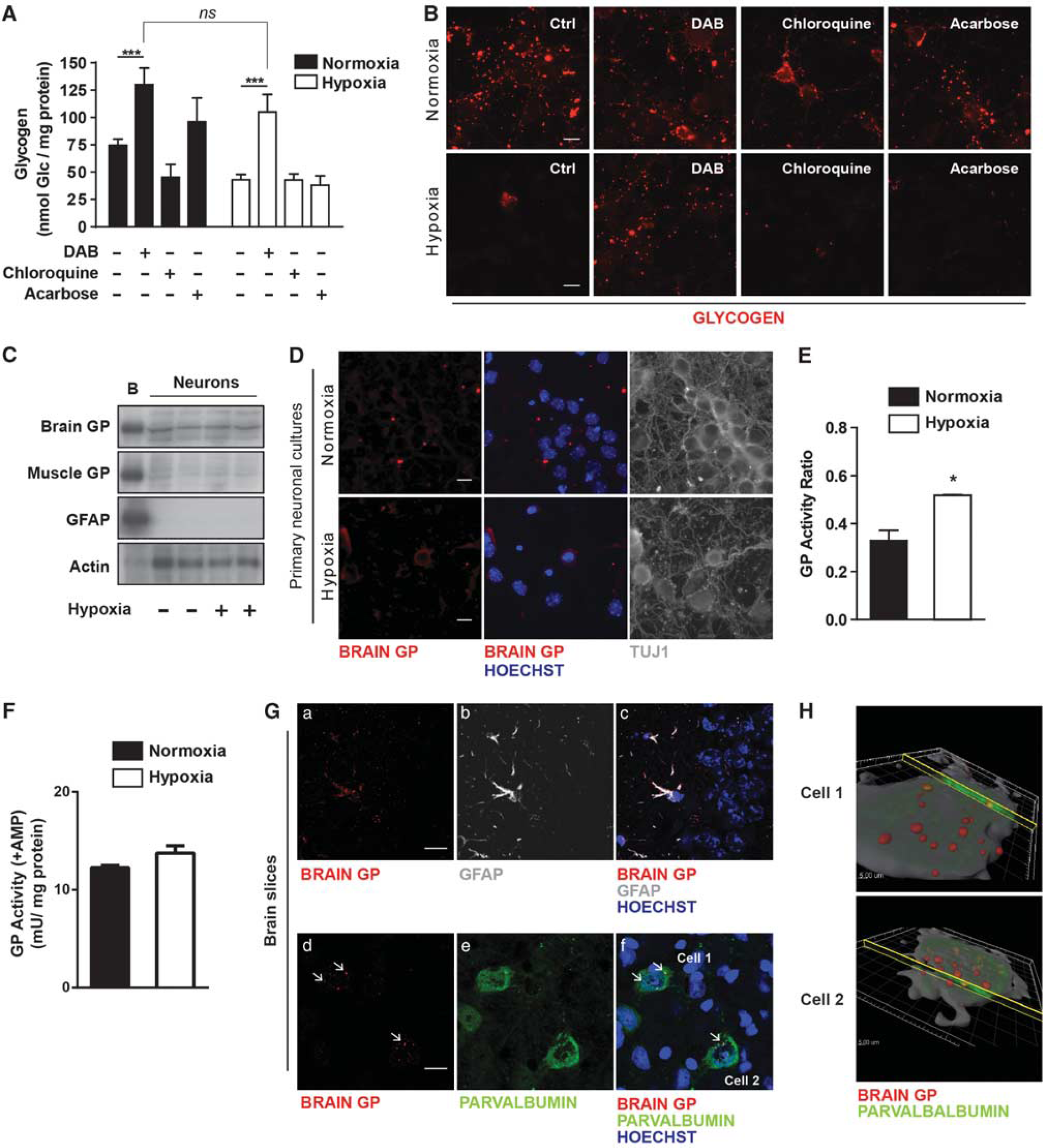
Neurons degrade glycogen using brain glycogen Phosphorylase (GP). (
Hypoxia Induces Muscle Glycogen Synthase Activation and Glycogen Synthesis But No Net Glycogen Accumulation
We next studied whether GS is regulated in hypoxia. Surprisingly, although glycogen was degraded in neurons exposed to hypoxia, the activation state of GS dramatically increased (Figure 4A, left panel). However, in contrast to what happens in other cell types, 35 total GS activity (a readout of the total amount of protein) remained unaltered (Figure 4A, right panel). Glycogen synthase showed increased electrophoretic mobility by WB analysis (Figure 4B, quantification of the WB can be found in Supplementary Figure S3), which is characteristic of the activated, dephosphorylated form of the enzyme. Accordingly, Serine 641 in GS was almost completely dephosphorylated in hypoxia-treated neurons, as measured with a specific antibody. Since both GS and GP were activated in hypoxia, we next tested whether glycogen synthesis and degradation occurred simultaneously. For this purpose, glycogen was measured in neurons exposed to hypoxia in the presence of the GP inhibitor DAB (Figure 4C). In agreement with the observed activation of GS, glycogen levels increased in hypoxia when GP was inhibited, thereby indicating simultaneous synthesis and degradation. In contrast, in normoxia, glycogen values with DAB practically remained unaltered. This observation would indicate a low turnover unless glycogen synthesis was influenced by its degradation. This is schematically represented in Figure 4G, in which low glycogen turnover is found in normoxia, but both glycogen synthesis and degradation increase in hypoxia as a result of the simultaneous activation of GS and GP.
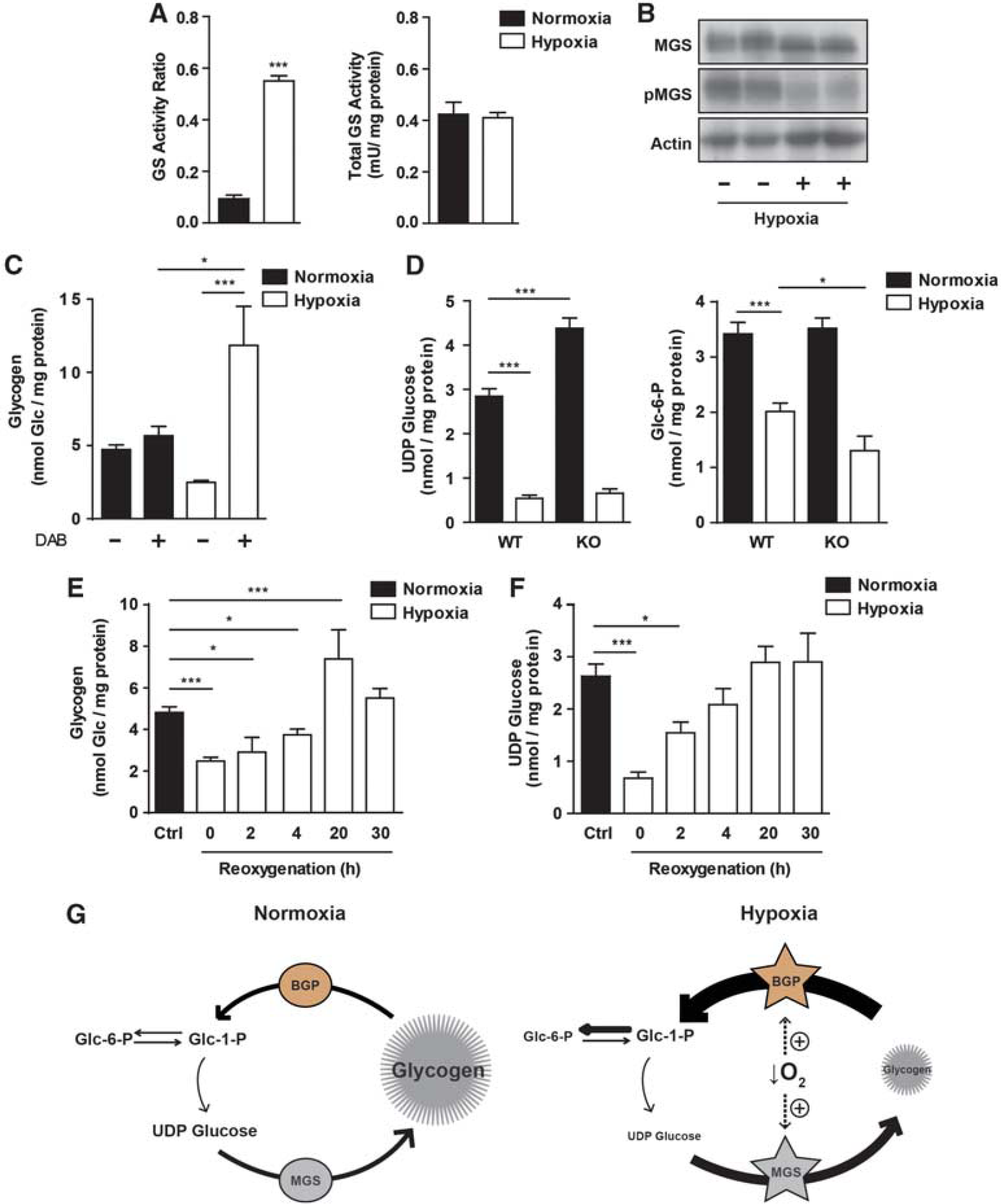
Hypoxia induces muscle glycogen synthase (MGS) activation and glycogen synthesis, but no net accumulation. Neurons were cultured for 5 days in neurobasal medium with antimitotics. At 5 days in vitro (DIV5) medium was replaced with fresh neurobasal and hypoxia was induced. (
To analyze whether additional regulatory factors influence the rate of glycogen synthesis, we analyzed the levels of GS substrate, UDP glucose. In hypoxia, a fivefold decrease in the concentration of UDP glucose was observed (Figure 4D, left panel). However, this decrease could be explained either as a consequence of the cellular adaptations to the lack of oxygen or as a result of the overactivation of GS. To solve this question, UDP glucose levels were measured in neurons lacking GS. Interestingly, basal levels of UDP glucose were significantly higher in GS KO neurons than in WT, thus supporting the hypothesis that neurons show active glycogen synthesis under resting conditions (Figure 4D, left panel). However, GS KO neurons subjected to hypoxia had equivalent levels of UDP glucose as the WT ones. This observation shows that the hypoxia-induced decrease in UDP glucose was not the consequence of a pull effect caused by increased GS activity. Since UDP glucose is formed from Glc-1-P (which is in equilibrium with Glc-6-P), the observed decrease in Glc-6-P in hypoxia (Figure 4D, right panel) may explain the decrease in UDP glucose. This reduction in Glc-6-P suggests that the entry and phosphorylation of glucose is not enough to fulfill the intracellular demand of the metabolite. Remarkably, GS KO neurons showed a greater reduction in Glc-6-P concentration in hypoxia, thereby indicating that glycogen contributed to maintain the levels of the metabolite during hypoxia. In this condition, Glc-6-P is used to fuel the glycolytic and Pentose Phosphate Pathway, thus being less available for glycogen synthesis. Therefore, decreased levels of UDP glucose may limit the rate of glycogen synthesis.
After a hypoxic episode, total brain glycogen levels are reported to rise above the levels registered before the insult. 36 This process is known as super-compensation and is a preparatory mechanism for further hypoxic stress episodes. 37 To test whether GS activation in hypoxia is a preparatory mechanism to resynthesize glycogen once the hypoxic exposure has ended, glycogen content was determined in neurons exposed to hypoxia for 4 hours and then incubated for increasing periods in normoxia (reoxygenation). Indeed, there was a twofold increase after 20 hours of reoxygenation compared with the basal glycogen levels (Figure 4E). The levels decreased to basal values after 30 hours of reoxygenation, as previously described for the whole brain. 36 UDP glucose concentration increased after reoxygenation and reached basal levels after 20 hours (Figure 4F). This finding suggests that UDP Glucose in reoxygenation was no longer a limiting factor for glycogen synthesis.
Glycogen Metabolism Has a Protective Role in Neurons Under Hypoxia
To address the functional relevance of neuronal glycogen metabolism, WT and GS KO neurons were exposed to increasing periods of hypoxia, and neuronal death was assessed (Figure 5A). A time-dependent increase in neuronal death was observed in WT cells. Importantly, GS KO neuronal mortality in hypoxia was clearly enhanced at 4, 6, and 8 hours, thereby indicating that GS was involved in the neuronal response to oxygen depletion. As a readout of the energetic status of the cell, we determined the levels of ATP of GS WT and KO neurons exposed to hypoxia (Figure 5B). As expected, ATP level gradually decreased in WT neurons under hypoxia. Interestingly, in GS KO neurons under basal conditions, ATP values were already lower than that in the controls, while the levels of both groups in hypoxia were equivalent.
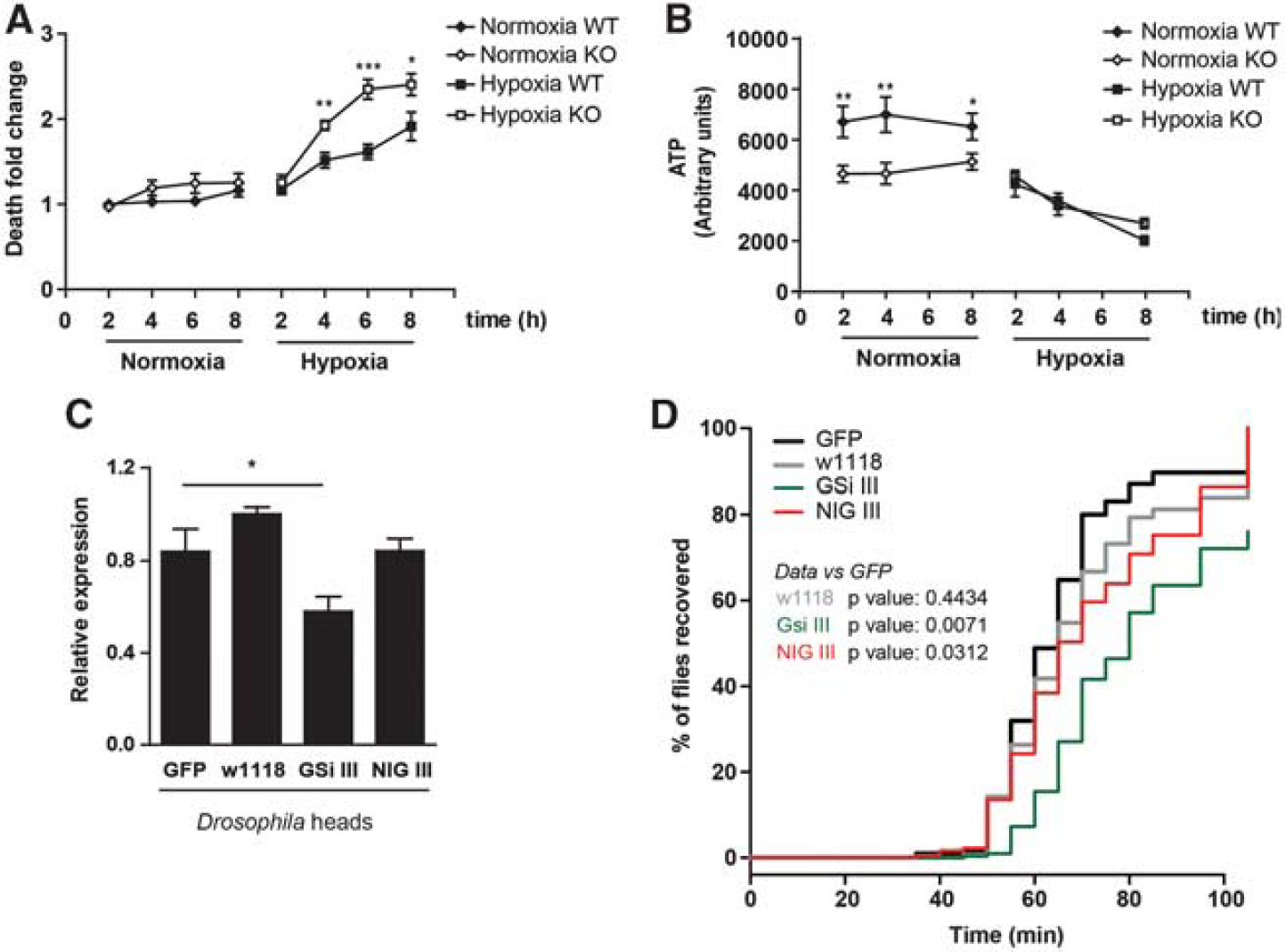
Glycogen synthase (GS) has a protective role in neurons under hypoxia. (
To assess the relevance of neuronal glycogen metabolism in the protection mechanism against hypoxia in vivo, we studied its role in D. melanogaster. We drove the expression of several shRNA against Drosophila GS (dGS) specifically in neurons (GSi III and NIG III). We first determined the efficiency of the dGS knock-down by measuring the messenger RNA levels of dGS from total heads (Figure 5C). Two control lines were used (a nontransgene expressing line—w1118 and a GFP-overexpressing line—GFP). The levels of GS were further reduced in the GSi III compared with the NIG III line. On the basis of previous studies on fly resistance to hypoxia, 38 we exposed flies to low oxygen (0.6% O2 for 1 hour) and determined their recovery latency in room air, a typical readout of the hypoxia tolerance of flies (Figure 5D). The dGS knock-down in adult neurons significantly delayed recovery relative to GFP-expressing flies, thus indicating that these flies were more sensitive to hypoxic stress. The severity of the phenotype correlated with the extent of dGS knock-down.
DISCUSSION
In this study we show that, against general belief, neurons do have an active glycogen metabolism and propose that it has a relevant role in neuronal survival in hypoxia. We show that neurons synthesize glycogen under basal conditions and have the full capacity to degrade it through GP. Our observations reveal the existence of a previously overlooked active glycogen metabolism in neurons.
This work builds on that of Vilchez et al, 17 whose main message was that neurons contain GS but that this enzyme must be kept essentially inactive, since its activation and the consequent glycogen accumulation cause apoptosis. As a result of that study, GS was deemed a ‘Trojan Horse’ for neurons. 39 Here, we refine that interpretation by showing that GS cannot be fully inactive since it is responsible for the synthesis of minute but functional amounts of glycogen far below the levels previously shown to induce neuronal death. In spite of these low concentrations of glycogen, here we show that glycogen metabolism is relevant for neuronal survival under conditions of hypoxia.
The cellular distribution of brain glycogen has been widely discussed, and it is generally accepted that in the brain glycogen is localized essentially in astrocytes, at a concentration of 60 to 200 nmol Glc/mg protein (see Supplementary Table S1). 2 Here, we show that glycogen is also present in immature cultured neurons and that its presence might have been overlooked because of its low concentration compared with that found in astrocytes. 40 This reinterpretation of the panorama has only been possible as a result of the optimization of the method used for glycogen measurement, which greatly increased its sensitivity. The method previously used by our group and others is not sensitive enough to detect the low levels of glycogen present in immature cultured neurons. The issue of sensitivity, together with the rapid degradation of glycogen in the postmortem state, probably hindered the proper detection of neuronal glycogen in previous studies.
A key finding of this study is that neurons express GP. Studies conducted many years ago reported the presence of GP in neurons.18,19 However, for the last decade, mainly after the immunofluorescence study performed by Pfeiffer-Guglielmi et al, 15 it has been accepted that these cells lack this enzyme. In fact, we also failed to detect GP in cultured neurons when measured by WB in Vilchez et al. 7 There are several possible explanations for this lack of detection. First, the detection of glycogen-bound enzymes by immunofluorescence is greatly influenced by the amount of glycogen present. As shown in this study, when bound to glycogen, both GS and GP can be clearly detected since they are in the form of aggregates. However, when glycogen is degraded, although total protein levels remain unaltered, the diffuse distribution of the enzymes makes them almost undetectable. This may have been the case when slices were prepared from whole brain postmortem. Second, since GP levels in neurons are far lower than in astrocytes (three times lower when comparing our results with Dringen and Hamprecht 41 ), it is plausible that the detection techniques used in previous reports were not sensitive enough. In our hands, endogenous GS and GP were detected by immunofluorescence only when using an amplification-based protocol. Similarly, recent technical improvements of WB have greatly increased the sensitivity of the method and explain why we now have been able to detect GP. Our results also show that GP is expressed in Parvalbumin-positive neurons in the adult brain. This finding suggests that GP expression is not restricted to embryonic neurons. The transcriptome analysis performed by Lovatt et al 42 comparing astrocytes and neurons from the brains of adult mice showed that the neuronal fraction is positive for both GS and GP. Thus, these results support our data indicating that GP is also present in adult neurons.
Our results show that neurons exclusively express the brain isozyme of GP and not the muscle one, while astrocytes express both forms. 15 These two isoforms differ in kinetic properties. The muscle isoform, activated mainly through phosphorylation, is tailored primarily to respond to extracellular signals, whereas the brain isoform, which is extremely sensitive to increases in AMP levels, is more adapted to respond to internal energy requirements. 43 Thus, the observation that neurons expressed only brain GP suggests that glycogen degradation in these cells is designed mainly to respond to their own energetic needs.
In contrast to what has been reported on muscle and hepatic cell lines, 35 total GS levels in neurons remain unchanged in hypoxia, but the enzyme is activated (dephosphorylated). In hypoxia, neuronal glycogen turnover is strongly enhanced by the simultaneous activation of GS and GP (shown in Figure 4G). The parallel activation of glycogen synthesis and degradation was first shown in exercised muscle and is referred to as the ‘glycogen shunt’. 44 Although energetically unfavorable, the shunt has been justified by the capacity of glycogen to rapidly generate energy at a specific time point, with a faster ATP production per molecule from glycogen-derived glucose than from extracellular hexose. 45 In the brain, the shunt hypothesis has been proposed for astrocytic glycogen; 46 here we show that it also takes place in neurons and that its absence makes these cells more vulnerable to oxygen depletion. However, the parallel activation of synthesis and degradation may be further modulated by other factors. First, although GS is activated in hypoxia, a number of factors may limit the rate of glycogen synthesis. Indeed, the concentration of both the allosteric activator of GS, Glc-6-P, and its substrate, UDP Glucose, decrease in hypoxia and could restrict the synthesis of the polysaccharide. Second, glycogen synthesis and degradation could be spatially separated, leading to the production of Glc-6-P as a result of glycogen mobilization in a specific subcellular region to respond local requirements, while elsewhere the polysaccharide would be synthesized. This scenario has been described in astrocytes with different lactate pools derived from extracellular glucose or from glycogen. 47 The hypoxia-induced activation of GS may also be a preparatory mechanism for the neuron to immediately resynthesize glycogen once the hypoxic stress is over, a process known as ‘super-compensation’. This process has been described in total brain glycogen after hypoglycemia 48 and hypoxia. 36 For super-compensation to occur, previous activation of GS is required. This situation would mimic a contracting muscle, in which there is glycogen super-compensation after the contraction. 49
A key conclusion of our study is that in a situation of hypoxic stress, glycogen metabolism protects immature cultured neurons from dying and flies from stupor. The exact mechanisms underlying this protection are unclear, but glycogen shunt-mediated protection may be involved. We have shown that the basal ATP value in cultured GS KO neurons is diminished. Thus, the amount of ATP available to face the hypoxic insult is lower in these cells. In addition, although glucose in the medium is in excess, intracellular Glc-6-P levels decrease in hypoxia. This observation suggests that under conditions of oxygen deprivation the extracellular uptake and phosphorylation of glucose do not fulfill the energetic requirements of the cell. Remarkably, Glc-6-P levels in GS KO neurons under hypoxia are lower than in those in GS WT cells, which may imply a survival disadvantage.
Since most of the experiments in this study were performed in immature neurons, we cannot rule out the possibility that glycogen metabolism in neurons has a protective role during brain development, which would provide protection, for example, for exposure to hypoxia during birth.50,51 Thus the impact shown here might be of greater relevance for the developing brain.
Here, we have shown that neurons have an active glycogen metabolism and that it has a biologically relevant function. To date, the diverse roles proposed for glycogen metabolism in the brain have been attributed exclusively to astrocytes. Although neurons have a much lower glycogen content than astrocytes (see Supplementary Table S1 for full comparison between cultured neurons, astrocytes, and whole brain), we propose that the contribution of neuronal glycogen to the above-mentioned brain functions to be reconsidered. Several questions are raised by this study, such as the biologic significance underlying glycogen protection, the function of the glycogen shunt—an ATP-consuming process—in an energy-demanding situation, and whether glycogen metabolism is important for other neuronal functions. Addressing these questions will help us to understand the differential regulation of glycogen metabolism between astrocytes and neurons and to unravel the complexity of glycogen metabolism in the brain.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Mar García Rocha for her help and Emma Veza and Anna Adrover for their technical assistance. Thanks also go to Evarist Planet for statistical analysis, Lídia Bardia for image processing, Joaquim Calbó for providing a critical review of the manuscript, and Tanya Yates for correcting the English manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.