
Abstract
Lactate is proposed to be generated by astrocytes during glutamatergic neurotransmission and shuttled to neurons as ‘preferred’ oxidative fuel. However, a large body of evidence demonstrates that metabolic changes during activation of living brain disprove essential components of the astrocyte-neuron lactate shuttle model. For example, some glutamate is oxidized to generate ATP after its uptake into astrocytes and neuronal glucose phosphorylation rises during activation and provides pyruvate for oxidation. Extension of the notion that lactate is a preferential fuel into the traumatic brain injury (TBI) field has important clinical implications, and the concept must, therefore, be carefully evaluated before implementation into patient care. Microdialysis studies in TBI patients demonstrate that lactate and pyruvate levels and lactate/pyruvate ratios, along with other data, have important diagnostic value to distinguish between ischemia and mitochondrial dysfunction. Results show that lactate release from human brain to blood predominates over its uptake after TBI, and strong evidence for lactate metabolism is lacking; mitochondrial dysfunction may inhibit lactate oxidation. Claims that exogenous lactate infusion is energetically beneficial for TBI patients are not based on metabolic assays and data are incorrectly interpreted.
INTRODUCTION
Traumatic brain injury (TBI) has a high impact on patients' lives, and major advances in clinical care have come from monitoring extracellular metabolite levels to help distinguish between major types of metabolic dysfunction. Linkage of changes in lactate level to the notion of lactate as a supplemental oxidative fuel in normal and post-ischemic brain gave rise to the concept that lactate may be beneficial after TBI. Because supplemental lactate may be useful, available data must be examined to evaluate the strength of supportive evidence. In this opinion article, metabolic aspects of TBI are first summarized to provide the context for lactate utilization. Then, evidence against astrocyte-neuron lactate shuttling is discussed because this concept is thought to be predictive of beneficial value of lactate as oxidative fuel after TBI. Last, alternative perspectives and interpretations are offered for data taken to support the notion of metabolism of lactate as preferred fuel over glucose after TBI. Strong evidence is lacking, and lactate utilization appears to be quantitatively small; it may be restricted by mitochondrial dysfunction after TBI.
BRAIN MICRODIALYSIS AFTER TRAUMATIC BRAIN INJURY
Biomarkers
Microdialysis provides continuous, real-time assays of extracellular fluid concentrations of glucose, lactate, and pyruvate as indicators of energy metabolism (Figure 1), glutamate as a measure of potential excitotoxic injury, and glycerol as an index of membrane degradation. 1 Under standard conditions (10mm microdialysis membrane and 0.3 μl/minute perfusion rate), biomarker recoveries (dialysate/interstitial fluid concentration) are ∼ 70%. 2 Because cytoplasmic lactate and pyruvate readily exchange with extracellular fluids (including blood and perfusion media 3 ), the lactate/pyruvate ratio (LPR) is considered a reliable index of intracellular cytoplasmic redox state, the [NADH][H+]/[NAD+] ratio of unbound nucleotides that participate in the lactate dehydrogenase reaction.3–5 Microdialysate LPRs are very useful but not diagnostically specific. 6 Poor outcome after TBI is associated with low oxygen, glucose, pyruvate levels, high LPR, and elevated glutamate and glycerol levels, particularly when changes have longer duration and higher magnitude.7–10
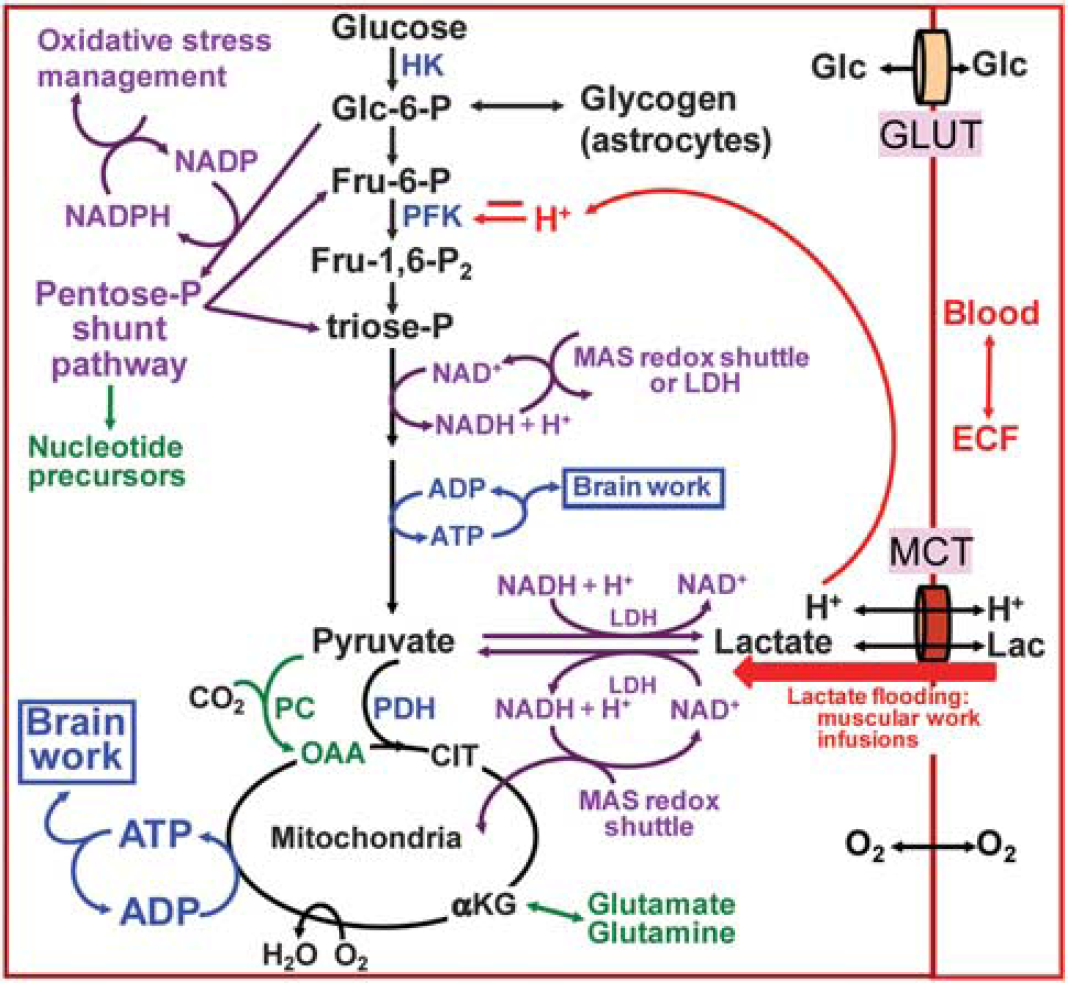
Major pathways for glucose and lactate metabolism. Glucose enters the glycolytic pathway via the hexokinase step and glucose-6-P can be diverted to the pentose-P shunt pathway from which carbon re-enters glycolysis as fructose-6-P and glyceraldehyde-3-P. Because these two molecules are upstream of the redox and ATP-producing steps, impairment of glycolysis by NAD+ depletion or phosphofructokinase inhibition may also restrict pentose shunt flux. NADH cannot cross the mitochondrial membrane and requires the malate-aspartate shuttle system or lactate dehydrogenase to regenerate NAD+. Flooding of brain cells with lactate owing to the intense physical work or lactate infusion can cause intracellular acidification at two steps (transport and oxidation to pyruvate). Phosphofructokinase is the major regulatory enzyme of glycolysis and it has a steep pH activity profile, being inhibited by more acidic conditions. Color schemes denote major aspects of glucose metabolism: blue, energy related; purple, redox related; green, biosynthesis related. Astrocytes are highly enriched with glycogen and its enzymes, pyruvate carboxylase, and glutamine synthase. Pyruvate carboxylase is the CO2-fixing enzyme that confers astrocytes with the ability to synthesize glutamate de novo from glucose by making a new molecule of OAA from pyruvate, which condenses with AcCoA from a second molecule of pyruvate to make a ‘new’ citrate and αKG molecule that can be converted to a new glutamate molecule that can be converted to glutamine by glutamine synthase. AcCoA, acetyl CoA; αKG, a-ketoglutarate; cit, citrate; ECF, extracellular fluid; Fru, fructose; Glc, glucose; GLUT, glucose transporter; HK, hexokinase; LDH, lactate dehydrogenase; MAS, malate-aspartate shuttle; MCT, monocarboxylic acid transporter; OAA, oxaloacetate; P, phosphate; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; PFK, phosphofructokinase. Modified from Dienel (2012), 45 with permission from the author.
Probe Site
Microdialysis catheter location is important, e.g., in gray or white matter ipsilateral or contralateral to the injury because metabolite level changes are larger in lesioned or penumbral zones compared with distant ‘normal’ tissue.10–12 Also, gray and white matter exhibit different magnitudes and time courses of responses (i) to hypoxia for lactate and pyruvate in immature brain, 13 and (ii) to ischemia and reperfusion for the decline in direct current potential and levels of extracellular [Ca2+], adenosine, inosine, hypoxanthine, glutamate, and GABA in adult brain, with white matter being usually less negatively affected.14–16 Because white matter has approximately two- to fourfold lower metabolic and blood flow rates than gray matter, synaptic activity and excitatory neurotransmission (and excitotoxic damage) predominate in neuropil, and white matter is enriched with axons and oligodendroglia, caution must be applied when extrapolating data derived from small extracellular fluid volumes sampled by microdialysis (frequently placed in ‘normal’ white matter) to other brain regions. Sedated-anesthetized TBI patients probably have blunted differences among metabolic markers in gray and white matter owing to the pharmaceutical depression of function, metabolism, and blood flow.
Injury Classification
Microdialysate biomarker levels help distinguish between disruption of energy metabolism after TBI by hypoxia/ischemia or mitochondrial dysfunction.1,17–19 Lack of blood flow during ischemia rapidly depletes tissue oxygen, glucose, pyruvate, and other metabolites, causing large increases in lactate and LPR. 5 Mitochondrial dysfunction impairs oxidative metabolism although tissue oxygen level is normal or elevated, causing glycolytic upregulation, large increases in lactate with normal or slightly elevated pyruvate levels, and increased LPR. 20 A non-ischemic metabolic crisis occurs frequently after TBI, and abnormal biomarker levels predictive of poor outcome are the following: LPR >40, lactate>1.5 mmol/L, glucose <0.2 mmol/L, pyruvate <25 μmol/L, glutamate >5 μmol/L, and glycerol >50 μmol/L, with a negative correlation between the global rate of oxygen consumption (CMRO2) and microdialysate LPR.8,19,21 A nonischemic rise in LPR above the normal range of 20 to 25 due mainly to low pyruvate is classified as type 2 LPR (contrasting type 1 LPR, associated with ischemia, high lactate, and low pyruvate), suggesting a hyperglycolytic state in which glucose demand exceeds supply, glycolysis may be impaired, glucose may be diverted to other pathways, and mitochondria are dysfunctional; metabolic crisis with normal oxygen and pyruvate levels suggests an increased rate of glucose utilization (CMRglc) via glycolysis, sufficient glucose supply, and mitochondrial dysfunction.9,17,18,22 In contrast, major metabolic features of activation in normal brain include glucose supply matches increased demand, oxygen delivery exceeds demand, CMRglc > CMRO2, lactate and pyruvate levels increase in parallel, LPR is normal, and lactate is released in blood.18,23 Notably, some TBI patients exhibit characteristics similar to normal brain activation, i.e., increased CMRglc, elevated pyruvate and lactate levels, and stable LPR. 24
Limitations
Many researchers recognize that metabolite concentrations do not reflect metabolic rates, as in the classic example that glucose concentrations are relatively uniform throughout brain in spite of a 10-fold range in local CMRglc,25,26 i.e., supply matches demand. In microdialysis studies, metabolic changes are inferred from extracellular biomarker concentration changes. Concentrations are the net result of input and output to a metabolite pool, and either or both processes can affect metabolite level. Interpretive complexity of data is compounded by heterogeneity of TBI type, location, and severity, and by standard-of-care clinical protocols that use drugs for sedation or anesthesia, e.g., barbiturates and propofol, that can inhibit GLUT-1-mediated glucose transport,27,28 as well as depress brain function, energy demand, blood flow, and metabolic activity, all of which influence metabolite levels.
Inaccurate Terminology
Unfortunately, not all publications use appropriate terminology to present, analyze, and discuss TBI findings. Some authors present microdialysate lactate or glucose concentration data using ‘lactate or glucose metabolism’ or ‘lactate metabolic patterns' as manuscript or topic heading titles. This is not correct because assays of metabolism require measurements of biochemical conversion of one compound to another and rates thereof, or measurement of net flux into or from brain using the Fick principle, CMR = CBF [A - V], where CMR is cerebral metabolic rate; CBF, cerebral blood flow rate; and A - V is the arteriovenous difference. Other authors measured changes in global CMRglc and CMRlactate after TBI and interpreted findings as evidence for metabolic coupling between astrocytes and neurons, but these data contain no information about cell-cell interactions, metabolic coupling, or preferential fuel use by a specific cell type. Metabolism of lactate by brain has been stated to support astrocyte-neuron lactate shuttling, but metabolism per se does not reflect directional cell-cell transfer. These are not semantic issues; data are overinterpreted.
Summary
Microdialysis is a valuable tool for bedside monitoring to distinguish ischemia from mitochondrial dysfunction. Terminology can reflect and influence mechanistic thinking, and inaccurate statements detract from understanding complex metabolic changes after TBI.
LACTATE AS NEURONAL FUEL: ASTROCYTE-TO-NEURON LACTATE SHUTTLE
Lactate receives attention in the TBI literature owing to the predictive value of the LPR for metabolic status and clinical outcome, and TBI reports have considered the possibility that lactate serves as neuronal oxidative fuel (Figure 1),9,24,29–34 in accordance with the astrocyte-to-neuron lactate (ANL) shuttle model (Figure 2A).35,36 Bouzat et al37,38,40 and Sala et al 39 emphasized that lactate may be a glucose-sparing energy substrate and neuroprotective for injured brain. Because these claims may influence future neurointensive clinical care, supporting data must be carefully examined. There is a lot of experimental evidence against lactate shuttling (Figures 2B and 2C), but much less is available to evaluate lactate metabolism after TBI.
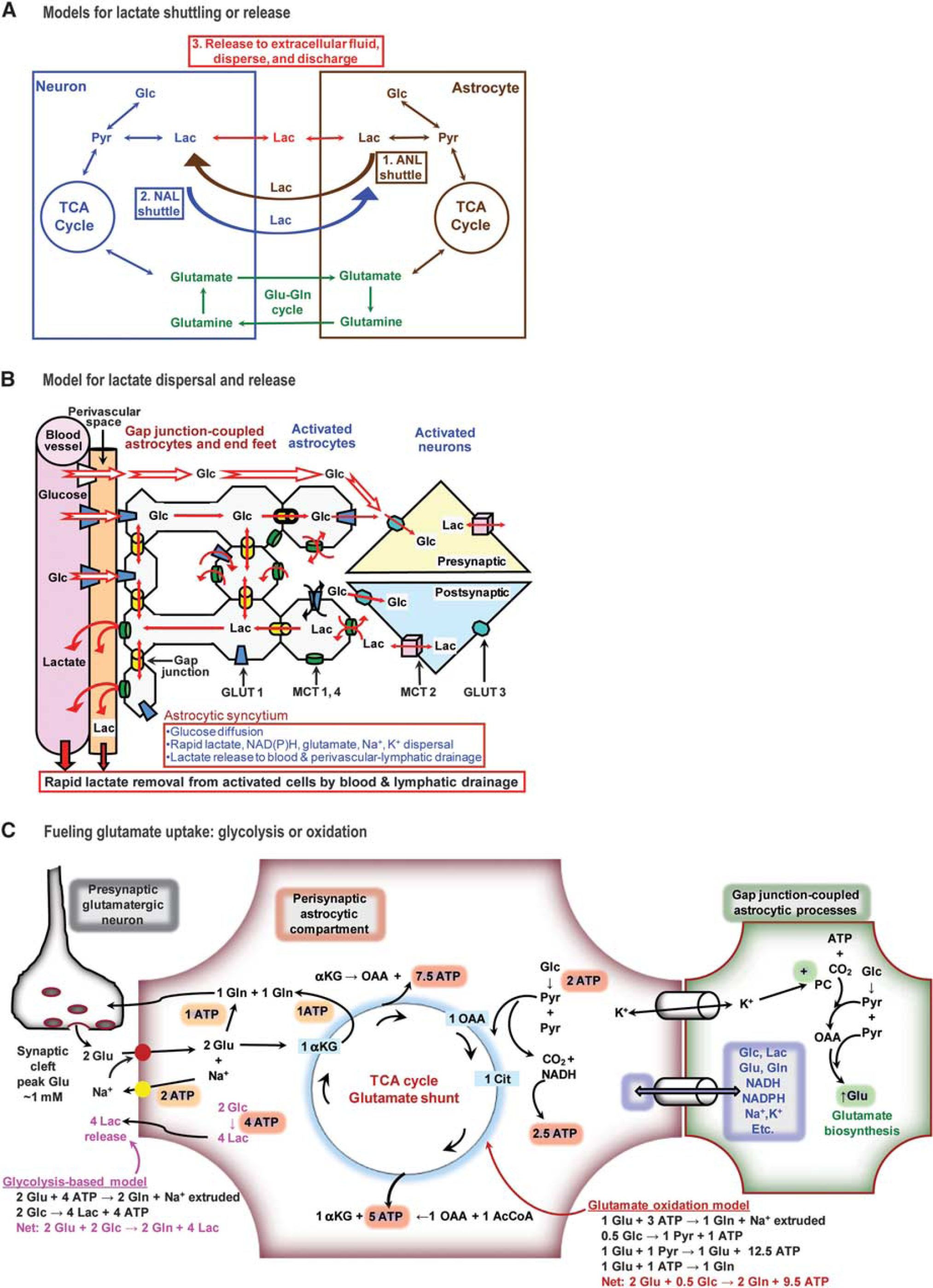
Models for lactate shuttling and release and fueling glutamate uptake. (
Lactate Shuttling
The ANL shuttle model is based on the experimental observation by Pellerin and Magistretti 36 of increased lactate efflux from cultured astrocytes after exposure to glutamate. The interpretive model (Figure 2A) predicts (i) metabolic compartmentation during brain activation coupled with glutamate-glutamine cycling, i.e., glutamate-evoked glycolysis to fuel Na+ extrusion from astrocytes (Figure 2C), lactate transfer to neurons, and neuronal lactate oxidation, with little or no rise in neuronal glycolysis; (ii) the rise in CMRO2 should stoichiometrically match the increase in CMRglc if lactate is locally oxidized; and (iii) metabolic assays of oxidative metabolism with labeled glucose or oxygen during activation should have a magnitude similar to assays of the hexokinase step using deoxyglucose (DG) (i.e., no loss of labeled lactate, which causes underestimation of calculated CMRglc and reduces CMRO2 owing to the substrate efflux). Recent reviews of this model by proponents35,38,41–44 do not adequately present its shortcomings, and many studies selectively cited in these reviews to support the model are not representative of similar studies in the neurometabolic literature. 45 Discordance between cited ANL-supportive studies and relevant uncited literature requires a more balanced presentation. Because (i) the magnitude of ANL shuttling has never been established and proven to be significant in vivo, and (ii) model predictions do not match experimentally established characteristics of brain activation in vivo, 23 statements such as the following by Bouzat et al 38 are very misleading: “However, emerging evidence demonstrated that in patients with TBI increased lactate seems predominantly non-hypoxic/ischemic and rather the consequence of increased cerebral glycolysis [2, 12, 13]. Lactate formed under such glycolytic conditions is shuttled from one lactate-producing cell (astrocyte) to another lactate-consuming cell (neuron) (astrocyte-neuron lactate shuttle) [14, 15].” Information documenting the rate and quantity of astrocyte-neuron lactate transfer in living brain is absent.
Strong Evidence Against the Astrocyte-to-Neuron Lactate Shuttle Model
Glutamate-evoked glycolysis, the keystone of the ANL model, is not a robust phenotype of cultured astrocytes. More laboratories find little or no effect of glutamate on astrocytic glycolysis and lactate release than those that observe the effect.45–47 The basis for these discrepant results is unknown but may arise from culture conditions causing differences in enzyme and mitochondrial development. In fact, glutamate is readily taken up and oxidized by cultured astrocytes (see McKenna et al 48 and cited references), glutamate uptake stimulates astrocytic CMRO2 (ref. 49) and glutamate uptake is accompanied by its oxidation and influences filopodial mitochondrial mobility and proximity to glutamate transporters.50–53 These features strongly support the conclusion that glutamate oxidation can provide ATP (Figure 2C) to support its uptake at the site and time when local energy demand increases during excitatory neurotransmission.54,55 Oxidation of neurotransmitter glutamate may also be important after TBI, because it is an energy-producing process, with glutamate turnover providing nearly as much ATP as glucose oxidation. 56 This process occurs in astrocytes, and astrocytic mitochondrial dysfunction after TBI would disrupt astrocytic energetics, glutamate-glutamine-GABA turnover, and neurotransmission.
During the 20 years since the ANL shuttle was proposed, lactate shuttling has never been directly demonstrated in the intact brain and the cellular origin of lactate in vivo is not known. Direct tests of the model are (i) rates of lactate uptake and shuttling, and (ii) neuronal upregulation of glycolysis. Rates of extracellular lactate uptake by astrocytes and rates of lactate transfer among gap junction-coupled astrocytes in adult rat brain slices were both four- to fivefold greater than neuronal lactate uptake and lactate shuttling to neurons, demonstrating low rates of lactate transfer to neurons and high capacity of astrocytes to take up and disperse lactate within the syncytium. 57 Increased neuronal CMRglc during activation in adult rat brain was recently demonstrated by co-authors of the lactate shuttling perspective, ‘Energy on Demand’.58,59 Parallel increases in glucose phosphorylation in synaptosomes and whole homogenate after in vivo labeling at rest and during activation suggest that the rise in neuronal glycolysis provides the pyruvate for oxidation. 58 These findings, along with many studies showing high capacity of synaptosomes and cultured neurons to increase glucose transport, glycolysis, and oxygen consumption with glucose as substrate (see Dienel 45 ) disprove the notion that neurons cannot increase glycolysis, a central element of the ANL shuttle model. Indeed, if neurons consume a lot of glucose during activation and are partially glycolytic, neurons may shuttle lactate to astrocytes (the NAL shuttle,60,61 (Figure 2A)) for dispersal and discharge from brain. The lactate release model (Figures 2A and 2B) is derived from studies to elucidate the basis for the large underestimation of CMRglc when assayed with [14C]glucose compared with [14C]DG. Lactate generated during activation and released to extracellular space is preferentially taken up by astrocytes, quickly dispersed throughout the syncytium, and discharged from astrocytic endfeet to perivascular fluid space for rapid clearance via the perivascular lymphatic drainage system and venous blood (Figure 2B). 23 To sum up, there are major fatal flaws with the ANL model: glutamate is oxidized, neurons upregulate glycolysis, CMRglc > CMRO2, and lactate is released.
Brain Slice Assays: Methods are Critical
Studies in brain slices by Schurr and Gozal 62 showed that lactate supports evoked potentials after ischemia (see Schurr and Gozal 62 and cited references). Similar findings have been obtained in some laboratories but not by others; the case for lactate as an important fuel is not open and shut. One reason for these discordant results is that outcomes of brain slice experiments are critically dependent on slice preparation procedures and assay conditions. Detailed studies by the Okada laboratory showed (i) lactate does not support neuronal function after rapid slice preparation, whereas it did after the slower procedure used by Schurr and others, and (ii) revealed that an unidentified calcium-dependent event is needed for lactate to support evoked potentials. 63 Furthermore, alternative substrates can maintain near-normal ATP levels in slices without preserving population spikes, indicating that glycolysis has essential functions besides providing pyruvate as fuel.63,64
Is Lactate a ‘Preferred’ Fuel For Neurons? Context is Important
Tissue culture and in vivo studies cited by ANL model proponents to document ‘preferential’ use of lactate compared with glucose are misleading if not presented in context. For example, lactate provided to cultured neurons within its range in normal and activated brain (∼0.5 to 2 mmol/L) is claimed to be the preferred substrate over glucose, contributing ∼ 50 to 80% of total oxidation.65,66 In sharp contrast, lactate contributes < 10% to total oxidation in brain of normal awake adult humans when assayed at < 3 mmol/L during lactate infusion under resting conditions or during moderate exercise.67,68 If lactate were truly ‘preferred’ fuel, one might expect that no lactate would be released from brain. However, normal, resting adult brain releases lactate equivalent to ∼ 5% of glucose uptake, and this fraction rises during activation69–71 (Table 1). Sensory stimulation or K+-induced spreading cortical depression is associated with rapid, substantial efflux of labeled and unlabeled lactate from rat brain (22% of glucose influx), the rise in CMRglc exceeds that of CMRO2, the CMRO2/CMRglc ratio falls, and CMRglc assays with [6-14C] glucose underestimate the increase by ∼ 50% compared with parallel [14C]DG assays. 23 A critical difference between adult brain and cultured neurons is the latter are from embryonic rodent brain, cultured in extremely high glucose (5 to 10 times that in diabetic brain) in the absence of astrocytes and oligodendroglia, and used for assays within a week, about the age at birth. Rodent brain is metabolically immature at birth and does not attain adult enzymes levels until after 30 to 40 days. 23 Immature cultured neurons are not a good model for adult brain metabolism, and they may be programmed to use substrates in milk during suckling when lactate and ketones are important fuel.72,73
Lactate fluxes to and from human brain relative to glucose uptake after TBI or normal awake at rest, during, or after mental testing
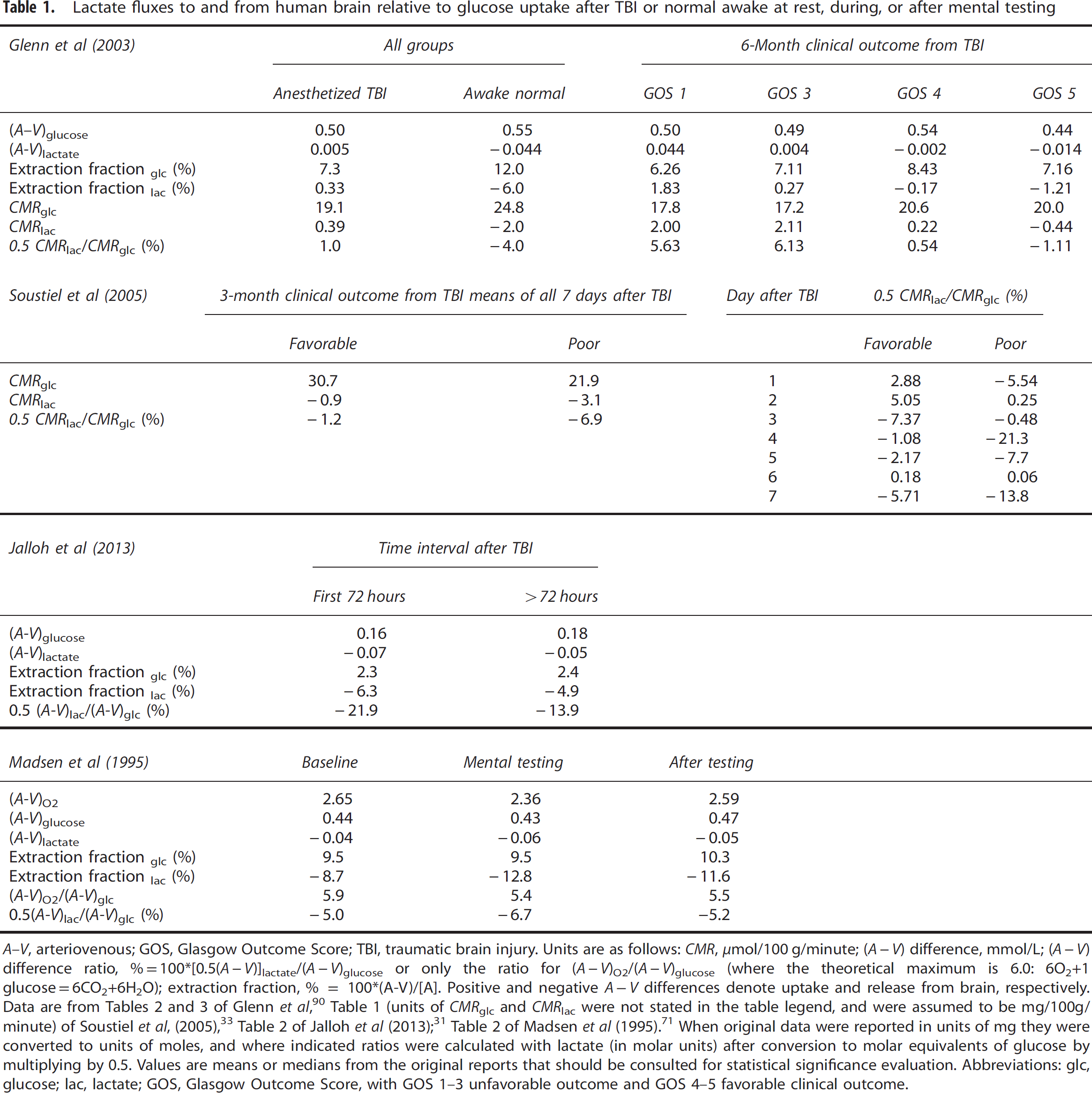
A-V, arteriovenous; GOS, Glasgow Outcome Score; TBI, traumatic brain injury. Units are as follows: CMR, μmol/100 g/minute; (A-V) difference, mmol/L; (A - V) difference ratio, %=100*[0.5(A-V)]lactate/(A-V)glucose or only the ratio for (A-V)O2/(A-V)glucose (where the theoretical maximum is 6.0: 6O2+1 glucose =6CO2+6H2O); extraction fraction, % = 100*(A-V)/[A]. Positive and negative A-V differences denote uptake and release from brain, respectively. Data are from Tables 2 and 3 of Glenn et al, 90 Table 1 (units of CMRglc and CMRlac were not stated in the table legend, and were assumed to be mg/100g/ minute) of Soustiel et al, (2005), 33 Table 2 of Jalloh et al (2013); 31 Table 2 of Madsen et al (1995). 71 When original data were reported in units of mg they were converted to units of moles, and where indicated ratios were calculated with lactate (in molar units) after conversion to molar equivalents of glucose by multiplying by 0.5. Values are means or medians from the original reports that should be consulted for statistical significance evaluation. Abbreviations: glc, glucose; lac, lactate; GOS, Glasgow Outcome Score, with GOS 1-3 unfavorable outcome and GOS 4–5 favorable clinical outcome.
Lactate Flooding Assays
The higher the arterial blood lactate level the greater its contribution to brain metabolism, and this phenomenon has a physiologic role of glucose sparing during strenuous muscular work and high glucose demand. In vivo studies cited for showing lactate is the ‘preferred’, glucose-sparing fuel involve either lactate infusions or intense physical exercise that elevate blood lactate to levels well above normal. This creates a large inward concentration gradient that drives lactate into the entire brain. Hyperlactemia increases lactate availability for all brain cells, and lactate contributes 19% and 27% of total oxidation at arterial lactate levels of 3.9 and 6.9 mmol/L, respectively, more at extremely high levels.67,68 The physiology of global hyperlactemia differs from activation when local brain lactate level rises approximately twofold and exceeds blood level. Similarly, blood ketone body levels are normally low and they are not ‘preferential’ fuel except during prolonged starvation when their levels rise and they become an important oxidative, glucose-sparing brain fuel. 74
Summary
The ANL shuttle has been an attractive, easy-to-understand metabolic model. However, cell-cell interactions are complex and the model is not compatible with results of many studies in normal adult brain, in brain slices, and in cultured cells. Recent studies disprove essential model elements, and the NAL model linked to lactate release (Figure 2A and 2B) may be more appropriate.23,45,54 There is no question that lactate is oxidized by neurons and astrocytes in varying amounts under different conditions, but lactate metabolism itself is not proof of astrocyte–neuron shuttling.
CHANGES IN LACTATE AND GLUCOSE LEVELS IN ACTIVATED BRAIN
Many microdialysis, microelectrode, and biochemical studies in experimental animals show that extracellular and total lactate levels rise ∼ 2-fold during activation. However, the cellular origin of lactate and the biochemical and physiologic processes that upregulate lactate production are unknown. The following examples illustrate some interpretive issues involved in use of lactate vs. glucose as fuel in vivo.
Percentage data
Hu and Wilson75,76 assayed extracellular lactate and glucose levels after electrical stimulation with enzyme-linked microsensors, calculated percent changes in lactate and glucose concentrations, and interpreted the larger percentage decreases in lactate level in terms of its preferential utilization. Recalculation of these data in units of moles to account for higher extracellular glucose concentration, molar glucose equivalents of lactate (lactate/2 = glucose), and continuous influx of glucose from blood to match CMRglc revealed that lactate use was a very small fraction of glucose utilization (for details, see Table 7 of ref. 45). This analysis underscores a serious problem arising from comparisons of percent changes that normalize and obscure differences in the magnitude of concentration changes and pathway rates.
Concentration change
Analysis of brain metabolites after sensory stimulation of non-fasted awake rats showed that brain glucose and lactate levels increased. 77 Without additional information, rise in brain glucose level might be interpreted as a reflection of reduced CMRglc and the increase in lactate as enhancement of glycolysis. Both increases were due, in part, to physical movement-induced increases in their arterial levels. Parallel assays showed that overall cortical CMRglc rose by ∼ 50% and brain:plasma glucose distribution ratios were similar during rest and activation indicating supply-demand coupling. Importantly, net lactate accumulation, a 100% increase, corresponded to only ∼ 5% of the calculated total amount of glucose converted to pyruvate during the experimental interval. Separate studies showed (i) increased lactate release from brain to blood and (ii) incomplete retention of 14C-metabolites of [6-14C]glucose.23,45 Thus, the quantity of accumulated lactate is trivial compared with glycolytic flux and its concentration change gives no information regarding the magnitude of glycolytic flux change. Metabolic and transport data are necessary for interpretation of concentration changes.
Metabolic fluxes
The above findings suggest a high flow rate of lactate from brain cells through the enlarged lactate pool to blood. This lactate trafficking is invisible when only lactate concentration is measured. For example, if TBI brain with a low CMRglc of ∼ 0.2 μmol/g per minute 24 became glycolytic and converted all glucose to lactate, the rise in microdialysate lactate level by 2.6 mmol/L (from normal of ∼ 2.9 mmol/L to ∼ 5.5 mmol/L after TBI 1 ) would only take ∼ 6.5 minutes (0.2 μmol glucose/g per minute × 2 lactate/glucose × 6.5 minutes = 2.6 μmol/g2.6 mmol/L, ignoring % brain water). With hourly microdialysate collections, this corresponds to ∼ 11% of the 24 μmol/g total pyruvate/lactate produced by glycolysis during the sampling interval (0.2 × 2 × 60 = 24). If CMRglc and dialysate lactate levels were constant, ∼ 89% of the lactate produced must have traversed through the lactate pool to blood. If a 6.5-minute transient glycolytic surge increases dialysate lactate level (pool input exceeds output), followed by normalization of CMRglc but equal input-output rates to the lactate pool, pool size would remain elevated even though CMRglc fell. The higher CMRglc and blood flow rates, the faster the response time and greater magnitude of change in metabolite pool size. These factors may differentially affect extracellular metabolite levels in gray and white matter in TBI brain.
Summary
Interpretation of concentration changes of extracellular metabolites as indicies of metabolic changes, while informative and diagnostically important, must be conservative. Comprehensive analyses of many pathways are required to understand the complex metabolic activities that are upregulated during activation and after TBI.
METABOLITE LEVELS, CMRGLC, AND CMRO2 AFTER TRAUMATIC BRAIN INJURY
Level and Rate Changes
Metabolic changes after brain injury are widely recognized by neurocritical-care researchers to be complex, with time, location, and injury independence. Common findings are initial increases in CMRglc shortly after TBI, followed by subsequent reductions that are usually smaller in magnitude than the decline in CMRO2 (see 34,78 and cited references). Thus, the CMRO2/CMRglc ratio falls after TBI and non-oxidative metabolism of glucose is more predominant. Reduced microdialysate glucose levels, elevated lactate levels and LPR, and lactate release to blood are common observations. Correspondence between local microdialysate metabolite levels and whole-brain arteriovenous measurements or regional positron emission tomographic metabolic assays of CMRglc or CMRO2 is difficult to evaluate owing to the small regions of interest surrounding a microdialysis probe and temporal relationships between data sets.19,24,79 Critically low extracellular glucose levels are often associated with a non-ischemic/non-hypoxic metabolic crisis after TBI, but glucose and lactate levels are not correlated, nor are CMRglc and glucose levels; CMRglc was positively, linearly correlated with lactate and pyruvate levels in one study, 24 but not in another. 19 Arterial glucose is maintained within set ranges by critical care protocols, and the reason(s) for low brain glucose levels are not known; the TBI literature suggests hyperglycolysis and/or transport deficits. Episodes of spreading depression may also contribute to metabolic dysfunction.80,81
Lactate and Clinical Outcome
Elevated extracellular lactate levels in ‘normal’ non-hypoxic white matter after subarachnoid hemorrhage were associated with better outcome compared with patients with hypoxic white matter. 82 Reference was made to lactate metabolism that was not measured in this and the authors' subsequent TBI study, 39 and the rise in lactate level must be due to glucose metabolism. This is an important distinction because the context presented in the introductions and discussions of these reports is that the lactate may be metabolized as neuronal fuel in injured brain. Instead, the primary factor influencing outcome may be increased glycolysis and pentose shunt fluxes to generate ATP, NADPH, and lactate. Moreover, their finding of normal or high pyruvate under nonischemic conditions and an LPR of ∼ 30 (ref. 39) suggests a metabolic crisis and mitochondrial dysfunction, 18 which should impair lactate metabolism. CMRglc and CMRO2 were not determined in the above TBI patients, but they were described as ‘hyperglycolytic’ (presumably based on elevated lactate), which, according to Soustiel et al, 33 occurs when CMRglc > 0.39 μmol/g per minute. At this rate, 23.4 μmol glucose/g is consumed during their 60-minute dialysate collection interval. 39 The mean level of ‘glycolytic lactate’ was 6.1 μmol/g, 39 and assuming a normal lactate level of 2.9 μmol/g, 18 there is a net gain of 3.2 μmol lactate/g, or 1.6 μmol glucose equivalents/g, which corresponds to only 7% of glucose consumed. Accumulated lactate is a trivial fraction of glucose metabolized. What happened to the remaining 93%? If mitochondria in TBI brain are dysfunctional, most of the lactate generated was probably released to blood. Quantitative metabolic/ transport studies are needed to evaluate these issues.
CMRglc Assays
Reduced brain glucose level is a critical issue for rate calculations in CMRglc assays with [18F]fluorodeoxyglucose or [14C]DG. The lumped constant of the DG method is the factor that converts DG phosphorylation to glucose phosphorylation, 25 and it contains the factor lambda that is sensitive brain glucose concentration; its value increases progressively as glucose level falls below ∼1 μmol/g.83,84 Changes in the lumped constant were proposed to explain the greater rise in calculated CMRglc when assayed with [18F]fluorodeoxyglucose compared with [1-14C]glucose after TBI. 85 Unfortunately, the authors did not measure glucose levels in their brain extract samples used to measure ATP, ADP, and AMP. They could have estimated the value for lambda even if glucose was not in a true steady-state after injury (brain glucose half life is ∼ 1.5 minutes 86 ). Also, increased flux of [1-14C]glucose-6-phosphate into the pentose shunt pathway after TBI (rising to 20% of glycolysis 87 ) would cause label loss as 14CO2 and contribute to underestimation of calculated CMRglc with [1-14C]glucose. Efflux from brain of labeled lactate causes proportionate underestimation of calculated CMRglc when labeled glucose is the tracer, and their 10- to 20-minute [18F]fluorodeoxyglucose-[1-14C]glucose assays 85 allowed sufficient time for [14C]lactate release to blood. Lactate efflux is fast during spreading depression and accounts for half of the 50% lower rates obtained in 5- to 7-minute assays with [6-14C]glucose compared with [14C]DG, using a corrected lumped constant.88,89 Discordance between these two CMRglc tracers also occurs after sensory stimulation and is due mainly to lactate release. 23
Arteriovenous Difference Ratios for Lactate and Glucose
A-V and CBF assays allow calculation of global CMR as CMR = CBF (A - V). Cerebral blood flow is not measured in most TBI studies, but the A - V ratio for two substrates represents their relative metabolic rates (CBF cancels out), and A - V ratios can be informative. Interpretation of relative utilization rates of oxygen, glucose, and lactate requires units of moles (not mg for glucose or lactate, or ml for oxygen) and lactate expressed as molar glucose equivalents (lactate/2 = glucose). Conversion of data in the Glenn study 90 to molar units revealed small mean (A - V)lactate (<0.05 mmol/L) values for all clinical outcome groups (Table 1), raising the question of their statistical significance owing to the <4% imprecision of lactate analysis. In the range of 2.7% to 4% imprecision the error margin is ± 0.05 to 0.08 mmol/L.91,92 Relative lactate uptake into brain (0.5*CMRlac/CMRglc) was ∼ +6% in TBI patients with worst outcome (GOS 1-3), whereas those with better outcome (GOS 5) showed small lactate release. Recalculation of data of Soustiel et al 33 revealed that lactate release from brain predominated during 7 days after TBI, averaging ∼ 1% or 7% of glucose uptake in the favorable and poor outcome groups, respectively, with daily variations in lactate fluxes (Table 1). Both studies 33,90 noted episodes of lactate uptake, but nearly all lactate uptake/efflux events were small, <6% of glucose influx (Table 1). Glucose was the predominant fuel.
In the study by Jalloh et al, 31 overall lactate efflux after TBI was 14% to 22% of glucose uptake, suggesting higher glycolytic rates in this cohort that were associated with relatively low extracellular glucose levels (0.54 to 0.77 mmol/L, uncorrected for dialysate recovery), low brain/plasma glucose ratios (0.08 to 0.11), and low glucose extraction fractions of ∼ 2% (i.e., [A - V]glucose/arterial glucose concentration; note: this ratio varies with CBF). Extraction fractions were higher in other studies, 6% to 8% after TBI and 12% in awake controls in the Glenn study 90 and ∼ 10% in the Madsen study 71 of normal humans before, during, and after mental testing (Table 1). Lactate release rose during mental testing and the oxygen/glucose ratio fell (Table 1). Although lactate efflux from TBI brain predominated in magnitude and frequency and (A - V)lactate values were generally small (see Jalloh et al 91 and Nordstrom et al 92 ), Jalloh et al 31 emphasized the frequency of periods of brain lactate uptake when microdialysate lactate levels were higher than those in blood and related this uptake to possible use of lactate as fuel according to the ANL shuttle model. Lactate uptake against a gradient were also observed by Meierhans et al 32 in TBI patients given lactate-containing fluid infusions for volume management. Estimates of (A - V)lactate/(A - V)glucose at arterial lactate levels > 2 mmol/L from their Figure 5 (∼ +17%) in Meierhans et al 32 suggest that lactate may contribute to brain energetics in spite of brain and blood glucose levels being sufficiently high to support CMRglc. In both studies, lactate uptake was greater when arterial lactate was higher, but its association with the magnitude of concomitant glucose uptake differed for unknown reasons (see discussion in Jalloh et al 31 ). Jalloh et al reported and stressed the relative daily frequency of lactate uptake and efflux events after injury, but this analysis would have been much more informative if (A - V)lactate/(A - V)glucose ratios were also analyzed to evaluate the metabolic significance of the relative rates/quantities of lactate uptake or release compared with glucose. Because (A - V)lactate/(A - V)glucose ratios calculated from overall median values reveal predominant, substantial lactate efflux (Table 1), quantitative use of lactate as supplemental fuel was probably minor.
In the study by Magnoni et al, 93 the LPR was 17 in TBI patients with extracellular glucose and lactate of 1.8 and 2.6 mmol/L, respectively, whereas LPR was 43 in patients with lower glucose and higher lactate levels,1.1 and 6.0mmol/L, respectively. Pyruvate levels were similar in both groups, 93 indicative of mitochondrial dysfunction in those with higher LPR. 18 A progressive rise in LPR as brain glucose level fell, particularly when glucose was below 1 mmol/L, was observed by the Stover group, 94 suggestive of increased glycolysis. This notion is consistent with their earlier report 95 revealing progressively more negative (A - V)lactate/(A - V)glucose ratios with decreasing arterial blood glucose levels, indicating greater release of lactate as arterial glucose became lower. Curiously, at the lowest arterial blood glucose levels, the CMRO2/CMRglc ratio exceeded the theoretical maximum of 6.0 (6O2 + 1 glucose 6CO2 + 6H2O), and some values were astonishingly high. The CMRO2/CMRglc ratio accounts for any oxidation of lactate generated in brain from glucose (i.e., as CMRglc), but oxidized substrates are not identified in CMRO2 assays. In these studies, more lactate was released when arterial blood glucose levels were lowest, and brain oxidation of non-glucose substrates increased.
Lactate Efflux During Pentose Shunt Assays in Traumatic Brain Injury Patients
Diversion of glucose from glycolysis to other pathways is considered as contributory to low pyruvate levels in type 2 elevated LPR during metabolic crisis after TBI. One diversion is flux through the pentose phosphate shunt pathway (Figure 1) that rises 2.8-fold from 6.9% to 19.6% of glycolytic rate in TBI patients, based on A - V differences for lactate isotopomers after intravenous infusion of [1,2-13C]glucose. 87 This important finding establishes the global magnitude of response to enhance oxidative stress management after TBI. Another striking aspect of this study is that the analysis was based on release of labeled lactate to blood after passage of labeled glucose directly through the glycolytic pathway to generate [1,2-13C]lactate and after detour through the pentose shunt and re-entry into the glycolytic pathway at the fructose-6-phosphate or glyceraldehyde-3-phosphate steps (Figure 1) to generate [1-13C]lactate. The quantities of lactate released relative to glucose uptake are not available, but sufficient lactate was continuously released to blood throughout the 2-hour assay period to enable isotopomer analysis and pathway flux assessment.
Glucose-6-phosphate that enters the pentose shunt can be used for biosynthesis or re-enter the glycolytic pathway (Figure 1). The biosynthetic fraction in normal adult brain is not known, but it is probably much less than during development and it may rise after TBI. Even if some shunted carbon were used for repair/regenerative processes, the remainder would re-enter the glycolytic pathway and produce energy (otherwise phosphate would be trapped in shunt intermediates and reduce its availability for ATP synthesis). If all of the glucose except carbon one that is lost by decarboxylation in the pentose shunt re-enters glycolysis, then only 1/6 of the glucose molecule (17%) is lost by shunting. In normal brain, regulatory mechanisms can rapidly increase glycolytic flux by three- to sixfold and easily compensate for this carbon loss. During acoustic stimulation of the awake rat, pentose shunt flux rose from ∼7% to ∼ 25% of glucose oxidative metabolism (Figure 6C in Dienel 23 ), and this increase was not associated with reduced microdialysate glucose level. 96 Stability of glucose level in normal brain is consistent with demonstration that glucose influx to rat brain exceeds demand by ∼ 60% (the rest diffuses back to blood) over a fourfold range of metabolic rates and wide range of plasma and brain glucose levels (Figure 3E in Dienel 23 ). Thus, detours from the glycolytic to pentose shunt pathway may not be a serious problem owing to the probable reentry of most of the remaining 83% of glucose carbon into the glycolytic pathway upstream of the ATP-producing steps (Figure 1). Even if half of the pentose shunt flux were used for biosynthesis after TBI, only ∼ 10% of glycolytic flux is lost from energy-producing pathways (i.e., 50% × 20%, the measured pentose flux as fraction of glycolytic rate in TBI 87 ).
Summary
Transient episodes of brain lactate efflux and influx occur in TBI patients, but excursions from zero net transport across the blood-brain barrier are generally small and lactate efflux predominates (Table 1). Mitochondrial dysfunction may restrict lactate oxidation, and available data indicate that lactate uptake is quantitatively small compared with glucose. Lactate does not robustly supplement glucose after TBI except in rare examples.
SOME SPECULATIONS: HYPOGLYCEMIC GLYCOGENOLYSIS, MITOCHONDRIAL UNCOUPLING, AND LACTATE UPTAKE AGAINST ITS CONCENTRATION GRADIENT
Glycogen
Taken together, the above human TBI studies often depict situations with (i) low brain glucose levels that are associated with greater lactate efflux (relative to glucose influx) than occurs at higher glucose levels, (ii) a rise in brain lactate level with normal pyruvate level, and (iii) a progressive rise in LPR ratio with decreasing brain glucose level. These findings suggests higher glycolytic rate when brain glucose levels are lowest, implying an endogenous source of glucosyl units. Glycogen is mobilized in astrocytes (Figure 1) during hypoglycemia,97,98 and glycogenolysis would help maintain astrocytic glycolytic rate, sustain their pyruvate level, increase lactate level, and increase LPR. Depending on TBI patient history (glucose level, anesthesia, and other factors that affect glycogen content99,100), glycogen can be an important endogenous fuel that contributes to lactate level and release during cerebral hypoglycemia. The cellular basis of extracellular metabolic biomarker levels is unknown, and astrocytes may make substantial contributions.
Metabolite Oxidation and Uncoupling
A CMRO2/CMRglc ratio below 6 is indicative of non-oxidative metabolism of glucose, whereas an increase above 6 reflects oxidation of non-glucose substrates. An elevated CMRO2/CMRglc ratio appears to be unusual after TBI, and the ratio >6 was not because of blood lactate consumption in the Holbein study 95 because there was a concomitant increase in net lactate release from brain when arterial glucose levels were lowest and ratio highest. These subjects probably had cerebral hypoglycemia, and endogenous metabolites oxidized during hypoglycemia include glycogen, glycolytic and tricarboxylic acid cycle intermediates, and amino acids (e.g., glutamine, glutamate, GABA, alanine). 5 Another possible explanation for the elevated CMRO2/CMRglc ratio after TBI is mitochondrial dysfunction with uncoupling. Respiratory control is the process in which CMRO2 is governed by the supply of ADP and phosphate (arising from brain work and ATP use, Figure 1) and is linked to ATP synthesis. Electron transport continues during uncoupling but does not lead to ATP production. Unregulated oxygen consumption continues and may contribute to a high CMRO2/CMRglc ratio.
Heterogeneous Brain Lactate Levels
Uptake of lactate into brain against its concentration gradient is puzzling because the process is passive and involves co-transport of lactate with one proton. 101 Possible explanations include the following. (i) Lactate transport driven against its uphill gradient by a downhill proton gradient. 31 (ii) The microenvironment of the microdialysis probe may not reflect the entire brain that is registered by global A-V assays. Jalloh et al 31 placed probes in white matter, whereas Meierhans et al 32 placed them within injured tissue while avoiding contusions. Oligodendroglia in white matter are considered to be glycolytic and supply lactate to axons. 102 Upregulation of their glycolytic rate after TBI may cause locally elevated white matter lactate levels compared with overall brain, particularly if white matter blood flow is lower than average brain and lactate is clearance slower. Regional heterogeneity of glycolytic rates in the penumbra may also give rise to discordant focal versus global brain lactate levels, e.g., owing to the local spreading depression episodes.80,81 Heterogeneity of lactate levels may explain apparent global lactate uptake against a focal concentration gradient.
Lactate Clearance Routes: A caveat
A-V assays may underestimate lactate efflux (and glucose and lactate influx), because they do not account for perivascular lymphatic drainage pathways. This system contributes to clearance of small molecules and proteins from interstitial fluid to lymph nodes in the neck and spine, and Johnston and colleagues have shown that it accounts for a large fraction of CSF clearance.103–106 Early studies established rapid distribution of tracers inserted into interstitial fluid or CSF throughout the entire brain via perivascular fluid, and rates of tracer delivery to lymph nodes varied with head position, intracranial pressure, arterial pulsations, and other factors.107,108 Amyloid-beta, 14C-metabolites of [14C]glucose, and fluorescent tracers are cleared via this pathway by mechanisms involving astrocytic water movements mediated by aquaporins.109–112 In our studies, lactate release to blood (22% of glucose influx) accounted for about half the magnitude of underestimation of CMRglc when assayed with [6-14C]glucose, and follow-up studies indicate that a similar quantity is probably lost via perivascular flow. 23 Clearance of some glucose and lactate from brain via lymphatic drainage after their uptake into brain but before they are metabolized may contribute to the ‘missing’ quantities of glucose and lactate taken up during exhaustive exercise and not accounted for by oxygen consumption or accumulation in brain. 113 Spreading of TBI biomarkers within brain via perivascular flow may help detect or obscure critical concentration levels, whereas CSF drainage to control intracranial pressure in TBI patients can remove metabolites, reduce biomarker levels, and affect A - V difference assays.
LACTATE AS SUPPLEMENTAL OXIDATIVE FUEL AFTER TRAUMATIC BRAIN INJURY
Rodent Models
Lactate is preferentially taken up at the injury site shortly after TBI, 114 lactate infusion reduces the post-injury decline in extracellular glucose level, 115 and in vitro assays of CMRO2 by percussion-damaged tissue harvested at 5.5 hours after injury showed that high levels of lactate, but not glucose, stimulated CMRO2. 116 Favorable effects of lactate infusion for 3 h after TBI include cognitive improvement and preservation of cerebral ATP.117,118 However, control ATP level (∼0.04 μmol/g) 117 is much too low (normally 2 to 3 μmol/g), suggesting inadequate methods to preserve labile metabolites. Lactate infusions for 6h after TBI did not prevent the reduced levels of a host of energy metabolites. 119 It is not clear exactly if or when mitochondrial dysfunction manifest after TBI in these studies, and these apparently conflicting reports suggest beneficial roles for lactate infusions during the early-recovery interval (< ∼ 6 hours) unrelated to preserving glucose levels and without restoring energetic deficits. For example, cognitive effects may arise from lactate-evoked redox signaling pathways that alter NAD+ and NADH levels and influence gene expression via their interactions with transcription factors.120–122
Lactate Oxidation after Traumatic Brain Injury
Gallagher et al 30 assessed metabolism of 13C-substrates after infusion into TBI patient brain by microdialysis and reported qualitative detection of oxidative and glycolytic metabolism, i.e., label incorporation into glutamine from acetate and lactate and into lactate (curiously, not glutamine) from glucose. Extension of this approach to establish quantitative substrate contributions to overall metabolism will be very difficult because of the low temporal sensitivity and inability to assess intracellular metabolites. Lactate and glucose are metabolized by neurons and astrocytes after infusion by microdialysis, 123 and assessment of cellular compartmentation requires assays of enrichment of total glutamate and glutamine. A caveat with direct delivery into brain is cell-type metabolic compartmentation can change as intracisternal injections of pyruvate, acetoacetate, and glucose favor astrocytic metabolism, whereas their intravenous delivery exhibits neuronal preference. 124
Lactate as ‘Preferred’ Fuel after Traumatic Brain Injury
To test their notion that providing additional lactate might improve outcome of TBI patients, Bouzat et al 38 infused lactate intravenously for 3h to increase arterial lactate from 1 to 6 mmol/L and monitored extracellular metabolites in subcortical white matter near ‘normal’ tissue. The infusion increased brain extracellular lactate, pyruvate, and glucose levels with no significant change in LPR. The authors claim to have shown that (i) exogenous lactate may be utilized aerobically as preferred substrate over glucose, (ii) increased pyruvate and glucose levels reflect improved energetics due to supplemental exogenous lactate during the early phase of TBI, and (iii) hypertonic lactate was associated with reduction of intracranial pressure and glutamate. These conclusions were criticized as misleading 125 and the rebuttal by Bouzat, Magistretti, and Oddo 37 is far from compelling. They have not “provided convincing data that exogenous supplemental lactate can be used as a preferential glucose-sparing substrate by the injured brain …‥”.37,38 Because this study had no direct measures of metabolism and likely alternative explanations for key findings were not taken into account, the authors' major claims are overinterpreted and misinterpreted for the following reasons.
Lactate infusions increase blood lactate, which is quickly interconverted with pyruvate in peripheral organs.126,127 A total dose of lactate similar to that used by Bouzat et al 38 given to initiate panic attacks in susceptible humans increased blood lactate (from 0.9 to 4.8 mmol/L) and pyruvate (0.08 to 0.23 mmol/L) levels in controls and nearly doubled the blood LPR (11.6 to 21). 128 These data suggest that the rise in brain pyruvate level in TBI brain after lactate infusion 38 was due to greater pyruvate influx from blood. Also, brain LPR during lactate infusion was probably influenced by blood lactate, pyruvate, and LPR. Elevated brain pyruvate is not proven to be evidence of ‘improved brain energetics' owing to the ‘preferred’ metabolism of lactate.
Metabolite concentrations were stated to be expressed as the maximal increase during lactate infusion, 38 so reported extracellular pyruvate and glucose changes are inflated compared with means of all values during the 3-h intervention, and any ‘benefit’ may be overrepresented. The group mean dialysate glucose level during lactate therapy plotted in their Figure 3 appears be closer to ∼ 2.8 mmol/L than 2.2 stated in text. 38 This plotted value exceeds all but 2 to 3 of the individual values and it may include the high outlier, suggesting the rise in glucose level after lactate infusion is overestimated. The baseline dialysate glucose levels in most patients (mean = 1.4 mmol/L, and all would be higher when corrected for recovery) exceed the level (∼0.7 to 1 mmol/L) at which hexokinase operates at maximal velocity (its Km for glucose is ∼ 0.05 mmol/L 129 ). Glucose supply appears to be adequate for its utilization, but CMRglc was not measured and any effects of lactate on CMRglc and sparing of glucose are not established.
An alternative explanation for the rise in brain glucose level during lactate infusion is prolonged, increased rate of co-transport of lactate and H+ into all brain cells (Figure 1). Lactate influx may cause sufficient intracellular acidification to inhibit phosphofructokinase that has a very steep pH activity profile.130–132 Intracellular acidification assays are used to measure lactate uptake into astrocytes and neurons 133 and other cell types.134,135 Inhibition of this key glycolytic regulatory enzyme could reduce CMRglc and increase brain glucose levels, regardless of whether lactate were metabolized after its transport.
Lactate conversion to pyruvate generates NADH that must be oxidized to NAD+ for lactate utilization to continue (Figure 1). This processes requires a redox shuttle to transfer reducing equivalents into mitochondria, functional mitochondria, and matching CMRO2. During metabolic crisis when mitochondria are damaged and CMRO2 is reduced, it is unlikely that lactate is an advantageous fuel. Furthermore, flooding the brain with lactate will ‘push’ the lactate dehydrogenase reaction toward NADH production, depleting NAD+ availability for glycolysis. Unless mitochondria and a redox shuttle are active, reduced NAD+ will not only depress metabolism of glucose via glycolysis (as will intracellular acidification), it may also affect pentose shunt flux by impairing metabolism of material re-entering glycolysis (Figure 1). Compromising oxidative defense via the pentose shunt and depressing glycolysis due to NAD+ depletion and acidification when mitochondria are dysfunctional are likely to have detrimental effects on clinical outcome. These issues must be evaluated before implementing lactate therapies.
The LPR is stated 38 to be a ‘marker of cerebral energy demand’ but this is not accurate because LPR can change owing to the altered fluxes in ATP-producing pathways. Assume CMRglc after TBI is 0.2 μmol/g per minute, with 2ATP/mol glucose from glycolysis and 30 from oxidation, giving rates of 0.4 and 6 μmol ATP/g per minute. If mitochondrial dysfunction causes a 30% decrement in oxidative ATP production (0.3 × 6 = 1.8 μmol/g per minute), then glycolysis would have rise by greater than fourfold to make up the difference, causing LPR to rise with the same energy demand.
Long-term clinical benefit of lactate in small cohorts is not robust because the percent favorable outcome for lactate-infused patients at 6 months (60%) (ref. 38) is similar to the 54% without lactate intervention. 39 If the six patients with poor outcome were the same ones with baseline LPR >30 (plotted in their Figure 5, Bouzat et al 38 ) mitochondrial dysfunction may be more severe and they may be least able to metabolize lactate/pyruvate.
To sum up, lactate infusions may confer various beneficial clinical outcomes, but the observed effects denoted as ‘improved energetics' and ‘preferential’ fuel probably arose for reasons other than lactate metabolism. Alternative explanations must be ruled out.
PYRUVATE AS AN ALTERNATIVE TO LACTATE AFTER TRAUMATIC BRAIN INJURY
One factor that contributes to metabolic dysfunction is activation of poly(ADP-ribose) polymerase-1, which consumes cytoplasmic NAD+, thereby impairing glycolysis and pyruvate supply. 136 NAD+ levels are reduced and ADP-ribose levels are increased after experimental TBI, 119 and this effect may underlie type 2 LPR with low pyruvate. Pyruvate oxidation does not need a redox shuttle system (Figure 1) and it by passes restrictions imposed by poly (ADP-ribose) polymerase-1. Furthermore, pyruvate is a ‘neuroprotective’ reagent, because it reacts non-enzymatically with reactive oxygen species 137 , and beneficial effects of pyruvate have been documented after TBI, subarachnoid hemorrhage, and glutamate excitotoxicity.138–142 Infusion of lactate provides a continuous supply of pyruvate that may contribute to beneficial effects of lactate, but pyruvate itself may be the best choice as metabolic substrate and neuroprotective agent. Infusions of sodium pyruvate would, like sodium lactate, be limited by increased Na+ levels in blood, and sodium pyruvate should help control increases in intracranial pressure.125,143,144
CONCLUDING COMMENTS
The notion of lactate shuttling is seriously flawed and use of lactate as supplemental fuel requires (patho)physiologic context. Provision of exogenous oxidative fuel supplemental to glucose may be a useful approach for treatment of TBI patients, particularly during cerebral hypoglycemia, but functional mitochondria are required. Many aspects of supplemental fuel use need to be carefully considered and potential deleterious effects ruled out. Because metabolic and CBF assays are difficult and expensive to carry out in neurointensive care wards, greater use of A - V ratios for substrate pairs may help evaluate their relative use after brain injury, and multiple dialysis probes could monitor different brain regions. Advances in understanding metabolic derangement after TBI will come from rigorous, detailed analysis of quantitative studies. Elucidation of the basis for mitochondrial dysfunction is central to this issue. Microdialysis is an extremely valuable technology, and innovative, complementary approaches need to be developed.
Footnotes
The authors declare no conflict of interest.