
Abstract
To date, only limited data are available on the effects of pretreatment with novel oral anticoagulants in the event of traumatic brain injury (TBI). We determined intracerebral hemorrhage volume and functional outcome in a standardized TBI model in mice treated with warfarin or dabigatran. Additionally, we investigated whether excess concentrations of dabigatran could increase bleeding and whether this was preventable by using prothrombin complex concentrate (PCC). C57 mice were treated orally with warfarin or dabigatran; sham-treated mice served as controls. Effective anticoagulation was verified by measurement of international normalized ratio and diluted thrombin time, and TBI was induced by controlled cortical impact (CCI). Twenty-four hours after CCI, intracerebral hemorrhage volume was larger in warfarin-pretreated mice than in controls (10.1 ± 4.9 vs 4.1 ± 1.7 μL; analysis of variance post hoc P = 0.001), but no difference was found between controls and dabigatran-pretreated mice (5.3 ± 1.5 μL). PCC applied 30 minutes after CCI did not reliably reduce intracerebral hemorrhage induced by excess dabigatran concentration compared with saline (10.4 ± 11.2 vs 8.7 ± 7.1 μL). Our data suggest pathophysiological differences in TBI occurring during warfarin and dabigatran anticoagulation. The reduced hemorrhage formation under dabigatran therapy could present a safety advantage compared with warfarin. An excess dabigatran concentration, however, can increase hemorrhage.
Keywords
INTRODUCTION
Approximately 1.7 million cases of traumatic brain injury (TBI) occur in the US annually, making it one of the leading causes of long-term disability and death. Approximately 12% of elderly patients and 4% of patients overall are treated with anticoagulants while suffering from TBI, which presents a diagnostic and therapeutic challenge. 1 The use of warfarin pre-injury has been described to be a major risk factor for more severe disease progression compared with patients with unaltered coagulation. 2 Very little clinical data, however, are available on TBI occurring during treatment with the new oral anticoagulants (NOACs), i.e., the direct thrombin inhibitor dabigatran and the factor Xa-inhibitors rivaroxaban and apixaban. In fact, in an extensive literature research, we were able to identify only five case reports3–7 and one small case series 8 on head trauma patients treated with dabigatran. All of them suggested increased bleeding complications due to anticoagulant intake.
Thus, there is an urgent need for exploring the pathophysiology of TBI occurring during treatment with NOACs, in the wake of their increased prescription for the prevention of thromboembolic events in patients with atrial fibrillation. A much closer focus on this problem is warranted not only for prognostic reasons, but also to assess potential therapeutic strategies in cases of bleeding, such as the rapid reversal of anticoagulation on using recombinant factor VII or prothrombin complex concentrate (PCC).
In a translational approach, we developed an experimental model of TBI during dabigatran anticoagulation. The first part of the study was designed to compare intracranial hemorrhage volumes between control, warfarin-, and dabigatran-pretreated mice. The second part evaluated long-term outcome and lesion size. The third part investigated the reversibility of excessive dabigatran anticoagulation after TBI using PCC.
MATERIALS AND METHODS
Animals
All experiments were performed in adherence to a protocol approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee in accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals. Adult C57/BL6 mice (Charles River Laboratories), aged 10–12 weeks, were used for all the studies. Mice were housed in 12-hour day/night cycles in a pathogen-free environment at the Massachusetts General Hospital Institutional Animal Facility.
Study design
In the first part of our study, to compare hemorrhage volume and neurologic outcome 24 hours after controlled cortical impact (CCI) between different anticoagulant-pretreated mice, we randomly and blindly assigned animals to three groups (control, warfarin, or dabigatran; 10 animals per group). In the second part we randomized mice in the same manner for analyzing long-term functional outcome up to 31 days after CCI. In the third part of the study, we compared control mice (n = 15) and mice dosed with excess dabigatran etexilate (DE, n = 18) that were randomized to receive either saline or PCC treatment (for anticoagulation reversal) 30 minutes after CCI. Twenty-four hours later brains were removed and hematoma volumes determined. Sample size was determined to be n = 10 using a power calculator for clinical trials based on data derived from an experimental TBI study following warfarin anticoagulation. 9
Pretreatment with oral anticoagulants and anticoagulation reversal
For administration of warfarin we followed a previously established protocol. 10 In brief, a 5-mg tablet of Coumadin (Bristol-Myers Squibb, New York City, NY, USA) was dissolved in 375 ml of tap water (warfarin concentration 0.013 mg/mL). Assuming a body weight of approximately 25g per mouse and an average water consumption of 15mL/100g over 24 hours, this leads to a warfarin uptake of 0.042 mg/mouse (1.67 mg/kg) over a feeding period of 20 hours. Measurement of coagulation parameters or CCI was performed directly after completion of warfarin feeding.
Anticoagulation with dabigatran was performed as previously described. 11 Contents of one 110-mg Pradaxa capsule (DE, Boehringer Ingelheim, Biberach, Germany) were dissolved in 5mL saline solution, and administered by oral gavage containing 0.085 mL three times in intervals of 8 hours (i.e., 3 × 75 mg/kg). The last gavage was given 1 hour before coagulation measurement or CCI. Control mice received a corresponding volume of saline solution in the same manner as the DE administration.
To achieve a supratherapeutic concentration for the third part of the study, DE (one 110mg Pradaxa capsule) was dissolved in 137.5 mL PBS, stirred for 1 hour at 35 °C, and injected at a concentration of 9 mg/kg intraperitoneal, as described in a recent paper with minor modifications. 12 For anticoagulation reversal, either 200U/kg of four-factor PCC (Beriplex, CSL Behring, Marburg, Germany) or an equal amount of saline (200μL) was injected into the tail vein 30 minutes after TBI in a masked fashion.
Measurement of coagulation parameters
The degree of anticoagulation was measured in three to four mice from each group. Mice were deeply anesthetized; a lethal amount of 0.6 mL whole blood was obtained via cardiac puncture with a 23-gauge needle and transferred into plastic tubes containing 67μL of 3.2% citrate, resulting in a 9:1 ratio. Prothrombin time was measured using a Destiny Max analyzer with TriniCLOT HTF reagent (Diagnostica Stago, Asnieres, France). International normalized ratio (INR) was calculated from prothrombin time. For the diluted thrombin time (dTT), 100μL of 1.5U/mL STA thrombin (Diagnostica Stago) was added to 100μL of diluted mouse plasma (mouse plasma diluted 1:4 in normal human plasma), and the clotting time was measured using a START-4 analyzer (Diagnostica Stago). The final concentration of thrombin was 0.75 U/mL. 13 The dTT gives results in seconds and, using a standard curve generated in our laboratory, results can also be given in ng/mL dabigatran concentration.
Controlled cortical impact
For inflicting TBI we used the CCI model as previously described with minor modifications.9,14 Mice were anesthetized with isoflurane distributed by a Fluotec vaporizer and positioned in a stereotactic frame. A 5-mm craniotomy was made using a portable drill and a trephine over the left parietotemporal cortex, and the bone flap was removed. Utmost care was taken to not disrupt the dura. CCI was performed using a pneumatic cylinder with a 3-mm flat-tip impounder, 5m/second velocity, 0.6 mm depth, and 150 milliseconds impact duration. Subsequently, the bone flap was not replaced in order to limit the confounding effects of intracranial hypertension. All mice were then allowed to recover under a heat lamp and were conscious and able to move after approximately 5 minutes.
Quantification of intracerebral hemorrhage volume
A photometric hemoglobin assay was used to quantify hemorrhagic volume 24 hours after CCI. For doing so, mice were transcardially perfused with 30 mL PBS under deep isoflurane anesthesia (5%). The brains were removed by the surgeon, who was unaware of the treatment assignment, and large epidural blood clots, if present, were carefully extracted and discarded without manipulating intraparenchymal bleeding. Brains were then placed in glass tubes containing 3mL PBS. After 30 seconds of homogenization, ultrasound was applied for 1 min to lyse erythrocytic cell membranes. After centrifugation for 30 minutes (13 000r.p.m. at 4°C), 250μL of supernatant was added to 1000μL of Drabkin's reagent. Using a photometer, absorption rates were determined at 540 nm, and hemorrhage volumes were calculated based on a standard curve.9,11
Functional outcome assessment and lesion size measurements
In the first part of the study, functional deficits were assessed 24 hours after CCI by using the modified Neurologic Severity Score, a 10-point score detecting neurologic deficits in motor, behavioral, and spontaneous locomotion tasks. 15 Additionally, a standard hanging wire test was conducted, and performance was assessed on a 5-point scale. 14 The latency until a fall from the wire occurred was recorded for three trials on each day (a maximum of 60 seconds was allowed).
In the second part of the study, functional outcome was determined by using the modified Neurologic Severity Score and hanging wire testing (as described above) on the following days after CCI: 1, 3, 5, 12, 16, 24, and 30 as compared with pre-injury testing. At the end of the study period mice were euthanized and brain sections of size 12 μm obtained at 0.5-mm intervals were stained with hematoxylin and eosin. Lesion size was quantified with a standard computer-assisted image analysis technique using NIH ImageJ Version 1.46r (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
IBM SPSS version 20 (IBM, Armonk, NY, USA) was used for statistical analysis. Parametric data (hemorrhage volume, hanging wire latency) were compared between the three treatment groups using analysis of variance (ANOVA). Post hoc testing was performed with the Bonferroni test. Non-parametric data were compared using the Kruskal-Wallis test. Mann-Whitney U testing was used for post hoc comparison between groups. P<0.05 was considered as a significant finding. Data are displayed using box plots, with the boxes indicating the median and the 25th and 75th percentile, respectively. Whiskers are extended to the maximum and minimum values.
RESULTS
Blood coagulation parameters
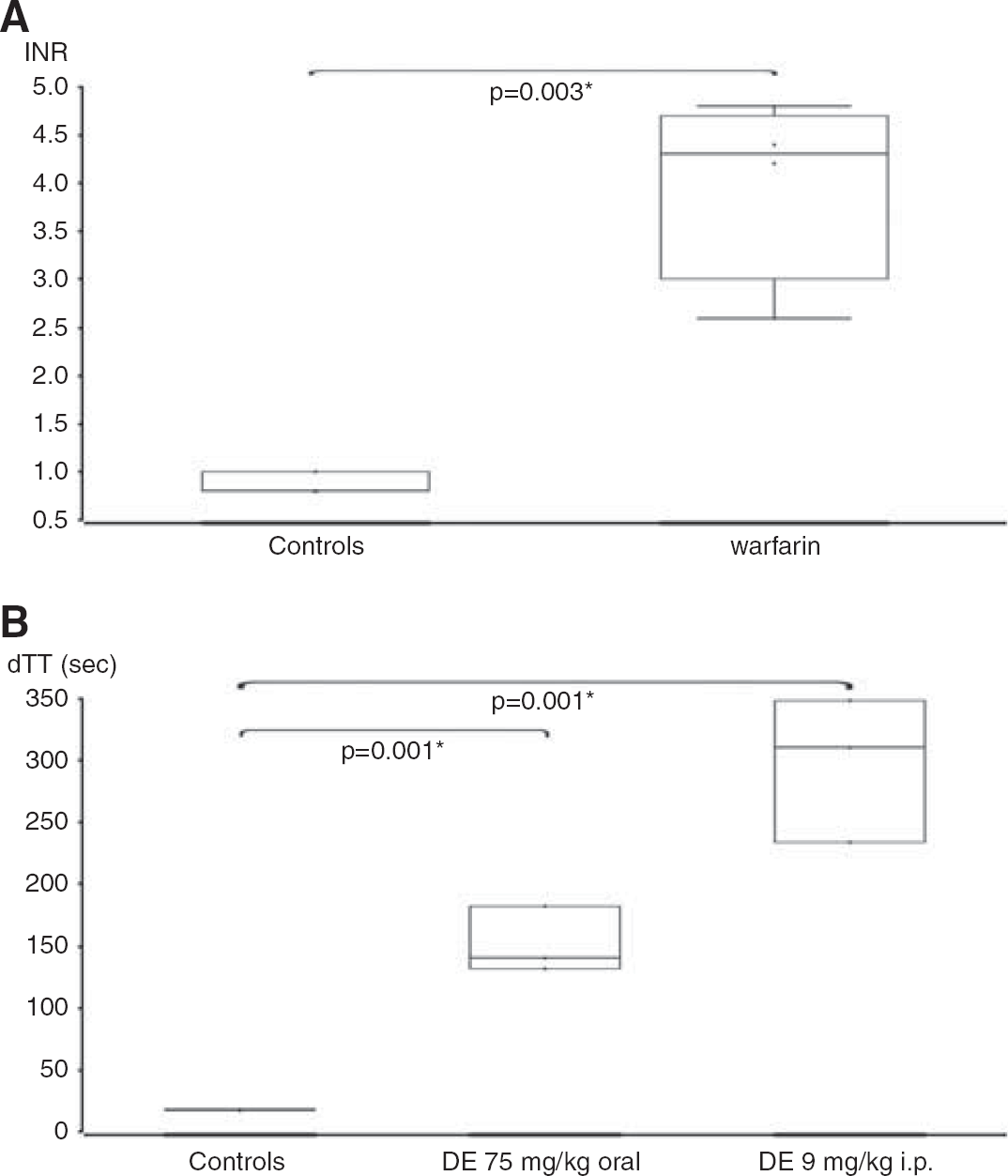
To ensure that anticoagulation was comparable to the therapeutic range in humans, we measured the standard coagulation parameters. The INR was 0.9 ± 0.1 (mean ± s.d., n = 3) in control mice. Oral administration of warfarin led to an increased INR of 4.0 ± 0.9 (n = 4, t-test, P = 0.003; Figure 1A). The dabigatran-sensitive dTT was 151.6 ± 26.6 seconds in the dabigatran group as compared with 17.7 ± 0.5 seconds in the control group (n = 3, P = 0.001; Figure 1B). The dTT of 151.6 seconds corresponds to 365.6 ng/mL dabigatran according to a standard curve, which is a high-therapeutic peak level. The dTT of 17.7 seconds in the controls corresponds to no dabigatran. In the excess dabigatran group a single intraperitoneal injection of 9 mg/kg produced a 16.8-fold prolongation of the dTT to 297.6 ± 58.5 seconds (n = 3, P = 0.001). The dTT of 297.6 seconds corresponds to > 526 ng/mL dabigatran (beyond the measuring capability of the assay, but an extrapolated estimate is 850.6 ng/mL), which is a very supratherapeutic level. PCC administration (applied for anticoagulation reversal) decreased the dTT in the excess dose dabigatran-pretreated mice to 89.1 ± 60.3 seconds (but none of the values returned to baseline), whereas mice that received saline had still largely elevated dTT values (221.7 ± 17.6 seconds; n = 3 per group, P = 0.020; Figure 2). The dTT of 221.7 seconds corresponds to > 526 ng/mL (extrapolated estimate 598.5 ng/mL), and the dTT of 89.1 seconds corresponds to 157.9 ng/mL dabigatran, a therapeutic trough level.
Intracerebral hemorrhage volume after CCI (part 1)
Two mice in the warfarin group died 24 hours after CCI injury; there was no mortality in the dabigatran and control groups. One mouse in the control and one in the dabigatran group were excluded, because their brains could not be extracted reliably enough to allow for precise hemorrhage measurement. However, both mice were alive after 24 hours. No significant differences between groups were discernible in the neurologic behavior evaluations 24 hours after CCI (5-point score, hanging wire latency). Figure 3 shows representative brain sections of each group 24 hours after CCI.
(
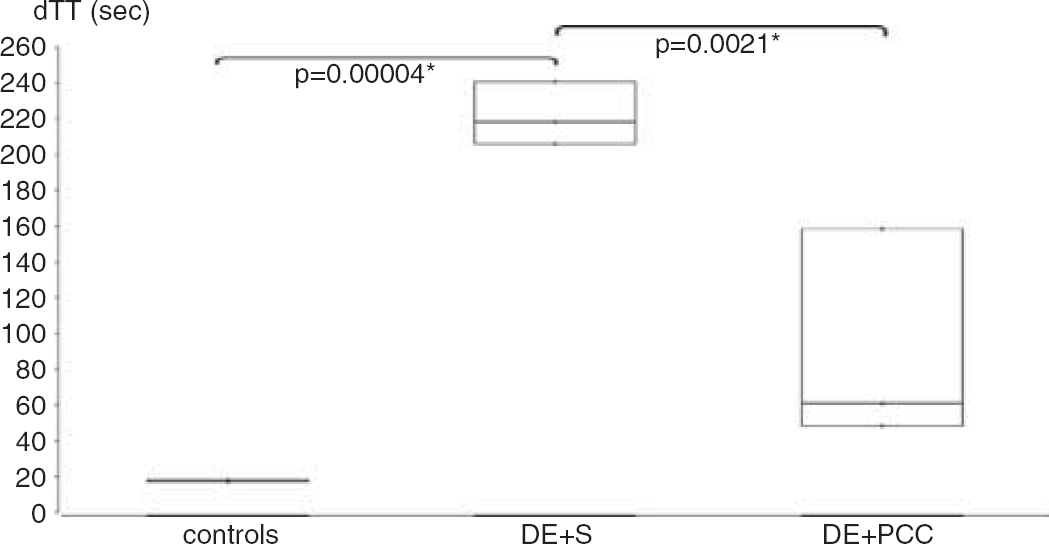
Reversal of excess dabigatran pretreatment (9 mg/kg intraperitoneally) is shown. One hour after dabigatran etexilate administration, mice were randomized to receive either 200 μL saline (DE + S) or 200U/kg PCC (DE + PCC) via tail vein injection. Blood was drawn 15 minutes later. * Indicates significant difference. dTT, diluted thrombin time.
Intracerebral hemorrhage volume was ˜2.5-fold larger in warfarin-pretreated mice than in controls (10.1 ± 4.9μL (n = 8) vs 4.1 ± 1.7 μL (n = 9), ANOVA P = 0.001; post hoc P = 0.001; Figure 3). In contrast, there was no significant difference between dabigatran-pretreated animals (5.3 ± 1.5 μL (n = 9)) and controls. Dabigatran-treated animals had a significant twofold smaller hemorrhage volume than the warfarin-treated mice (P = 0.010). The highest hemorrhage volume in the warfarin group was 15.7 μL, which exceeded the maximum volume in the control group by 2.3-fold (6.7 μL) and the dabigatran group by 2-fold (7.6 μL; Figure 4).
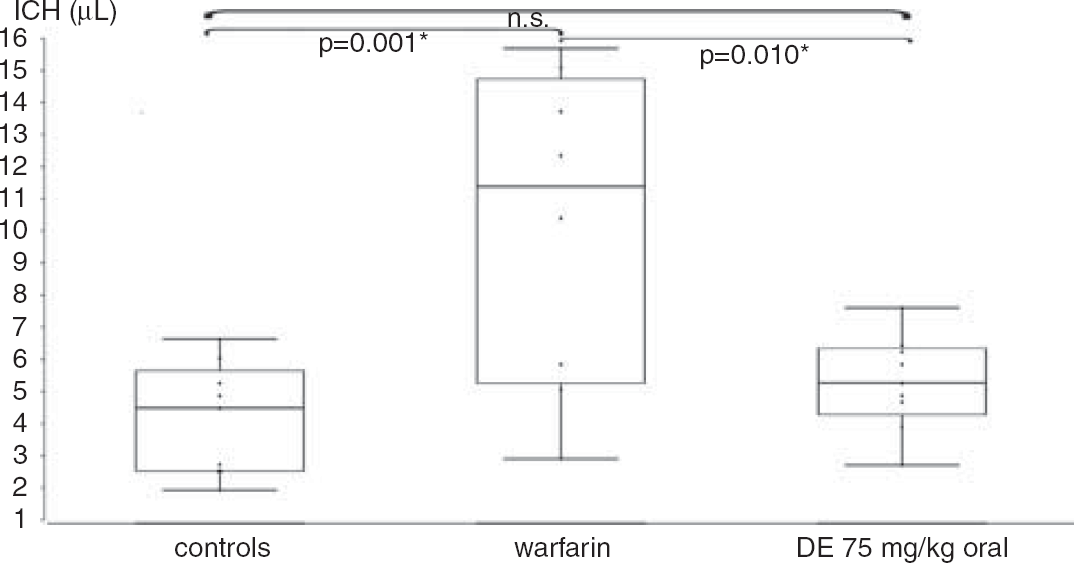
Intracranial hemorrhage (ICH) volumes 24 hours after controlled cortical impact for control mice (n = 9), warfarin mice (n = 8), and dabigatran etexilate (DE) mice (n = 9), measured by using a photometer and a standard curve. * Indicates significant difference, n.s., not significant.
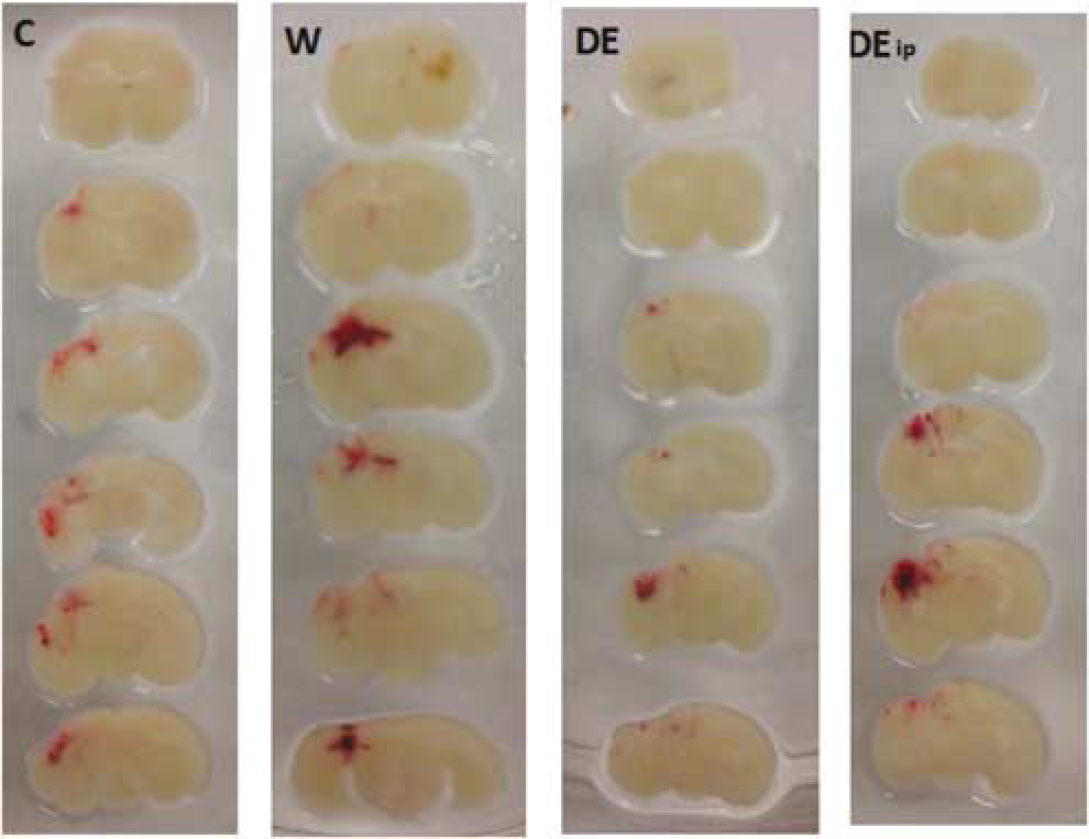
Representative brain sections depicting intracranial hemorrhage 24 hours after controlled cortical impact in control mice (C), warfarin mice (W), dabigatran etexilate mice (DE), and excess dabigatran etexilate mice (DE ip).
Long-term outcome after CCI (part 2)
One mouse in the warfarin group and one mouse in the control group died on day 5 and 12 after CCI, respectively. One mouse in the dabigatran group was lost on day 1 after CCI owing to an accident involving behavioral testing. All other animals survived the entire study period. Regarding body weight gain, hanging wire grip score and latency, and the modified Neurologic Severity Score, no significant differences were observed between groups, although changes over time pointed toward a tendency for a delayed functional recovery in the warfarin group (Figure 5).
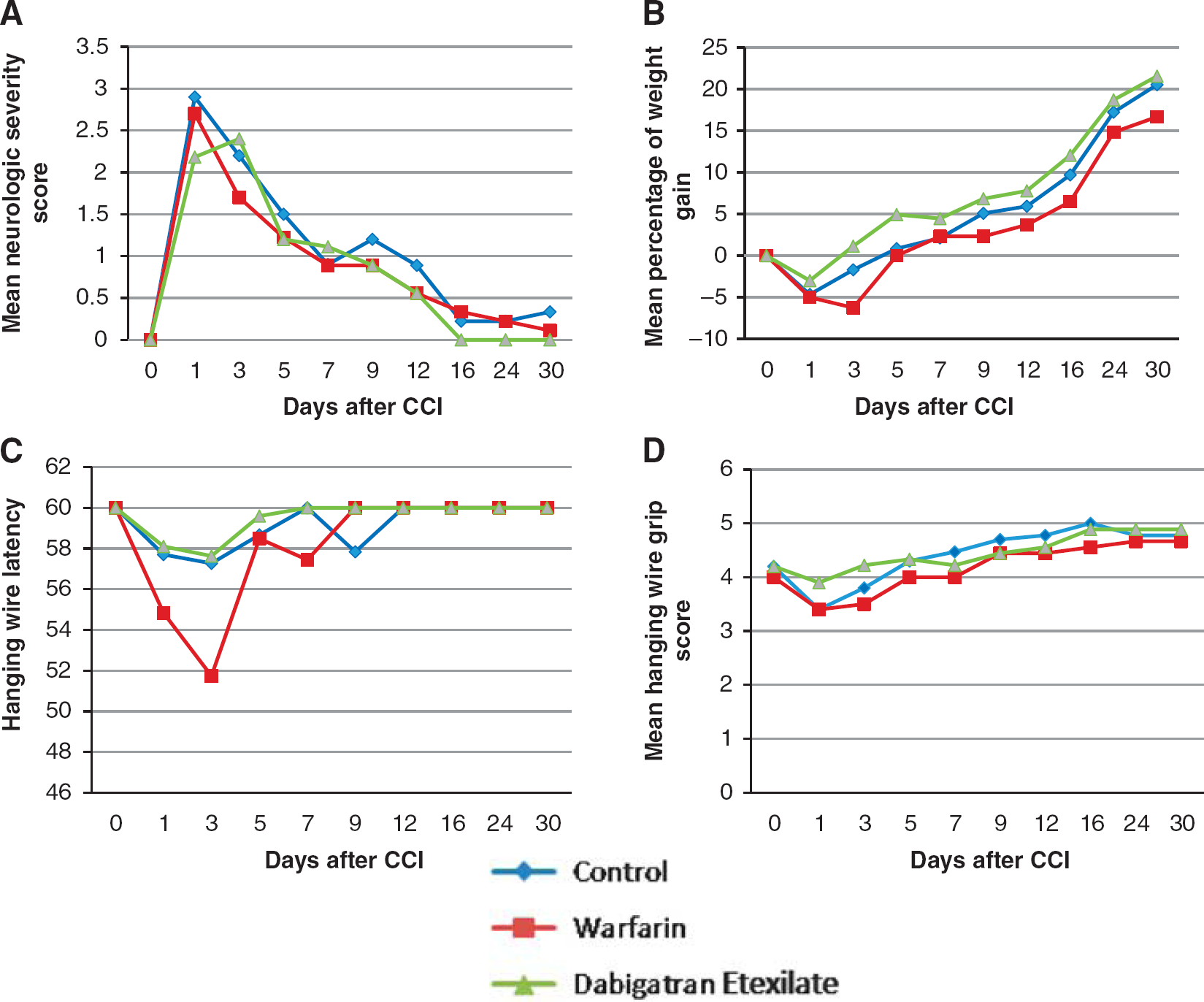
Long-term functional outcome. (
Lesion size at 31 days after CCI did not significantly differ between controls (7.7 ± 3.5 mm3, n = 9), warfarin- (8.4 ± 4.7 mm3, n = 8) and dabigatran-pretreated mice (5.5 ± 3.5 mm3, n = 9).
Excess dabigatran and anticoagulation reversal (part 3)
The only mortality during the 24-hour period was observed in the dabigatran group receiving saline after CCI. Hemorrhage volume 24 hours after CCI injury was increased approximately twofold in excess dabigatran animals receiving saline as compared with non-anticoagulated control mice receiving saline (8.7 ± 7.1 μL (n = 8) vs 4.6 ± 1.1 μL (n = 9); Kruskal-Wallis P = 0.041, post hoc P = 0.046; Figure 6). Excess dabigatran mice receiving PCC had a hemorrhage volume of 10.4 ± 11.2 μL (P = 0.077 vs control). No difference in hemorrhage volume was found between dabigatran mice receiving saline and dabigatran mice receiving PCC. Whereas the highest value in control mice receiving saline was 6.6 μL, one animal in the dabigatran group receiving saline and one animal in the dabigatran group receiving PCC had excessive bleeding 24 hours after CCI (25.7 and 39.0 μL, respectively).
DISCUSSION
The findings of our translational study suggest important pathophysiological differences in experimental TBI induced during warfarin and dabigatran anticoagulation. No difference in intracerebral hemorrhage volume was observed in mice with an unaltered coagulation status and mice receiving therapeutic doses of DE before the induction of CCI. In contrast, warfarin pretreatment induced significantly elevated hematoma formation.
Despite the fact that NOACs are increasingly used since their approval for the prevention of stroke and embolic events in atrial fibrillation patients, very little is known about their effects in TBI occurring during treatment. It is unclear whether hematoma volumes are increased in NOAC-treated trauma patients, and whether the rapid reversal of anticoagulation is beneficial in this setting. Furthermore, elderly patients are more prone to fall, which is often referred to as a reason to not anticoagulate, despite very little evidence for bleeding complications. In view of the lack of reliable clinical data, this field of research particularly qualifies for a translational approach.
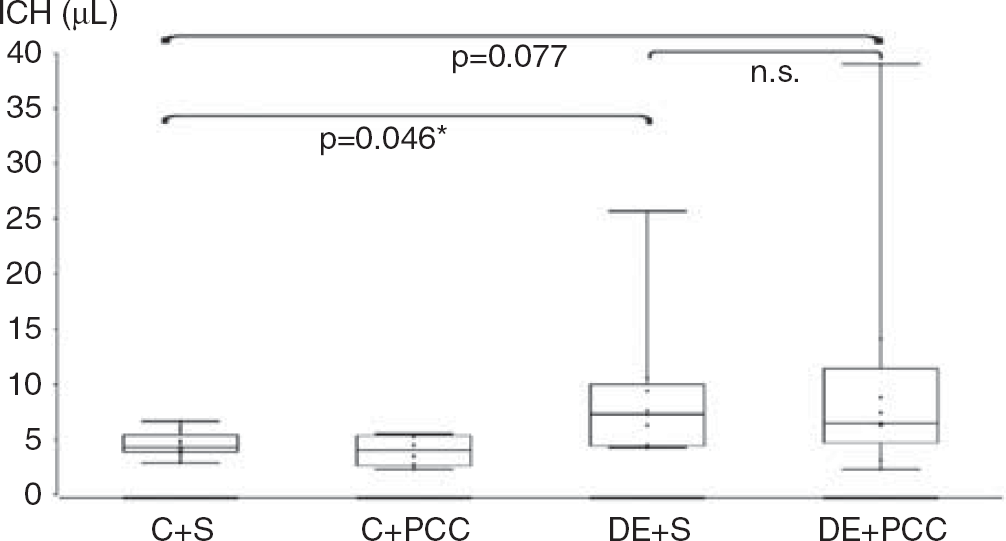
Intracranial hemorrhage (ICH) volumes 24 hours after controlled cortical impact (CCI) for control mice with subsequent saline injection (C + S, n = 9), controls with subsequent prothrombin complex concentrate injection (C + PCC, n = 6), excess dabigatran pretreatment with subsequent saline injection (DE + S, n = 8), and excess dabigatran pretreatment with subsequent PCC injection (DE + PCC, n = 9). * Indicates significant difference.
In order to provide for a comparable concentration of the two anticoagulants in our study, we evaluated the dabigatran plasma levels and the coagulation measures as derived from the randomized evaluation of long-term anticoagulation therapy (RE-LY) study. 16 In these patients, the peak dabigatran concentration was 100–310 ng/mL (10th–90th percentile) and trough dabigatran concentration was 50–160 ng/mL 17 The dTT of 151.6 seconds in our murine 75 mg/kg dabigatran group corresponds to 365.6 ng/mL dabigatran according to a standard curve, which is a high-therapeutic peak level. The target INR in the RE-LY study was 2–3, as it is in clinical practice during anticoagulation treatment with warfarin. In the RE-LY study, INR values fell into this range only 64% of the time. Our oral administration of 1.67 mg/kg warfarin led to an INR of 4.0 ± 0.9. Hence, anticoagulation parameters measured in our experiment indicate that the effect of both types of medication fell into a moderately supratherapeutic range.
Our finding that warfarin anticoagulation increases hemorrhage volume in TBI is in line with observational clinical data 2 and the results from one experimental study by our group in mice. 9 Furthermore, the absence of increased intracerebral bleeding under dabigatran anticoagulation in therapeutic dosages is similar to the observations from experimental studies of primary intracerebral hemorrhage. 11 Additional evidence stems from a subgroup analysis of the RE-LY study, which has observed significantly fewer traumatic intracranial hemorrhages in dabigatran patients compared with warfarin patients. 18 This raises the challenge how to explain the difference in hematoma volumes between dabigatran and warfarin. One possibility might be that warfarin has a more global effect on the coagulation cascade by impairing normal synthesis of the vitamin K-dependent factors II, VII, IX, and X, so they are no longer functional, whereas dabigatran selectively, yet reversibly, only inhibits factor IIa. In particular, the considerably large presence of tissue factor (factor III) in the perivascular system of the brain may be part of a possible pathophysiological explanation. 19 Under dabigatran treatment, the activated tissue factor-factor Vila complexes, which are formed after traumatic rupture of blood vessels, may provide enough local initiation of clotting via the extrinsic pathway to limit hemorrhaging. During warfarin anticoagulation factor VII levels are decreased and this may contribute to larger hematoma formation. Another explanation may be that dabigatran only binds to the active site of the thrombin molecule, leaving the two exosites functional for platelet activation, thereby allowing a local ‘thrombin burst’ and swift hemostasis. 20
In contrast, vastly supratherapeutic plasma levels of dabigatran (achieved via intraperitoneal injection) led to a significant increase in hemorrhage volumes after TBI. This is consistent with the results obtained by Zhou et al. 12 in a primary intracerebral hemorrhage model. In human patients, supratherapeutic dabigatran concentrations can be reached by acute or severe chronic kidney insufficiency 21 or dabigatran overdose. Pathophysiologically, it could be assumed that excess levels of dabigatran result in a continual suppression of factor IIa, whereby the ability for stable hemostasis by fibrin formation could be impaired.
It has been suggested that PCC might be an effective therapeutic strategy to limit excessive bleeding under supratherapeutic dabigatran dosages. 12 According to some guidelines, patients with bleeding occurring under dabigatran treatment should generally be treated with hemodialysis (in order to remove the drug), activated PCC, or factor VIII inhibitor bypass agent. 22 Other guidelines did not reach a consensus regarding the use of PCC. Effects on outcome are not yet proven, since no data from randomized, controlled studies in this setting are available. 23 There are some case reports pointing toward a better recovery after PCC administration. Chang et al. 5 report a patient with a successful recovery after TBI under dabigatran anticoagulation. 8U/kg of factor VIII inhibitor bypass agent was applied, followed by a 3-hour hemodialysis. It remains unclear whether the factor VIII inhibitor bypass agent had a significant impact on the clinical outcome. In a different case, blood loss due to hematemesis and melana under dabigatran anticoagulation appeared to be promptly reduced after receiving 40 IU/kg of PCC (Beriplex). 24 Taken together, there is a lack of data regarding this clinical problem and an evidence-based routine reversal strategy for dabigatran in trauma patients is not available at this point of time.
Zhou et al. 12 showed in the aforementioned study that PCC reduced bleeding expansion in mice with high levels of dabigatran. We investigated whether a similar effect could be achieved in the event of TBI. Our coagulation studies revealed that application of a high dose of PCC partially reduced, but did not completely normalize, the dTT after excessive dabigatran treatment. This phenomenon could possibly be explained by the excess dabigatran dosage, which may intercept and continuously inhibit any thrombin made available through the injection of PCC. It is important to note, however, that coagulation parameters such as dTT and activated partial thromboplastin time may not reliably predict a successful anticoagulation reversal for dabigatran. In experimental studies, diminished bleeding times have been observed in the absence of measurable parameter changes after PCC administration.12,25
In our third experimental series, the application of PCC 30 minutes after TBI for anticoagulation reversal did not prevent the development of larger bleeding volumes in some of the mice with excess dabigatran levels as compared with saline injections. Apart from an incomplete anticoagulation reversal, another explanation might be that the PCC injection occurred at a time that was too late to substantially change the hemorrhage volume after 24 hours. In contrast to the intracerebral haemorrhage model, in which hematoma formation develops over more than 30 minutes, CCI produces an immediate injury to the blood vessels, causing sudden hemorrhage. Hence, profound bleeding may occur until the time of anticoagulation reversal. Notwithstanding, a single intraperitoneal injection of DE did not result in remarkably enlarged hemorrhage volumes in all mice, raising the possibility that our model was not sensitive enough to detect a significant decrease in intracranial hemorrhage volume after PCC administration.
The evaluation of long-term outcome over a 31-day study period in dabigatran-, warfarin-, and placebo-treated mice did not yield any significant differences. There was a trend toward lower body weight, lower hanging wire scores, lower hanging wire latency, and larger lesion size in the warfarin-pretreated animals. Potentially, the initial damage inflicted upon the cortex after impact could only be minimally exacerbated by prolonged bleeding and any additional detrimental effects are therefore below the threshold for detection by the behavioral tests we employed. An additional issue with the murine CCI model was already raised in reports about a dissociation between lesion size and functional recovery. 14
Histologic assessment included lesion size after 31 days. Although no overall significance was detectable, a lower mean lesion volume was found in dabigatran-treated mice compared with the warfarin-pretreated animals (5.5 ± 3.5 vs 8.4 ± 4.7 mm3, ANOVA P = 0.16). The lack of a significant correlation between larger hemorrhage volume and greater lesion size between the different groups could be due to bleeding into an already preformed contusion cavity and as such the hemorrhage could not augment the already pre-existing lesion. Furthermore, the decompressive effect of craniectomy might have limited further enlargement of the parenchymal lesion through hematoma formation, as we did not replace the bone flap after CCI, thereby allowing blood to exit the cranium.
In a direct comparison, dabigatran appears to provoke less bleeding than warfarin at the doses used and hence shows a superior safety profile after traumatic cortical impacts. Our finding that excessive plasma levels of dabigatran could lead to larger hemorrhage formation after CCI with little effect of anticoagulation reversal in a relatively short time frame leads to the conclusion that careful observation of the degree of anticoagulation in patients treated with the NOACs who present in the clinic with head trauma is imperative.
Our study has some limitations, one being that the murine anticoagulation model can never fully mimic the human system, but has been shown to be extensively comparable in its properties and reaction to oral anticoagulants. Additionally, TBIs in the clinical setting present themselves more heterogeneously, whereas the CCI model only imitates a penetrating cortical impact. However, this inherent simplification presents a reliable and standardized model for the comparison of different anticoagulation methods in regard to their effect on intracranial hemorrhage after TBI. This approach allows to proceed without external confounding factors such as age, gender, mechanism of injury, and comorbidities.
Considering the trends toward a higher mortality, a lower weight gain, and a larger lesion size in the warfarin group compared with the dabigatran group, there is a possibility that a larger sample size would have conferred a significant finding.
Summary
Compared with warfarin pretreatment, therapeutic dabigatran anticoagulation led to lower intracranial hemorrhage volumes 24 hours after TBI. The application of PCC 30 minutes after TBI for anticoagulation reversal did not decrease increased hemorrhage volumes caused by an excess dose of dabigatran.
Footnotes
This is an investigator-initiated study. It was supported by Boehringer Ingelheim with a restricted grant. The study was designed independently of the sponsor. The experiments as well as the analysis of the data were performed without involvement of the sponsor. With regard to her expertise in the field, Joanne van Ryn helped in interpreting the dabigatran plasma concentrations in the experimental setting and helped in editing the manuscript as a native English speaker.