
Abstract
Neovascularization is an innate physiologic response by which tissues respond to various stimuli through collateral remodeling (arteriogenesis) and new vessel formation from existing vessels (angiogenesis) or from endothelial progenitor cells (vasculogenesis). Diabetes has a major impact on the neovascularization process but the response varies between different organ systems. While excessive angiogenesis complicates diabetic retinopathy, impaired neovascularization contributes to coronary and peripheral complications of diabetes. How diabetes influences cerebral neovascularization remained unresolved until recently. Diabetes is also a major risk factor for stroke and poor recovery after stroke. In this review, we discuss the impact of diabetes, stroke, and diabetic stroke on cerebral neovascularization, explore potential mechanisms involved in diabetes-mediated neovascularization as well as the effects of the diabetic milieu on poststroke neovascularization and recovery, and finally discuss the clinical implications of these effects.
INTRODUCTION
The simple fact that the mammalian brain, which accounts for only 2% of the body mass, actually receives 20% of the cardiac output makes one realize the complexity and the importance of the cerebrovascular network in proper brain function.1, 2 It is estimated that the capillary network of the brain runs ∼400 miles long and that there are up to 100 billion vessels in the brain.2, 3, 4 This elaborate vascular system, especially the cerebral microvasculature, quickly adapts and responds to physiologic, pathologic, and microenvironmental stimuli in a very dynamic manner. For instance, under hypoxic conditions, blood vessel networks expand to meet the growing oxygen demands and brain capillary density can double in 3 weeks. 5 This neovascularization response requires new vessel formation through sprouting angiogenesis as well as remodeling of the existing vasculature to form new collaterals. Both these processes are tightly modulated by environmental cues and in this context, it is highly likely that the neovascularization response of the brain may differ under physiologic and disease conditions.
Diabetes increases the risk of a number of neurologic disorders including stroke, vascular cognitive impairment, and Alzheimer's disease, in all of which the cerebrovasculature has an important role in disease onset, progression, and treatment. 6 On the basis of the fact that diabetes is the most rapidly increasing risk factor for stroke, stroke is the leading cause of disability, and reparative angiogenesis is being pursued as a therapeutic strategy, the purpose of this review is to take a closer look at cerebral neovascularization in diabetes and stroke.
I. Neovascularization Processes: Vasculogenesis, Angiogenesis, and Arteriogenesis and Vascular Remodeling
Neovascularization (new blood vessel formation) occurs through vasculogenesis, angiogenesis, and/or arteriogenesis. Although all three can occur in response to tissue hypoxia and injury, they differ in the molecular triggers and underlying mechanisms. Because excellent recent reviews describe these concepts in detail,2, 7, 8, 9, 10, 11 we will briefly describe these processes and focus on the key players that are involved in cerebral neovascularization in diabetes and stroke described in next sections.
Vasculogenesis is the formation of a primitive endothelial network from mesenchymal stem cells or endothelial progenitor cells in response to local cues. This unorganized and undifferentiated plexus is further developed by angiogenesis not only mediating embryonic blood vessel formation, but also contributing to neovascularization in the adult.12, 13
Angiogenesis is defined as the formation of new capillaries from preexisting vessels in a multistep process (Figure 1). Hypoxia is a key stimulus for angiogenesis and through the activation of hypoxia inducible factor-1α, pro-angiogenic molecules such as vascular endothelial growth factor-A (VEGF-A) and VEGF receptor 2 (VEGFR-2) (flt-1), angiopoietins (Ang-1 and −2) and cognate receptor Tie-2, neuropilin-1, and basic fibroblast growth factor are stimulated. These growth factors activate otherwise quiescent endothelial cells to start the angiogenic cascade. When there is VEGF-A and the Ang-2/Ang-1 ratio is high, sprouting angiogenesis occurs.14, 15 Specialized endothelial cells (the so-called ‘tip’ cells) lead the process along the VEGF-A gradient.7, 8, 16 It was recently shown that endothelial cells are in a constant competition to assume the tip cell role and there is a dynamic exchange from a stalk cell to a tip cell or vice versa to ensure that the sprout is guided in the correct direction toward highest VEGF-A levels.17, 18 In this regard, vessel sprouting is similar to axonal sprouting where guidance signals regulate sprout direction and elongation.19, 20 Roundabout-4 (Robo4), an endothelial cell-specific member of the neuronal guidance molecules,19, 21, 22 has been shown to inhibit endothelial cell migration and has a role in angiogenesis and vascular patterning, vascular stability, and directional endothelial cell migration.23, 24, 25 Tip cells also inhibit stalk cells from becoming tip cells via the Notch-Delta like ligand (deltall) system and again this is dynamically regulated. These tube-like structures are stabilized and become capillaries by Ang-1-mediated recruitment of pericytes and basement membrane deposition.8, 16, 26
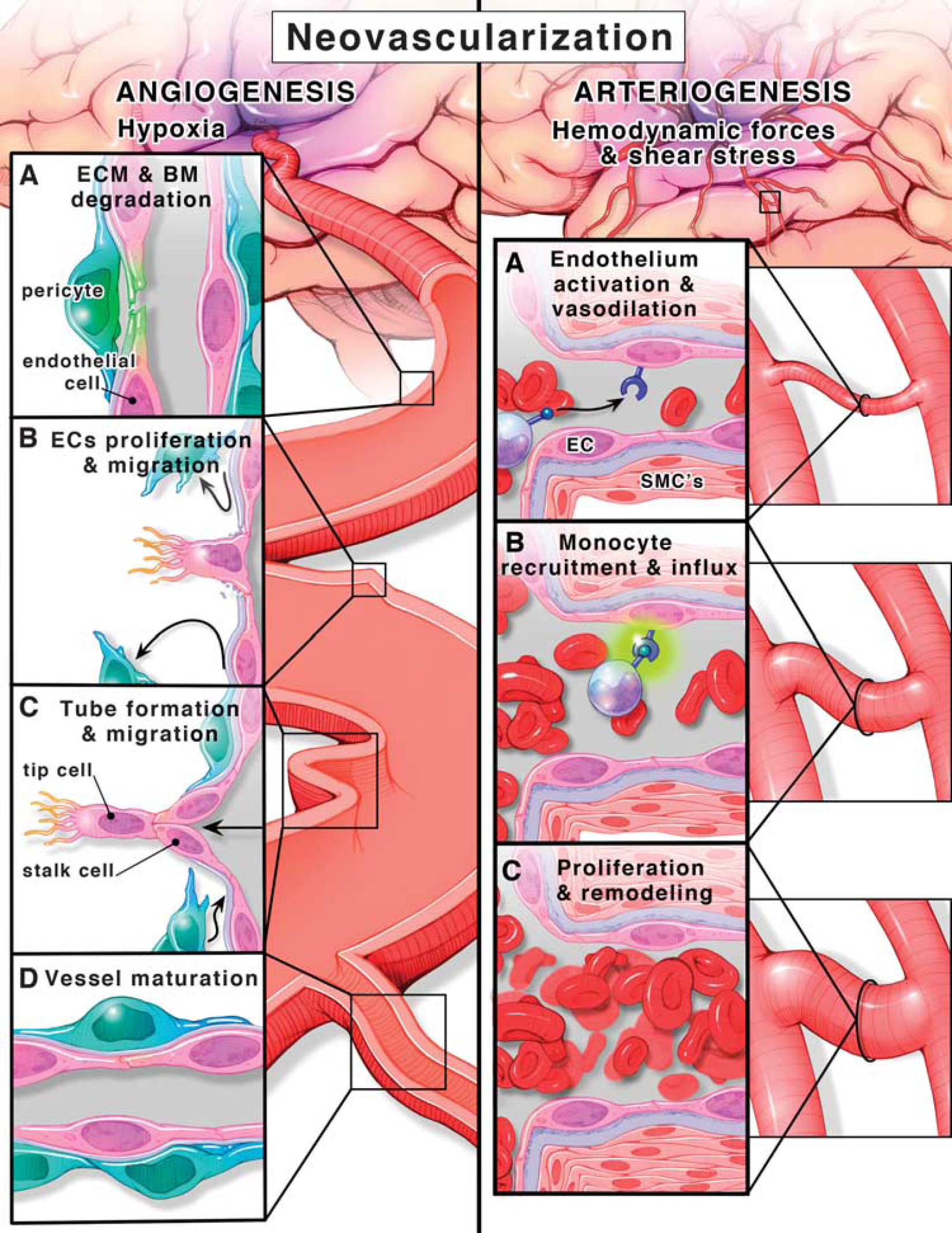
Angiogenesis, formation of new vessels from existing ones (left panel). (
Arteriogenesis, however, is a further development of these capillaries into larger vessels like arterioles and venules or remodeling and maturation of already existing collateral arterioles that are subjected to low flow conditions under normal conditions. Unlike angiogenesis, arteriogenesis occurs in the normoxic tissue surrounding the ischemic area, triggered by increased fluid shear stress as a result of blood flow redistribution after vessel occlusion. Physical forces and increased shear stress result in endothelium activation (Figure 1). 27 Together with vasodilation and increased permeability, upregulation of adhesion molecules and monocyte chemoattractant protein-1 results in monocyte recruitment and migration to the endothelium. These events trigger a proliferative phenotype in all layers of the developing vessel.28, 29 Smooth muscle cells (SMCs) switch from the contractile to the proliferative phenotype and start to divide and increase the wall thickness to accommodate the increase in lumen diameter then redifferentiate into the contractile phenotype in the later steps of the arteriogenesis cascade resulting in overall collateral vessel maturation and perfusion. 28 A visual comparison between angiogenesis and arteriogenesis is depicted in Figure 1.
II. Diabetes and Neovascularization
The impact of diabetes on neovascularization, in particular on angiogenesis, is most widely studied in the eye and peripheral vasculature. Thus, we will first review the effect of diabetes on cerebral neovascularization and then compare with other vascular beds to provide a perspective.
A. Cerebral neovascularization
In the cerebral circulation, the impact of diabetes on neovascularization was not explored until recently. We have shown that diabetes causes increased, yet dysfunctional, neovascularization in the cerebrovasculature.30, 31, 32 There is increased arteriogenesis (greater number of collaterals and increased vascular tortuosity) in the pial vasculature of type 2 diabetic Goto-Kakizaki (GK) rats. 30 A follow-up study provided evidence for increased cerebral angiogenesis and arteriogenesis. 31 Vascular density, volume, and surface area in the brain parenchyma were greater in diabetic animals. These indices of neovascularization were greater in the cortex and progressively increased from front to the back of the brain. However, this augmented angiogenesis was associated with poor vessel wall maturity as indicated by reduced pericytes and increased nonperfused vessels and permeability. 31
Glycemic control prevents this dysfunctional cerebral neovascularization in diabetes, suggesting that hyperglycemia is a major player driving the angiogenic response. 32 Comparative studies with the db/db mouse model of type 2 diabetes showed that augmented cerebral neovascularization is not unique to the GK model of diabetes. Goto-Kakizaki rats had an increase in both microvessel and macrovessel densities suggestive of angiogenesis and arteriogenesis, whereas db/db mice had an increase only in the microvasculature. While branch density and tortuosity of penetrating arterioles were increased in both models of diabetes, lumen diameter of penetrating arterioles was increased only in GK rats. The fact that these models of type 2 diabetes are different species with different disease severity strongly suggests that diabetes has a profound effect on brain microvasculature. Using the same GK model, Beauquis et al 33 reported decreased vascularization and capillary branching in the dentate gyrus of the hippocampus, an area associated with memory and learning processes. It is also possible that there are differences in the angiogenic response in very specialized areas of the brain and needs further evaluation especially with respect to disease severity and duration.
This cerebral neovascularization is similar to pathologic angiogenesis that occurs in diabetic retinopathy. 16 The retina is like the brain in the sense that it has its own neurovascular unit. Retinal ischemia is a complex event and diabetes-mediated neurodegeneration and glial inflammation contribute to increased apoptosis in pericytes and endothelial cells in retinal microvessels leading to acellular capillary formation and vascular regression. 34 Collectively, these changes lead to upregulation of angiogenic molecules VEGF-A, erythropoietin (EPO), and other vascular growth factors34, 35, 36, 37 and result in pathologic angiogenesis, increased vascular leakage, and bleeding.38, 39, 40
Diabetes-induced dysfunctional cerebral neovascularization response is vastly different from the neovascularization observed in other vascular beds. In the coronary circulation, diabetes alters the balance between pro- and antiangiogenic growth factors, impairs endothelial function, and mediates an imbalance in microenvironment redox state of the coronary circulation 41 resulting in impaired coronary collateral growth and cardiac angiogenesis.42, 43, 44, 45, 46 In the peripheral circulation, there is again impaired neovascularization in experimental models.31, 47, 48, 49 In the renal circulation, there is increased renal angiogenesis in early stages of diabetes in both clinical and experimental studies.50, 51, 52, 53, 54 These early vascular changes were attributed to increased VEGF expression and mild inflammation.51, 55 At later stages of diabetes, vascular regression occurs, where chronic inflammation leads to increases in vascular permeability, thickening of the glomerular basement membrane, endothelial cell apoptosis, and loss of peritubular capillaries.
B. Potential mechanisms for diabetes-induced dysfunctional neovascularization
1. Cerebrovascular dysfunction and decreased cerebral blood flow. Constant cerebral blood flow is critical for neuronal function and the brain quickly responds to hypoxia by increasing capillary density.9, 56 Numerous studies have reported cerebrovascular dysfunction in various diabetes models at the large artery or small penetrating arterioles levels as we recently reviewed. 6 We have also shown that in the GK model, there is cerebrovascular dysfunction and decreased cerebral blood flow, 57 which develops shortly after the onset of diabetes. This is accompanied by upregulation of hypoxia inducible factor-1 (unpublished data), suggesting that early vascular dysfunction and decreased blood flow create a hypoxic environment that may be the initial trigger for increased cerebral neovascularization. 58 As discussed above, in the retina, hyperglycemia-induced changes in the neurovascular unit and capillary drop-out contribute to pathologic angiogenesis. In the brain, the initial cause of hypoxia seems to be different and improvement of vascular function may be a good therapeutic target to prevent dysfunctional angiogenesis (Figure 2).
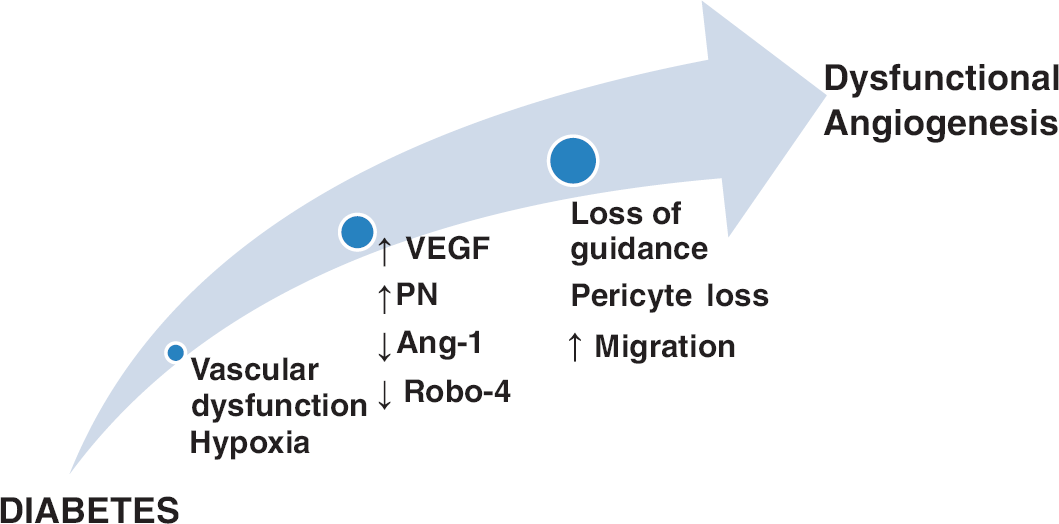
Cerebral angiogenesis in diabetes. A schematic illustrating the mechanisms by which diabetes induces cerebral dysfunctional angiogenesis. Diabetic hyperglycemia induces vascular dysfunction that creates a state of cerebral hypoxia. Diabetes-induced hypoxia triggers a series of events including (1) increased production of vascular endothelial growth factor (VEGF), (2) increased oxidative and nitrative stress (increased peroxynitrite (PN) formation), (3) decreased angiopoietin-1 (Ang-1), and (4) decreased expression of guidance molecule, Roundabout-4 (Robo-4). These events lead to pericyte loss, increase endothelial migration with loss of guidance that cumulatively increases dysfunctional cerebral angiogenesis.
2. Augmented vascular endothelial growth factor-A signaling: involvement of oxidative and nitrative stress. While physiologic angiogenesis represents a fine balance between numerous anti- and pro-angiogenic growth factors, it is widely accepted that VEGF-A has a central role in the regulation of neovascularization. Vascular endothelial growth factor-A is important for endothelial cell proliferation, survival, migration, and tube formation, as well as matrix degradation and vessel permeability. 59 Vascular endothelial growth factor-A primarily binds to VEGFR-2, 60 then activating extracellular signal-regulated kinases 1 and 2, Src, and phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) to stimulate cell survival and migration. 61 The neovascularization process is affected not only by VEGF and VEGFR-2 levels but also by the redox state of the microenvironment. Mild, but not severe, increases in reactive oxygen and nitrogen species are needed to transduce VEGF's angiogenic signal. This concept is now known as the redox window 42 (Figure 3) and might be one explanation for the angiogenic paradox (increased versus impaired angiogenesis) in different vascular beds in diabetes as well as the reason why ‘therapeutic angiogenesis trials' failed.42, 62
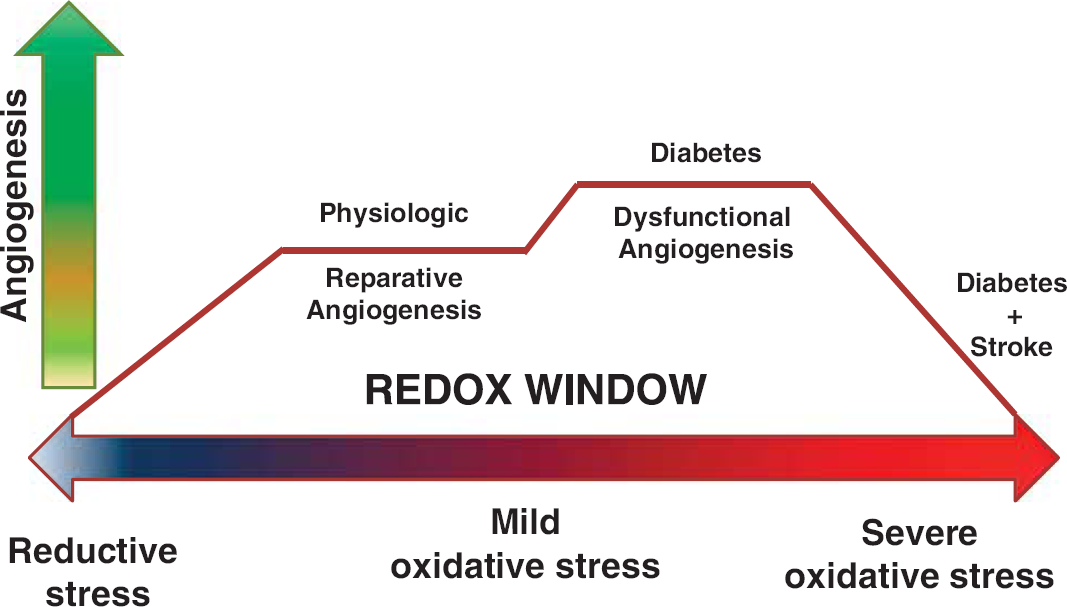
Redox window and angiogenesis. A diagram representing the redox window concept: the tissue redox state ranging from reductive to oxidative levels is depicted on the X axis. Y axis is the angiogenesis process. Physiologic/reparative angiogenesis requires the tissue microenvironment to express mild levels of oxidative stress. Extreme levels of reductive or oxidative stress impair angiogenesis. Oxidative stress levels in diabetes depend on disease severity and promote dysfunctional angiogenesis. Adding stroke to diabetes greatly increases oxidative stress and corrupts the reparative angiogenic process after stroke.
Several studies provided evidence as to how the redox window modulates VEGF-A signaling in diabetic retinopathy.63, 64, 65, 66 Low levels of peroxynitrite, which is rapidly generated from the interaction between superoxide and nitric oxide, sustain and amplify VEGFR-2 signaling leading to pathologic angiogenesis. However, high levels of peroxynitrite nitrate the p85 regulatory subunit of the PI3 kinase and divert the pro-survival effects of VEGF-A to apoptosis in retinal endothelial cells, again emphasizing the redox window concept in angiogenesis. 63 We have extended these findings to the brain microvasculature. Endothelial cells isolated from brain microvessels of diabetic GK animals interestingly retain their angiogenic properties in culture and exhibit augmented cell migration and tube formation as compared with cells isolated from control animals. These cells have increased basal VEGF-A and phosphorylated VEGFR-2 levels. The angiogenic properties of endothelial cells that are isolated from diabetic animals can be blocked when cells are treated with a VEGF-A neutralizing antibody or the peroxynitrite scavenger FeTPPs [5,10,15,20-tetrakis (4-sulfonatophenyl) porphyrinato iron (III)]. 31 These cells also exhibit a defect in their barrier function as measured by transendothelial resistance. They take a longer period of time to establish barrier function as compared with control cells and they are more susceptible to peroxynitrite-mediated loss of barrier function. As will be discussed below, when ischemia/reperfusion is overlaid on this pathology, the redox window is shifted to the right toward excessive oxidative stress and endothelial cell death.
3. Impaired vessel guidance and maturation. As discussed under the general description of angiogenesis, the robo is a family of proteins that act as guidance receptors and were originally identified in the nervous system. Activation of robo1 to 3 by slit ligands (Slit1 to 3) provides repulsive signals for axons. 8 Robo4 is uniquely expressed in endothelial cells and its ligand is Slit2.23, 25 The Robo4/Slit2 signaling pathway has recently been identified as a regulator of microvascular maturation, endothelial permeability, and angiogenesis.67, 68, 69 Our ongoing studies suggest that Robo4 protein is significantly decreased in the cerebral microvasculature of diabetic GK rats that develop erratic and dysfunctional angiogenesis (unpublished data). Interestingly, crosstalk between VEGF receptor tyrosine kinases and integrin signaling has been reported. There is a protein–protein interaction between Robo4 and β3 integrin that is associated with a reduction in Robo4/Slit2 signaling leading to vascular hyperpermeability. 70 A better understanding of how diabetes impacts Robo4 regulation, especially by VEGF-A, may provide novel targets to prevent and/or treat dysfunctional cerebral angiogenesis in diabetes.
Another important step in angiogenesis is vessel maturation. Angiopoietin-1 promotes migration, sprouting, and survival of endothelial cells through activation of Tie-2 tyrosine kinase receptor14, 15 and it is critical for vessel stabilization. Angiopoietin-2 acts as an antagonist for Ang-1 and inhibits Ang-1-promoted Tie-2 signaling and vessel maturation and stabilization. An Ang-1 peptide mimetic treatment was reported to accelerate wound healing in diabetic animals 71 as well as preserve the renal microvasculature. 72 An increase in Ang-2/Ang-1 ratio was found to be associated with angiogenic activity in patients with diabetic retinopathy. 73 Whether this system is altered in the brain microvasculature in diabetes needs to be established.
Pericytes, located at the periphery of the microvessel wall, communicate with endothelial cells and other cells of the neurovascular unit and are very important for neovascularization and vessel maturation. 74 At early stages of angiogenesis, pericytes migrate away as an initial step to allow endothelial cell proliferation and migration. 74 At later stages of vessel formation, pericytes increase the stability of newly formed vessels via the prevention of angiogenesis and promote vessel stability via Ang-1 and platelet-derived growth factor-B.75, 76, 77 In the brain, Wnt/β-catenin signaling, a critical pathway in developmental angiogenesis and vascular differentiation, promotes vessel maturation by increasing endothelial platelet derived growth factor-B expression and recruiting pericytes. 78 Hyperglycemia-induced dysfunction causes loss of pericytes, which is a hallmark of diabetic retinopathy and other diabetes-induced vascular disease. 79 Diabetic rats have less pericytes along microvessels of the brain and increased cerebral angiogenesis. 31 However, whether the loss of pericytes gives way to angiogenesis or newly formed vessels are unable to recruit pericytes for maturation is yet to be determined. In this context, similar to the Ang-1/Tie-2 system, the regulation of the platelet derived growth factor-B/platelet derived growth factor receptor-β system in the brain microvasculature in diabetes is still unknown and warrants further research.
4. Chronic inflammation. Diabetes causes a state of chronic inflammation 80 and increases VEGF production and other inflammatory cytokines that activate NF-κB, inducing the secretion of several factors including interleukin-1, interleukin-6, tumor necrosis factor-α, chemokine C–C motif ligand-5, and transforming growth factor-β, all of which stimulate angiogenesis. 81 Oxidative stress seems to be the link between inflammation and angiogenesis.
In response to inflammation, not only adaptive but also the innate immunity is activated. In this regard, toll-like receptors have a critical role in regulation of the innate immune response. Toll-like receptors may be involved in the regulation of endothelial cell survival and the angiogenic response as well. Lipopolysaccharide, a well-established ligand for TLR4, induces endothelial sprouting. 82 In addition, both TLR2 and TLR3 have been reported to promote angiogenesis. 83 While there is strong evidence for the key role of inflammation and inflammation-associated oxidative stress plays in angiogenesis, most, if not all, of this evidence comes from pathologic angiogenesis associated with tumor growth and our understanding of the role of this pathway in dysfunctional angiogenesis of the brain is yet to be shown.
5. Uncharted mechanisms and remaining questions. As discussed above, VEGF-A and Ang-1 are the main pro-angiogenic factors in the brain and the eye but undoubtedly other factors are involved. There are two newly identified molecules that seem to be uniquely involved in pathologic angiogenesis. While searching the retinal microvessel transcriptome for factors contributing to the erratic neovascularization that occurs in diabetic retinopathy, Wang et al 84 discovered a protein called leucine-rich alpha-2-glycoprotein 1 of previously unknown function. Leucine-rich alpha-2-glycoprotein 1 mediates angiogenesis through the regulation of endothelial transforming growth factor-β signaling and this novel protein may be predominantly involved in uncontrolled angiogenesis. The other intriguing protein is ataxia telangiectasia mutated kinase or simply ATM kinase, which is involved in DNA repair and damage. Activation of ATM by oxidative stress suppresses the p38MAP kinase pathway and leads to excessive neovascularization in the retina. 85 Further research is needed to determine how diabetes impacts the expression and action of these proteins across vascular beds and especially in the brain.
The net result of angiogenesis depends on the balance of pro- and antiangiogenic factors. Angiopoietin-2, angiostatin, endostatin, thrombospondin-1, and soluble VEGF receptor (sFlt-1) are among antiangiogenic factors that have been shown to impact the neovascularization process in other tissues.86, 87 However, the regulation and the impact of these antiangiogenic factors in the brain in diabetes are not fully understood.
In summary, diabetes stimulates dysfunctional neovascularization of the brain. Potential underlying mechanisms that are briefly discussed above are summarized in Figure 2. Unstable, leaky and dysfunctional vessels cause increased blood–brain barrier permeability and cannot meet the demands of the brain for proper blood flow and nutrient delivery. Dysfunctional angiogenesis of the brain in diabetes is a new concept. Our knowledge of pathologic angiogenesis comes from tumor angiogenesis and diabetic retinopathy. Given that diabetes is an exponentially growing risk factor for stroke and neurodegenerative disorders with cognitive impairment including dementia and Alzheimer's disease, there is an urgent need for further studies involving the effect of the type of diabetes and the degree/duration of hyperglycemia on spatial and temporal regulation of cerebral angiogenesis and maturation.
III. Stroke and Neovascularization
Angiogenesis genes are upregulated within minutes of the onset of cerebral ischemia in rodents.88, 89 There is now mounting evidence that angiogenesis occurs in concert with neurogenesis and synaptogenesis in experimental models of recovery after ischemic brain injury. 90 These processes take place in response to a disparate range of interventions, from cortical stimulation 91 to antidepressant therapy. 92 Although the time course of angiogenesis and neurogenesis overlaps, many investigators have now concluded that the angiogenesis occurs first and leads to axonal remodeling 93 and neuroblast migration along new blood vessels. 94 A recent study showed that endothelial cells transplanted into the brain promote vasculogenesis and enhance neurogenesis further providing support for this concept. 95 This neuroplasticity is important for meaningful functional recovery, but may be diminished by aging and other comorbidities. 93
There is emerging evidence that neuroplasticity and recovery after brain injury involve areas remote from the injury itself.96, 97 In an investigation of the beneficial effects of EPO on motor recovery, EPO was administered after temporary middle cerebral artery occlusion (MCAO) in a rodent model and was shown to induce improved perilesional remodeling that was accompanied by increased axonal sprouting from the contralesional hemisphere. 96 This concept was supported by an investigation showing an early increase in brain-derived neurotrophic factor expression in both hemispheres after experimental ischemia in rats, and this was followed by a rise in synaptophysin (a marker of synaptogenesis) ipsilaterally. 97 Since it is clear that neuroblasts migrate along blood vessels in areas of angiogenesis after stroke, 94 it is logical that the plastic changes in the contralesional hemisphere are accompanied by contralesional angiogenesis. In fact, it has been shown that blockade of VEGFR-2 can prevent postischemic neurovascular remodeling by preventing neuroblast migration along blood vessels. 98 Most investigators have focused their efforts on quantification of angiogenesis after stroke in the peri-infarct areas, however. 90
Recently, we showed that the angiotensin II type 1 receptor antagonist, candesartan, when administered at reperfusion in a rat model of temporary MCAO, promoted recovery and angiogenesis in the contralesional striatum at 7 days after the stroke. 99 This was confirmed in a more recent investigation where normoglycemic rats showed increased angiogenesis and recovery after stroke compared with diabetic animals, where vascular regression after stroke accompanied a much poorer functional outcome. 100 In summary, angiogenesis occurs after stroke and is closely linked to recovery. A lack of consensus on the contribution of angiogenesis to stroke recovery may be due to a failure to look beyond the peri-infarct area and the use of the contralesional hemisphere as a convenient control. The impact of premorbid vascular diseases on cerebral angiogenesis after stroke needs to be further investigated.
IV. Neovascularization after Stroke in Diabetes
Angiogenesis can improve functional recovery from stroke as discussed above. However, it has to be recognized that most experimental studies used young and healthy animals without confounding factors that are commonly found in patients. As discussed above, diabetes stimulates dysfunctional and uncontrolled angiogenesis in the cerebral vasculature. If these animals are subjected to ischemic stroke, then they develop greater vascular injury including hemorrhagic transformation, especially around the infarcted area, and edema. Ultimately, animals exhibit poor functional outcome.101, 102, 103 To determine the impact of diabetes on cerebral neovascularization after an ischemic event, we compared and contrasted various indices of cerebral neovascularization in the ipsilateral ischemic and contralateral hemispheres of control and diabetic rats subjected to sham or stroke surgery. Several important observations were made. While there was reparative neovascularization in control animals in both ischemic and nonischemic hemispheres as compared with the sham group, diabetic animals developed a significant vasoregression in both hemispheres (Figure 4). 100 This was associated with increased astrocytic swelling and poor functional recovery. Glycemic control during the recovery phase after stroke partially prevented the robust decline in vascularization and improved outcome. Other studies also showed that type 1 and type 2 diabetes impair angiogenesis after stroke.104, 105 Vessel density in the ipsilateral hemisphere 14 days after stroke is increased but these vessels are not mature as indicated by reduced diameter, arteriolar density, and SMCs.104, 105 These studies were conducted in stroked animals and there were no sham control animals thus it is not possible to comment on how stroke injury affected the neovascularization response in the nonischemic hemisphere.
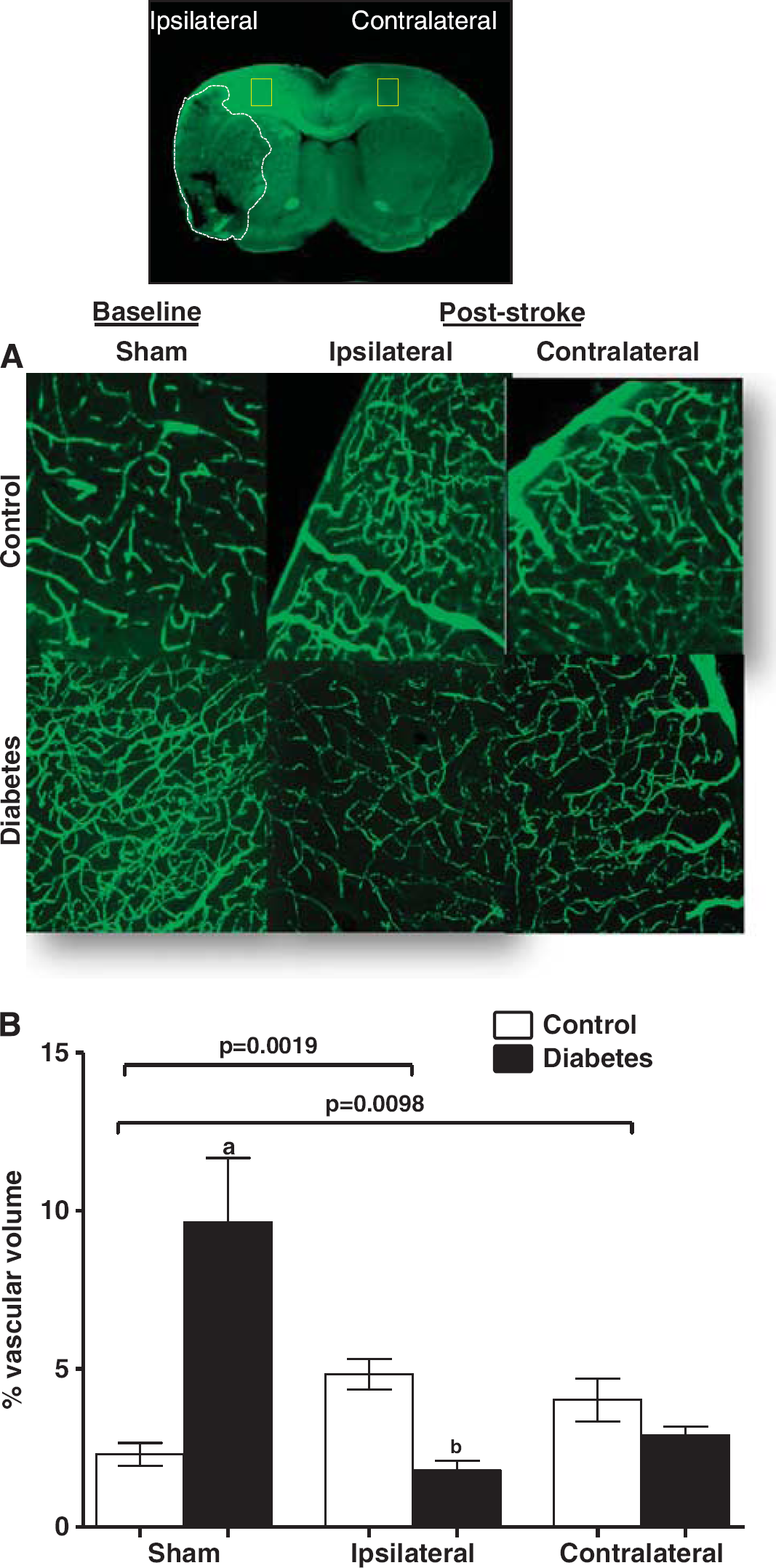
Diabetes impairs poststroke neovascularization in the ipsilateral and contralateral hemispheres. Vascularization was assessed in the ipsilateral and contralateral hemispheres in control and diabetic animals 14 days after 90-minute occlusion of the middle cerebral artery. Sham animals were exposed to anesthesia and neck dissection was performed and sutured without occluding middle cerebral artery. Three-dimensional reconstruction of the fluorescein isothiocyanate (FITC)-stained vasculature was achieved analysis of the z-stack confocal images by the Volocity program. (
The mechanisms by which diabetes impairs the repair process and causes this dramatic decline in the cerebrovascular network after stroke are unknown and likely to be multifactorial (Figure 5). One potential mechanism may be the redox microenvironment as discussed above. It appears that in diabetic stroke, there is a nitrative switch. In diabetic animals, there is even greater peroxynitrite formation in the cerebrovasculature after stroke and this is associated with greater endothelial apoptosis (unpublished data). There is a significant decrease in Akt signaling and a concomitant increase in p38 signaling in brain microvascular endothelial cells isolated from diabetic animals when exposed to hypoxia and reoxygenation which can be prevented by peroxynitrite scavenging.
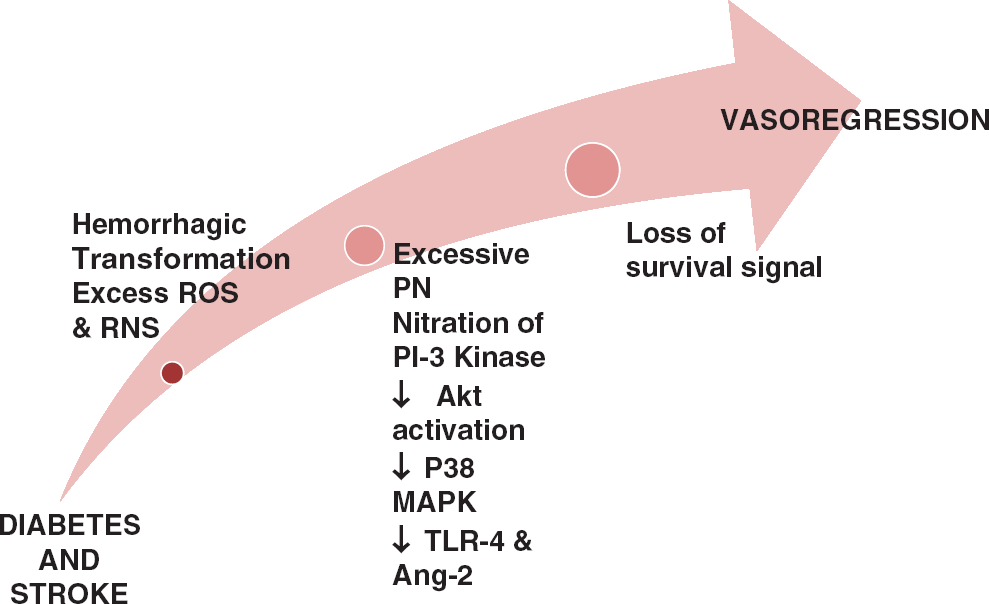
Cerebral neovascularization in diabetes after stroke. A representative diagram illustrating the mechanisms by which vasoregression of cerebral vessels occurs in diabetes after stroke. When stroke is overlayed on diabetes pathology, there is greater hemorrhagic transformation and free iron accumulation that induces cell death. In addition, adding stroke injury to diabetes dramatically increases peroxynitrite (PN) formation and nitration of p85 regulatory subunit of PI3 kinase that downregulates the downstream pro-survival Akt pathway and activates the pro-apoptic p38MAP kinase pathway. Finally, increases in angiopoietin-2 (Ang-2) and activation of toll-like receptor 4 (TLR4) increase endothelial death and promote vasoregression in diabetes after stroke. ROS, reactive oxygen species; RNS, reactive nitrogen species.
It is also of great interest that in both type 1 and type 2 diabetes models, in which repair and recovery are impaired, there is increased bleeding into the brain after ischemic stroke.101, 102, 103, 104, 105 However, the impact of bleeding on vascular repair has not been fully studied. Evidence from intracranial hemorrhage models suggests that hemoglobin and heme released from red blood cells enter the brain parenchyma and free iron from further degradation of the heme molecule, disrupts cellular integrity and function via increased oxidative stress.106, 107, 108, 109 It is also intriguing that heme upregulates TLR4, 110 a gate keeper of the innate immune system and TLR4 mediates disruption of endothelial barrier function. 111 These observations collectively raise the possibility of bleeding and TLR4 being additional mechanisms involved in the vasoregression and impaired repair process after diabetic stroke.
The possibility of increased antiangiogenic molecules mediating vascular regression cannot be overlooked. An interesting study showed that angiogenesis is impaired in the GK diabetic model after stroke and this is associated with decreased VEGF and increased angiostatin signaling. 112 In other studies where immature vessel formation was observed,104, 105 authors reported increased Ang-2 and decreased Ang-1 expression in the brain sections of diabetic rats.103, 104
V. Clinical Relevance
On the basis of experimental evidence, cerebral neovascularization response differs in diabetes, stroke, and diabetic stroke (Figure 6). Important questions remain unresolved with respect to the clinical relevance of these studies: (1) How can we promote adaptive brain neovascularization in health and disease? and (2) Is cerebral angiogenesis always good or attainable?
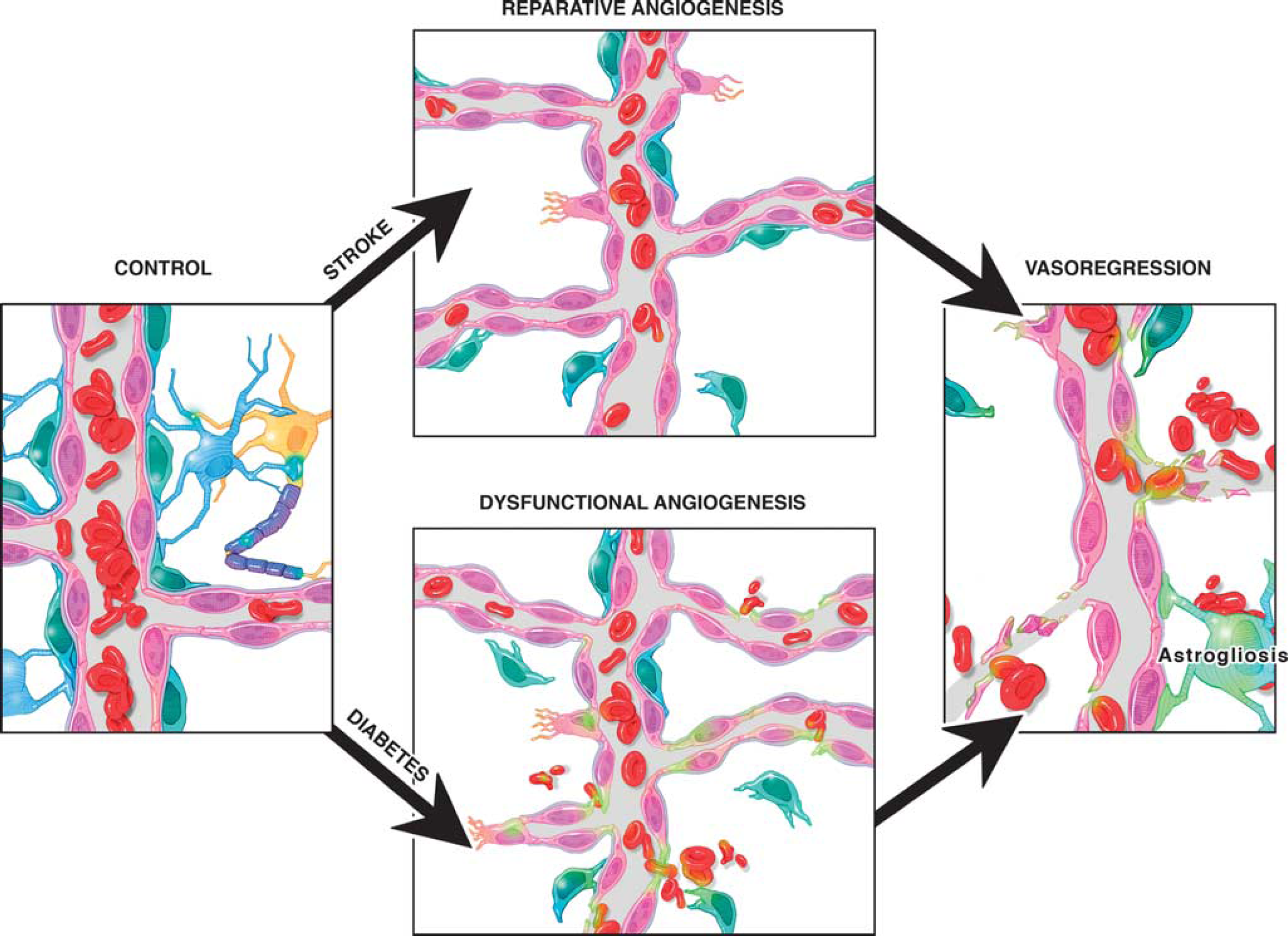
Cerebral neovascularization in diabetes and stroke. Diabetes causes dysfunctional angiogenesis. Stroke stimulates reparative angiogenesis in the nondiabetic state. However, when stroke occurs in diabetes, survival signals are lost leading to vasoregression.
From a diabetes standpoint, the first strategy is to evaluate the impact of glycemic control. We have shown that regulation of blood glucose is an effective strategy to prevent pathologic neovascularization of the brain and improves vessel maturity. 32 We now have evidence that glycemic control with metformin can also reverse established remodeling (Abdelsaid et al, Life Sciences, in press). When one looks at the clinical trials including DCCT (Diabetes Control and Complications Trial) and UKPD (United Kingdom Prospective Diabetes), glycemic control does not decrease stroke incidence that is considered to be a macrovascular complication of diabetes. Given that tight glucose control prevents microvascular complications such as retinopathy and nephropathy, the impact of glycemic control on microvascular disease of the brain such as cognitive impairment and the recovery phase after stroke remains to be determined.
From a stroke perspective, examples of different therapeutic interventions to promote arteriogenesis and angiogenesis are listed in Tables 1 and 2. Three of these agents (candesartan, EPO, and granulocyte colony stimulating factor) showed extremely promising results in experimental studies and made their way to humans, but findings from these clinical trials were disappointing. While they passed initial small-scale phase I clinical safety trials, they failed to show improvement or further worsened stroke outcome in larger multiphase II/III trials.113, 114, 115 These failures are not only due to the vast genetic differences between human and rodent brains, but could also be attributed to the lack of full characterization of these agents experimentally to allow for rigorous design of clinical trials with appropriate patient population, dosing regimen, and end points. A more complete understanding of the mechanisms of actions of these drugs is imperative for translating them from bench to bedside.
Arteriogenic interventions in experimental stroke research
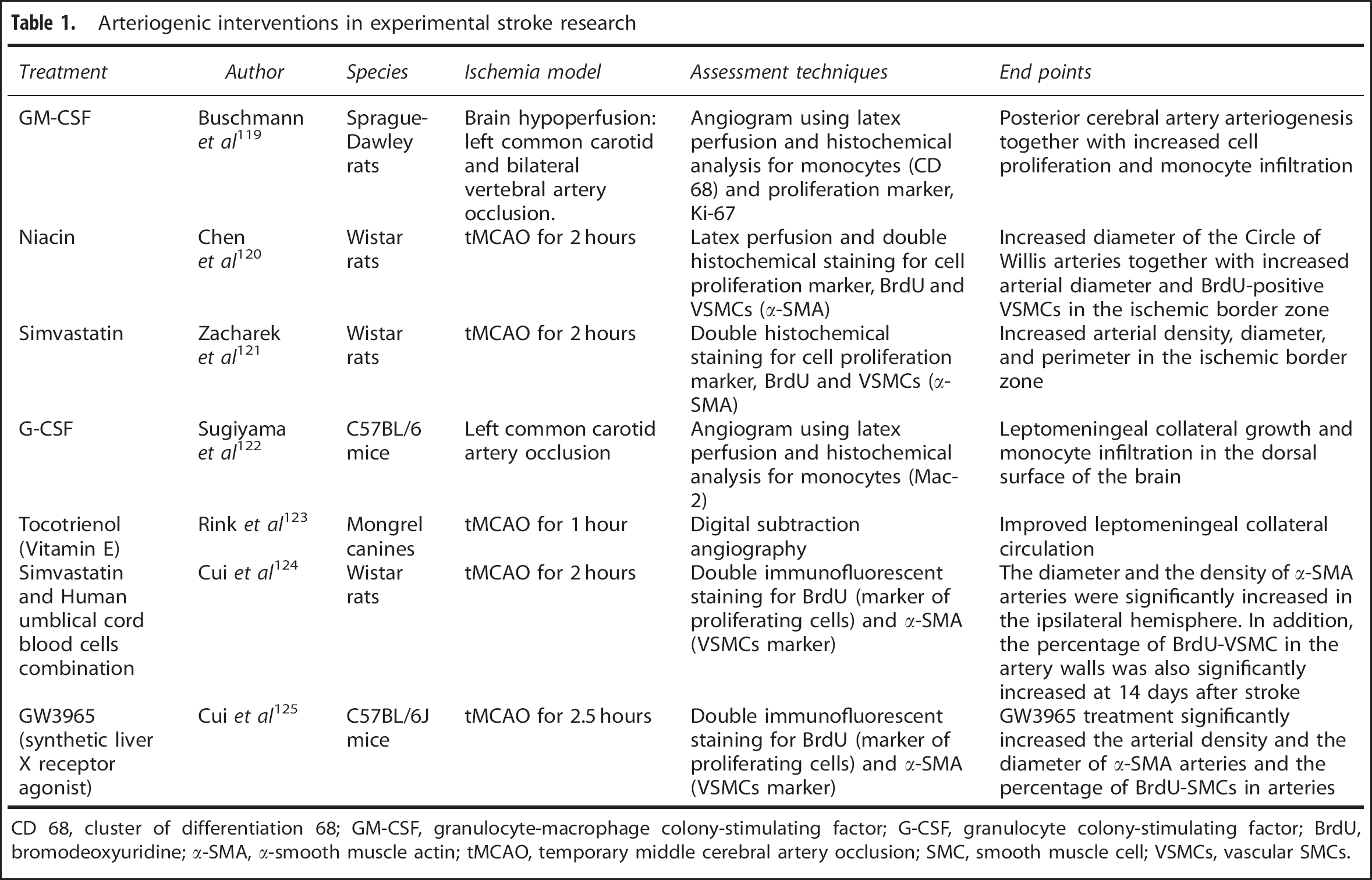
CD 68, cluster of differentiation 68; GM-CSF, granulocyte-macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; BrdU, bromodeoxyuridine; α-SMA, α-smooth muscle actin; tMCAO, temporary middle cerebral artery occlusion; SMC, smooth muscle cell; VSMCs, vascular SMCs.
Angiogenic interventions in experimental stroke research
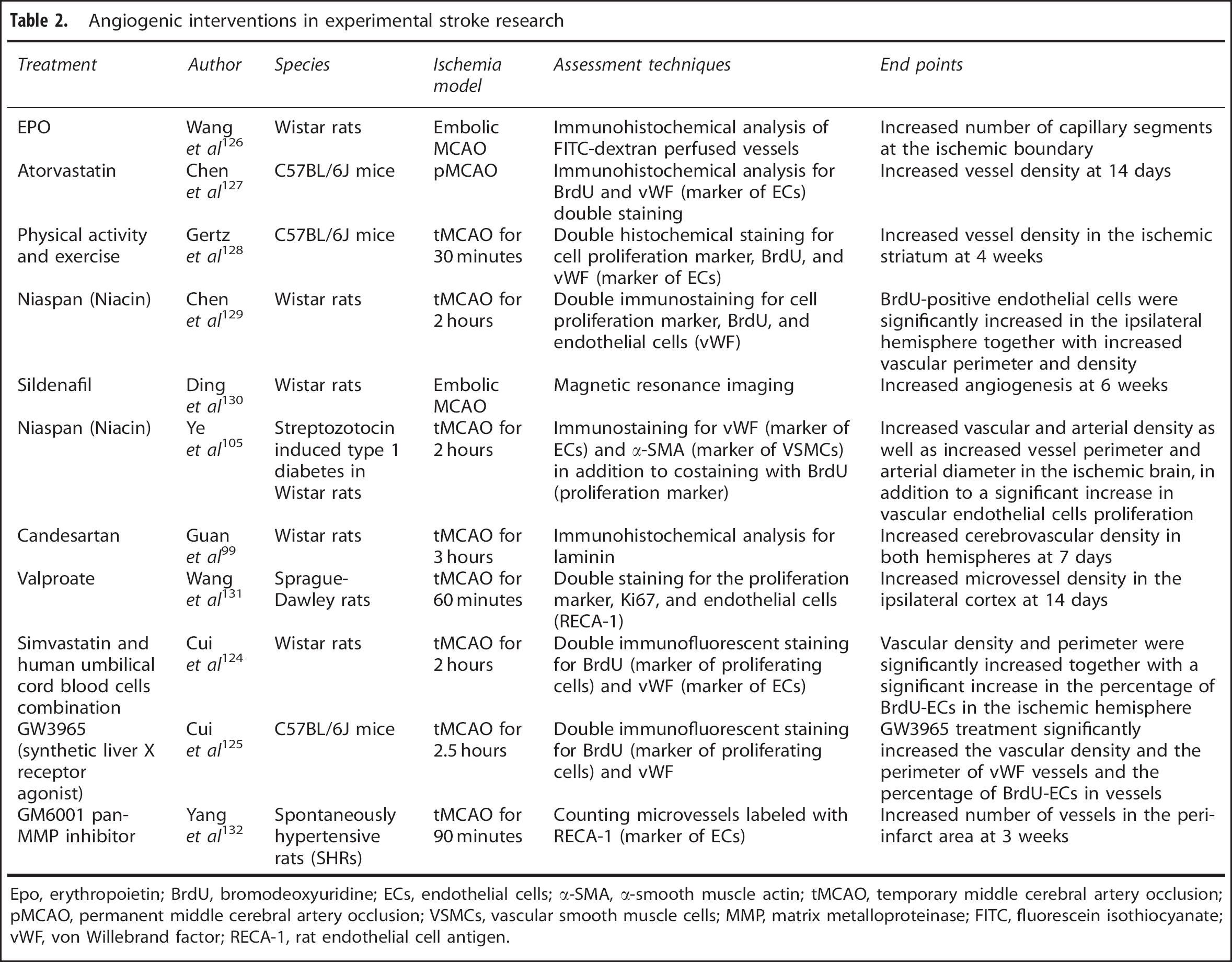
Epo, erythropoietin; BrdU, bromodeoxyuridine; ECs, endothelial cells; α-SMA, α-smooth muscle actin; tMCAO, temporary middle cerebral artery occlusion; pMCAO, permanent middle cerebral artery occlusion; VSMCs, vascular smooth muscle cells; MMP, matrix metalloproteinase; FITC, fluorescein isothiocyanate; vWF, von Willebrand factor; RECA-1, rat endothelial cell antigen.
From a diabetic stroke perspective, stimulation of angiogenesis does not seem to be a good strategy, at least for now. Chen et al 116 showed that cell therapy with bone marrow stromal cells improved repair and functional recovery in control but not in diabetic animals. On the contrary, this approach worsened blood–brain barrier integrity. As elegantly reviewed, therapeutic revascularization and vascular repair strategies face many challenges in other tissues involved in diabetic complications.117, 118 Studies focusing on the impact of these strategies in the brain to prevent and treat cerebral complications of diabetes are only beginning.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
AE is a research pharmacologist at the Charlie Norwood Veterans Affairs Medical Center in Augusta, Georgia. Authors would like to thank Ms Colby Polonsky for her assistance with Figures 1 and ![]() .
.