
Abstract
Most patients who die after traumatic brain injury (TBI) show evidence of ischemic brain damage. Nevertheless, it has proven difficult to demonstrate cerebral ischemia in TBI patients. After TBI, both global and localized changes in cerebral blood flow (CBF) are observed, depending on the extent of diffuse brain swelling and the size and location of contusions and hematoma. These changes vary considerably over time, with most TBI patients showing reduced CBF during the first 12hours after injury, then hyperperfusion, and in some patients vasospasms before CBF eventually normalizes. This apparent neurovascular uncoupling has been ascribed to mitochondrial dysfunction, hindered oxygen diffusion into tissue, or microthrombosis. Capillary compression by astrocytic endfeet swelling is observed in biopsies acquired from TBI patients. In animal models, elevated intracranial pressure compresses capillaries, causing redistribution of capillary flows into patterns argued to cause functional shunting of oxygenated blood through the capillary bed. We used a biophysical model of oxygen transport in tissue to examine how capillary flow disturbances may contribute to the profound changes in CBF after TBI. The analysis suggests that elevated capillary transit time heterogeneity can cause critical reductions in oxygen availability in the absence of ‘classic’ ischemia. We discuss diagnostic and therapeutic consequences of these predictions.
Keywords
INTRODUCTION
Ninety percent of patients who die after severe traumatic brain injury (TBI) show histologic evidence of ischemic brain damage, even after excluding infarctions in relation to contusions, and brainstem infarctions, which can be attributed to increased intracranial pressure (ICP).1,2 The ischemic lesions are typically localized in the cortex (distributed diffusely or according to either vascular territories or their boundaries), in the subcortical gray matter (basal ganglia and the hippocampus), or in the cerebellum.1,2 These findings are consistent with extended periods of hypoxia, hypotension, or elevated ICP, 2 but even after the introduction of optimized guidelines to avoid pre- and in-hospital hypoxic episodes in TBI victims, the post mortem frequency of ischemic damage has remained largely unaffected.3,4
It has proven surprisingly difficult to demonstrate where, when, and why cerebral ischemia occurs in TBI patients. The extent to which cerebral blood flow (CBF) is reduced early after severe blunt head injury depends on the size and location of the resulting tissue injury. Adjacent and ipsilateral to large contusions or hematoma, hypoperfusion is typically more severe than in smaller lesions, whereas CBF is often globally affected by diffuse brain swelling. 5 In addition to this spatial heterogeneity, CBF varies considerably over time after the injury. Within the first 12 hours after TBI, one-third of TBI patients reveal CBF values that are consistent with cerebral ischemia (< 18–20 mL/100 mL/minute),5,6 but the parallel increases in the arteriovenous oxygen concentration difference are lower than those found in cerebral ischemia. 6 Positron emission tomography studies within 8–14 hours of injury show reduced CBF and global cerebral metabolic rate of oxygen (CMRO2), 7 but also that brief reductions in CBF by hyperventilation (HV) are compensated by increased oxygen extraction fraction (OEF), both globally 7 and regionally. 8 Accordingly, the metabolic needs of brain tissue appear to be met within the first 12 hours after TBI.
After this initial hypoperfusion, the coupling between CBF and the metabolic needs of brain tissue is seemingly lost. After 12 hours, many patients develop hyperemia, with CBF values as high as twice those of normal reference tissue, whereas others maintain either low or normal CBF.6,9–14 Later, typically between days 4 and 15, as many as 50% of TBI patients develop signs of vasospasm and hypoperfusion.15,16 Although these vasospasms are associated with infarction on subsequent computerized tomography images in some patients, CBF and OEF values recorded during this late phase remain higher than those recorded during the initial hypoperfusion.13,15,16 Because of these conflicting findings, the time at which ischemic damage occurs after TBI remains uncertain.
The apparent uncoupling of CBF and oxygen metabolism develops not only over time, but also across the brains of individual patients. Using positron emission tomography data acquired in TBI patients 9–23 hours after injury, Coles et al 17 showed that the distribution of voxel OEF values after injury was much broader in TBI patients than in controls. Accordingly, some brain tissue show abnormally low OEF, consistent with either tissue damage or relative hyperperfusion, whereas a large proportion of brain tissue shows OEF values above thresholds that, in acute stroke, would characterize ischemic, infarction-prone tissue. 17 Positron emission tomography studies conducted 2 to 3 days after injury show that although the CMRO2 threshold for subsequent infarction is similar to that found in acute stroke, the corresponding CBF and OEF thresholds vary greatly, 18 suggesting that the uncoupling of CBF from the metabolic needs of the tissue varies across the injured brain.
The origin of the apparent flow-metabolism uncoupling remains poorly understood. Verweij et al 19 reported impaired mitochondrial function in pericontusional tissue, suggesting that ATP production may be critically low, although oxygen consumption is only moderately reduced. Menon et al 20 found that regional OEF showed little dependence on local tissue oxygen tension (PtO2) and changes in CBF, and proposed that diffusion from blood to tissue may be hindered by changes in capillary wall morphology. Stein et al. 21 found an association between the presence of microthrombosis and selective neuronal loss, suggesting that intravascular thrombosis may contribute to the findings of widespread ischemic changes after TBI.
Capillary flow heterogeneity has been shown to reduce the extraction of diffusible solutes from the capillary bed,22–24 and animal models of elevated ICP show profoundly disturbed capillary flow patterns with evidence of ‘shunt flow'. 25 In this review, we identify potential sources of capillary flow heterogeneity after TBI and examine whether the changes in CBF over time are consistent with coupling to the metabolic needs of the tissue under varying degrees of capillary flow disturbances. Then, we discuss how therapeutic interventions such as hyperventilation (HV), therapeutic hypothermia, the use of vasopressors to augment cerebral perfusion pressure (CPP), and edema management, affect brain oxygenation if capillary flow patterns are disturbed.
CHANGES IN THE MICROCIRCULATION AFTER TRAUMATIC BRAIN INJURY
We first review the evidence of changes in capillary morphology and blood viscosity in the early phases after brain injury, and of changes in capillary flow patterns in conditions of elevated ICP.
Astrocytic Endfeet Swelling and Capillary Compression
Glial swelling is a prominent feature of tissue specimens obtained early after cerebral contusions in humans. In tissue removed between 3 h and 3 days after injury, disruption and gross swelling of pericapillary astrocytic endfeet is a consistent finding, and this swelling is observed to cause compression of the capillary lumen 26 —see Figure 1. Astrocytic endfeet swelling is limited after day 3, but microvascular compression is typically still observed within the first week. 26 Astrocytic endfeet swelling has been observed as early as 1 hour after TBI in rats, 27 and results in baboons further suggest that this abnormality is an early and transient phenomenon that peaks 6hours after injury. 28 In biopsies obtained the first week after injury, the capillary lumen is also affected by capillary endothelial swelling, microvascular collapse, and perivascular edema. 20
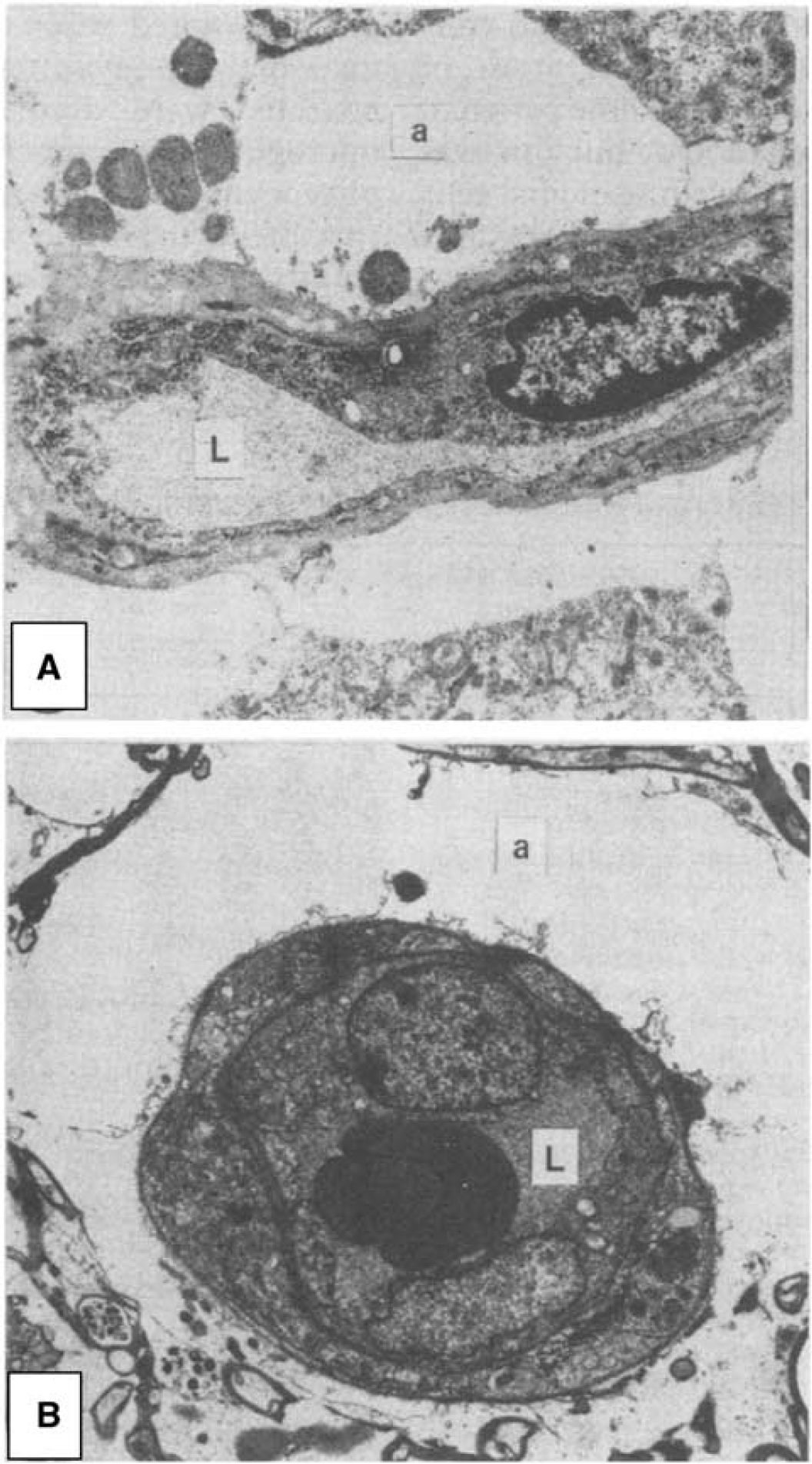
Capillary compression by astrocytic endfeet swelling after cerebral contusions. Astrocytic endfeet swelling and membrane disruption is a typical finding in humans 3 hours-3 days after cerebral contusions. (
Pericyte Dysfunction
The capillary endothelium is surrounded by a basement membrane in which contractile capillary pericytes are embedded. 29 Pericytes are involved in the regulation of capillary diameter during functional activation, 30 in blood-brain barrier (BBB) function,31,32 angiogenesis, 33 and in aspects of the brain's immune response. 34 After experimental head trauma, as many as 40% of capillary pericytes leave their pericapillary location within the first hour of the insult. 35 The extent to which the loss of pericyte coverage impairs the normal control of capillary flow patterns, facilitates the formation of brain edema, or initiates inflammatory processes in the surrounding parenchyma, remains poorly understood.
Dore-Duffy et al 36 recently reported widespread reductions in arteriolar and capillary diameter 4 to 8 hours after experimental TBI, coinciding with increased expression of contractile smooth muscle actin, of endothelin-1, and of its receptors (ET-A and ET-B) in pericytes. Both smooth muscle actin expression and the arteriolar and capillary constrictions were partly reversed by an ET-A antagonist. 36 Importantly, the upregulation of ET-A and ET-B receptors coincided with hippocampal and sensory cortex hypoperfusion until 48 hours after TBI. 37 Blockade of ET-A (but not ET-B) receptors reduced the degree of hypoperfusion in these regions at 24hours, inducing mild hyperperfusion at 48 hours, and reducing the overall degree of neuronal damage. 37
The origins of these pericyte changes remain poorly understood. Experimental cerebral ischemia has been shown to result in the constriction of cerebral pericytes, seemingly owing to the increased levels of oxidative and nitrosative stress.29,38 Tissue that experience periods of hypoxia, critically reduced CPP, or oxidative stress at some point after TBI are therefore also likely to develop pericyte constrictions.
Intracranial Hypertension, Brain Edema, and Cerebral Hemodynamics
Increased ICP is a frequent cause of death after TBI, and the degree of brain swelling on the initial computerized tomography scan is a powerful predictor of patient outcome. 39 Cerebral edema is associated with cerebral hypoperfusion, and with reductions of both cerebral blood volume (CBV) and CBF in tissue with radiologic evidence of edema.40–42 Diffusion-weighted magnetic resonance imaging has been employed in animal models of TBI to address whether the edema early after injury represents cell swelling due to acute ischemic injury (cytotoxic edema) or extravasation of fluid due to BBB leakage (vasogenic edema). Although both types of edema may cause local microvascular compression, the presence of cytotoxic edema would suggest preexisting, possibly irreversible cell injury. At 2 hours after injury (the first common time point for these studies) some studies report reduced ipsilateral apparent diffusion coefficient, which is interpreted as the presence of cytotoxic edema and a reduced extracellular space.43–47 Indices of fractional diffusion anisotropy and mean kurtosis (an index of non-Gaussian diffusion) agree with this interpretation. 47 Other studies, however, report elevated apparent diffusion coefficient in the hours after injury.48,49 This is believed to reflect vasogenic edema47,50 and an enlarged extracellular space. This is supported by histology studies that show increased BBB permeability in the same period.49,51 The BBB leakage is transient 49 and these studies show apparent diffusion coefficient reductions later within the first 24 hours after TBI, indicating that cytotoxic edema is also present in these models. The etiology of edema after TBI in humans, its temporal course, and its management, however, is still not fully understood. 52
In brain tissue, increased ICP and local pressure from edema or hematoma might be expected to compress capillaries and thereby disturb capillary flows: in peripheral tissue, half of the tissue capillaries are thus closed as the interstitial pressure reaches half the critical closing pressure of arterioles. 53 Bragin et al 54 studied the heterogeneity of capillary flow velocities during reductions in CPP by either increased ICP or reduced mean arterial pressure (MAP). For similar reductions in CPP, increased ICP caused redistribution of capillary flows to an extremely heterogeneous pattern, whereas reductions in MAP seemingly caused a reduction in capillary flow heterogeneity—see Figure 2.
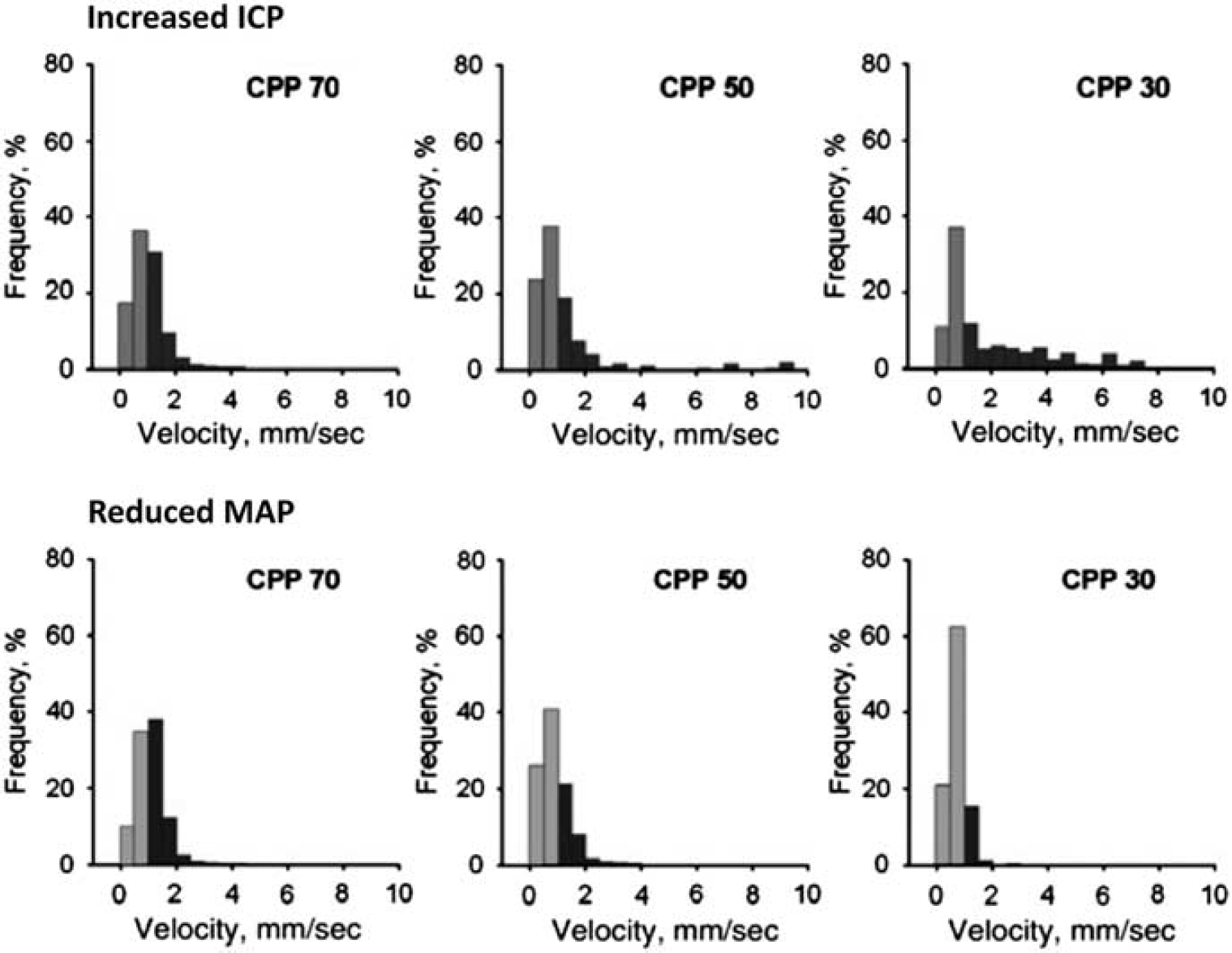
Capillary flow heterogeneity during reduced cerebral perfusion pressure (CPP): Increased intracranial pressure (ICP) versus reduced mean arterial pressure (MAP). Bragin et al, 54 recorded cortical capillary erythrocyte velocities in rats at three CPP levels. In the top row, CPP reductions were the result of increased ICP, in the bottom row of reduced MAP. Note that intracranial hypertension lead to increased flow heterogeneity, with some capillaries displaying extremely high-flow velocities. During systemic hypotension, similar CPP values lead to homogenization and reductions of capillary flows, which is predicted to provide efficient oxygen extraction. Reproduced from ref. 54 with permission from the publisher.
Vasoactive Effects of Blood Breakdown Products
Hematoma and bleedings are frequent after TBI, and erythrocytes are found outside microvessels in biopsies taken around contusions throughout the first 2 weeks after injury. 26 Evidence from subarachnoid hemorrhage suggest that hemolysis of subarachnoid blood peaks after ~1 week, 55 and that the period of the most intense angiographic vasospasms thereby coincides with peaking levels of oxyhemoglobin (HgbO). 55 The vasoactive properties of hemoglobin breakdown products are described in detail in refs. 55, 56 and only briefly summarized here: first, the spontaneous autooxidation of HgbO to methemoglobin, and the iron released from hemoglobin, cause the release of highly reactive superoxide radicals.55,56 Superoxides are thought to cause vasoconstriction by depleting vascular nitric oxide (NO) levels57,58 and to cause lipid peroxidation, which in turn causes vasoconstriction and structural damage to cerebral microvessels, including the endothelial cell layer. 59 Second, the breakdown of heme into bilirubin under such oxidative conditions results in the formation of bilirubin oxidation products that change the contractility, signaling, and metabolism in large vessels—see ref. 60 for a review. Third, HgbO has very high affinity for the NO and acts as a sink for this important vasodilator. 55
The release of vasoactive substances into cerebrospinal fluid (CSF) or the interstitial space is likely to have widespread effects on both arteriolar and capillary tone. Interstitial fluid and solutes such as hemoglobin from brain parenchyma are removed along the basement membranes of arteries and capillaries to the cervical lymph nodes. 61 Recent studies show that after intracisternal injection, molecules the size of hemoglobin distribute rapidly along penetrating arteries into brain tissue, along the basement membranes of arterioles and capillaries after which they drain into the cervical lymph nodes. 62 It is therefore likely that the exposure of smooth muscle cells and pericytes to hemoglobin and other vasoactive substances is widespread during the period of vasospasm after TBI. 62 In vitro experiments suggest that NO acts as a pericyte dilator63,64 whereas the oxidative and nitrosative stress that result from the release of superoxide radicals have been shown to cause pericyte constrictions in vitro 38 and in vivo. 38
Blood Viscosity
Increased count and endothelial adhesion of leukocytes are known to disturb capillary flow patterns and lead to functional ‘shunting’ of erythrocytes through the capillary bed. 65 In biopsies acquired from TBI patients, adhesion of both erythrocytes and leukocytes to the vascular endothelium is observed throughout the first weeks after injury, suggesting that abnormal endothelial adhesion of blood cells may indeed disturb capillary flow patterns. 26 The notion that elevated blood viscosity and impaired capillary blood passage impair brain oxygenation is consistent with findings that leukocytosis is associated with poor outcome and secondary increases in ICP after TBI.66–68 Also, administration of corticosteroids, a frequent cause of leukocytosis, in the early phases after TBI is associated with poor outcome 69
Increased blood lipid levels increase blood viscosity and would be expected to increase the heterogeneity of capillary perfusion patterns. Statin use at the time of injury has been shown to correlate with better survival and functional outcome in older head-injured individuals. 70 The lipid-lowering effect of statins becomes significant 2 to 4 days after initiation of treatment in normocholesterolemic subjects, 71 and any benefits of early, postinjury administration of statins are therefore likely to depend on mechanisms other than their viscosity-lowering effects.
NEUROVASCULAR COUPLING IN THE PRESENCE OF CAPILLARY TRANSIT TIME HETEROGENEITY
Oxygen transport in tissue is traditionally described by combining the classic flow-diffusion or Crone–Renkin equation72,73 with accepted properties of oxygen binding to hemoglobin, 74 under the assumption that OEF is identical for all tissue capillaries. In the extension of this formalism to take capillary transit time heterogeneity (CTH) into account, which is derived in ref. 22 and briefly summarized below, the Crone–Renkin formalism corresponds to the special case of negligible CTH. The maximum OEF (OEFmax) and metabolic rate of oxygen (CMRO2max) that can be supported for a given CBF in this case are shown as a green curves in Figure 3A. Note that CMRO2max measures tissue oxygenation (oxygen availability) and equals tissue CMRO2 if CBF remains perfectly coupled to the metabolic needs of the tissue. The plot was calculated for a PtO2 of 26mm Hg 75 where hemoglobin is half saturated with oxygen. The highest OEFmax value is therefore 0.5 in the plot. Note that OEFmax is high at low CBF values, whereas oxygen extraction becomes increasingly inefficient toward high CBF values.
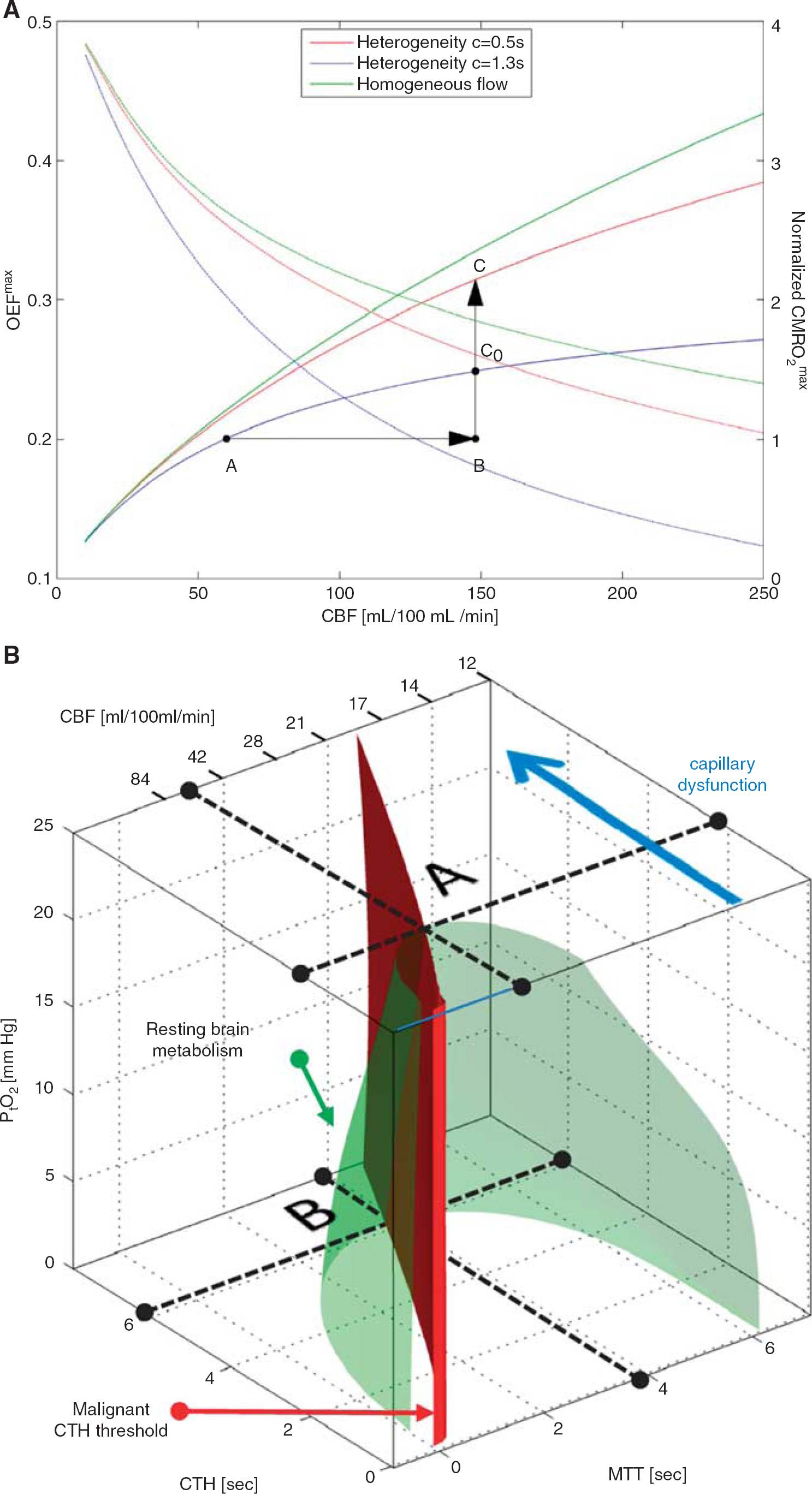
The effects of capillary transit time heterogeneity (CTH) of tissue oxygenation. In
The low OEFmax toward high flow values is the very reason why capillary flow distributions matter: if capillary flow patterns are distributed according to a certain distribution around the mean flow (CBF), then fast-flowing blood introduces a net reduction in OEFmax, and thereby CMRO2max, relative to the homogenous case assumed by the traditional Crone–Renkin equation.22,23 The red and blue curves in Figure 3A show the relations between CBF, OEFmax, and CMRO2max, respectively, for CTH values estimated during rest (CTH = 1.3) and during functional activation (CTH = 0.5) in rat brain.22,76 Note how the combination of increased CBF and reduced CTH (A → C) efficiently increases oxygen availability during functional activation, unlike an increase in CBF without concomitant reductions in CTH (A → C0).
Perhaps the most important property of the curves in 3A is that if CTH is high, and fails to decrease during increases in CBF (so-called capillary dysfunction) then vasodilation gradually fails as a means of increasing oxygen availability in tissue. Note that if CTH is maintained at the level of resting tissue, then a 250% increase in CBF (A → B) is needed to increase brain oxygenation by 50% (B → C0). If CTH increases further, our analysis shows that for a fixed PtO2, even this degree of hyperemia cannot improve tissue oxygenation sufficiently to support the metabolic needs of brain tissue. 22 Instead, the extraction of sufficient oxygen to meet metabolic needs become contingent on the suppression of flow responses to (i) limit the number of erythrocytes that pass through the microvasculature too fast to exchange oxygen with the tissue, and (ii) permit oxidative metabolism to reduce PtO2 such that blood–tissue oxygen concentration gradients, and thereby OEF, increase.
If the suppression of flow responses fails, a critical condition dubbed malignant CTH develops: 22 Here, high CTH and CBF combine to cause a reduction in OEFmax that outweighs the normal benefits of increased CBF. As a result, CBF increases will, paradoxically, either fail to improve tissue oxygenation, or cause it to deteriorate. 22 In the brain, this would manifest itself as a paradox reduction in PtO2 during episodes of increased blood flow, and be associated with extreme risk of tissue injury. We note that such changes are characteristic of the so-called luxury perfusion syndrome, which is associated with extreme risk of infarction after cerebral ischemia. 77
The Combined Effect of Cerebral Blood Flow, Capillary Transit Time Heterogeneity, and Tissue Oxygen Tension on Brain Oxygenation
Figure 3B shows how CBF, CTH, and PtO2 (along the three axes) in combination can secure sufficient oxygen to support normal brain function—see also ref. 78. In addition to CBF, capillary blood mean transit time is provided as x-axis, assuming fixed capillary blood volume. Mean transit time is given by the central volume theorem 79 as the ratio between capillary blood volume and CBF. The green surface shows all combinations of mean transit time (or CBF), CTH, and PtO2 that make 2.5 mL oxygen available to each 100mL of tissue per minute. This rate of oxygen delivery is a conservative estimate of the metabolic needs of cortical gray matter, and the interior of the green half-cone therefore represents hemodynamic conditions that can support normal brain function provided that arterial hemoglobin concentration and oxygen saturation are maintained at normal values.
The figure illustrates why, theoretically, reductions in PtO2 and in resting CBF are the only alternatives to immediate neurologic symptoms and tissue damage if CTH increases and fails to homogenize during hyperemia. Label A in Figure 3B shows the theoretical maximum for the increase in CTH (indicated by a broken line parallel to the mean transit time and CBF axes) that brain tissue can sustain at the PtO2 of normal brain tissue (25mmHg) 75 before neuronal metabolism is predicted to become impaired. As CTH approaches this limit, oxygen availability gradually approaches the consumption by tissue, causing PtO2 to decrease. At lower PtO2, oxygen extraction is more efficient, and the green cone therefore wider. Therefore, tissue metabolism can be maintained despite increasing CTH, until oxygen tension cannot be reduced further (label B). Note that as CTH increases, CBF must be attenuated (mean transit time prolonged) in parallel to reduce the loss of oxygenated blood through ‘functional shunts’ (as opposed to ‘true’ anatomic shunts) through the capillary bed. When CTH approaches a critical threshold (6 seconds for the transit time distribution adapted in ref. 80), and PtO2 zero, CBF is predicted to approach 21 mL/100 mL/minute. 80 If CBF is indeed attenuated to optimize brain oxygenation as capillary flow patterns become increasingly disturbed, CBF values would therefore be predicted to be close to the classic ischemic threshold of 20 to 21 mL/100 mL/minute when neurologic symptoms ensue. 81 Accordingly, the classic ischemic threshold may not only be characteristic of thromboembolic events, but also of CBF adaptations to secure sufficient oxygenation in the face of critical disturbances of capillary flow patterns. 78
Cerebral Blood Flow, Cerebral Blood Flow Responses, and Oxygen Extraction Fraction for Three Levels of Capillary Transit Time Heterogeneity Increase
Figure 4A illustrates the dynamics of CBF and capillary flow patterns during rest and during increased metabolic needs in normal brain. The flux of erythrocytes through cortical brain capillaries is highly heterogeneous during rest, but become more homogenous during functional activation. This phenomenon maintains relatively high OEFmax during functional activation, even without changes in PtO2.
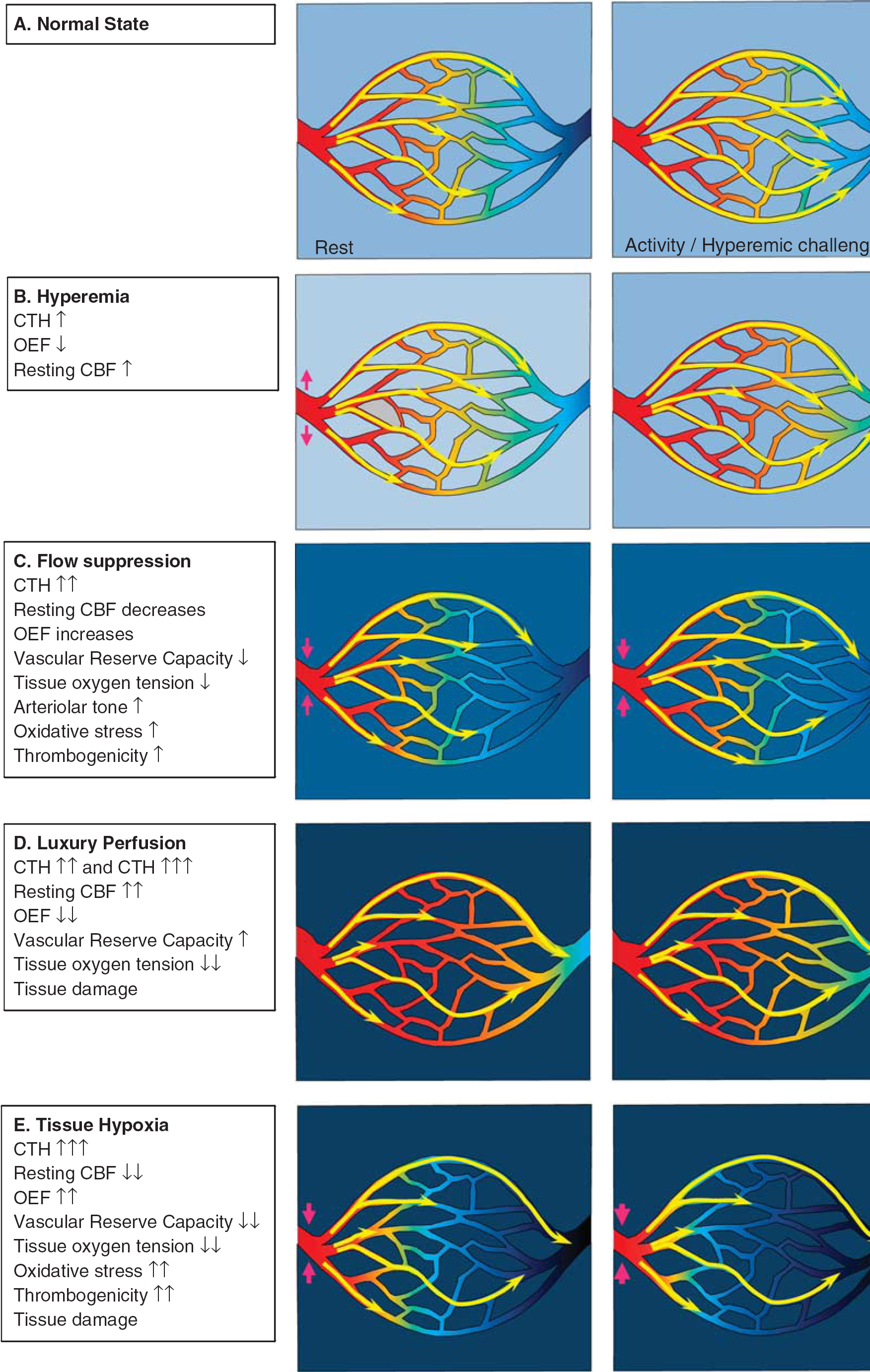
Levels of capillary dysfunction (capillary transit time heterogeneity (CTH) increase). The yellow arrows indicate erythrocyte flows with different velocities through the capillary bed. The color within the vessels indicates oxygen saturation, and the background color outside indicates tissue oxygen tension. (
Mild Capillary Dysfunction: Hyperemia
Figure 4B illustrates the metabolic consequence of mild capillary dysfunction: capillary flows are more heterogeneous than in normal tissue during rest, and cannot homogenize during hyperemia. The elevated CTH reduces the OEF that can be attained for a given PtO2, but the condition is defined by the fact that the metabolic needs of tissue can be met by slight increases in CBF, both during rest and during hyperemia (e.g., functional activation or hypercapnia). We therefore refer to states of mild CTH increases as hyperemic. The various capillary dysfunction stages, and their predicted degree of metabolic impairment, are summarized in Table 1.
Relation between dynamic changes in CBF, PtO2, and blood oxygenation under various degrees of capillary dysfunction and neurovascular coupling
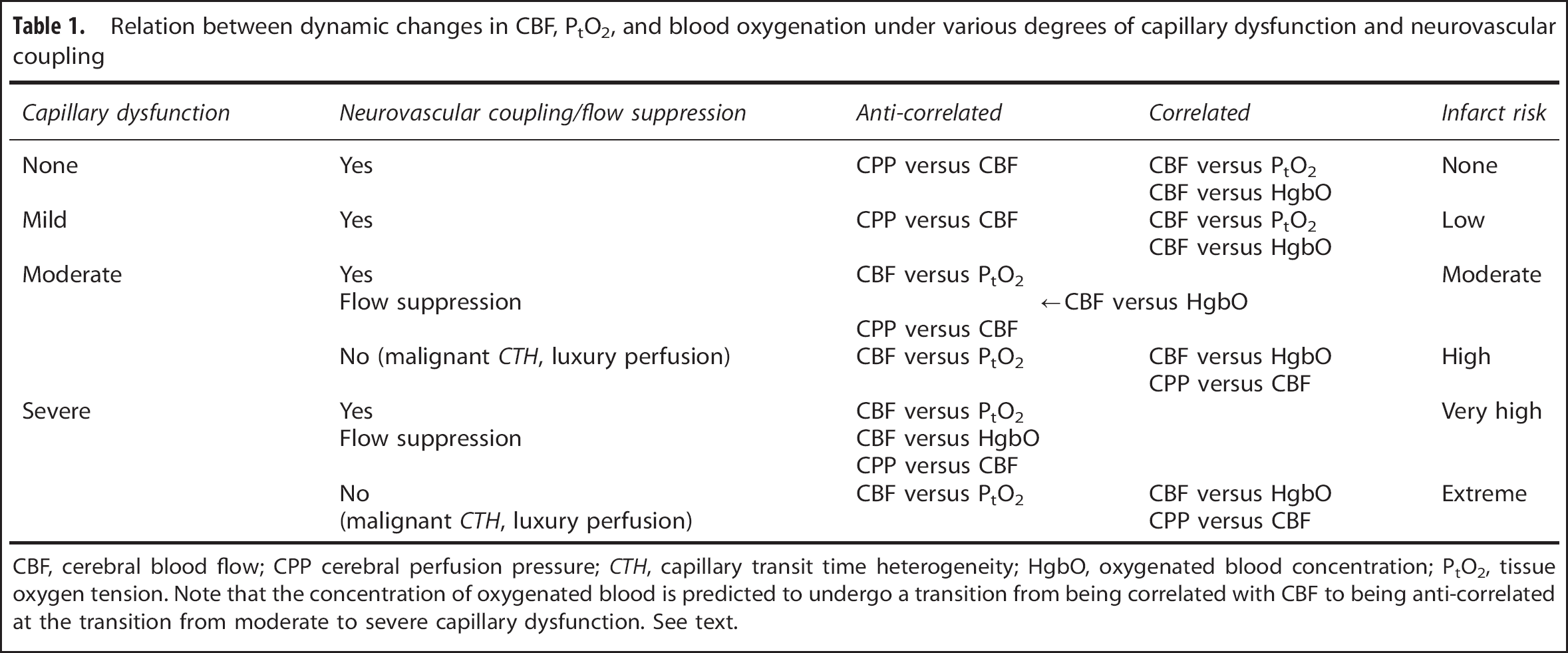
CBF, cerebral blood flow; CPP cerebral perfusion pressure; CTH, capillary transit time heterogeneity; HgbO, oxygenated blood concentration; PtO2, tissue oxygen tension. Note that the concentration of oxygenated blood is predicted to undergo a transition from being correlated with CBF to being anti-correlated at the transition from moderate to severe capillary dysfunction. See text.
Moderate Capillary Dysfunction: Flow Suppression or Luxury Perfusion and Tissue Damage
At higher CTH levels, hyperemia can no longer compensate for the parallel reduction in OEFmax during episodes of increased metabolic needs—or even during rest. Tissue function and survival in this stage depend on mechanisms that suppress normal, vasodilatory responses—see Figure 4C. As we argued above, failure to inhibit CBF or CBF responses may lead to uncontrolled hyperperfusion, tissue hypoxia, and tissue injury similar to that observed in luxury perfusion syndrome 77 —see Figure 4D. Note that an improvement in CTH from moderate to being only mild is expected to result in a transition from hypoperfusion to hyperperfusion and improved tissue oxygenation if CBF remains coupled to the metabolic demands of the tissue. The hyperperfusion and attenuated neuronal damage observed by Dore-Duffy et al. after ET-A blockade in their TBI model are therefore consistent with improved capillary flows and a therapeutic reduction of CTH as capillary constrictions resolve. 37
Microvascular responses to a range of vasodilators are impaired after TBI in patients 82 and in experimental models of TBI.83–86 According to the analysis above, this phenomenon is in fact predicted to improve tissue oxygenation under conditions of elevated CTH, by suppressing flow increases that would otherwise fail to improve, or even reduce, tissue oxygenation. We have speculated that if ‘normal’ vasodilation results in tissue hypoxia because of elevated CTH, this would increase hypoxia-inducible factor 1 (HIF-1) levels (see discussion of hypoxia-inducible transcription factor 1 alpha - HIF-1α - in TBI below), 87 thereby upregulating nicotinamide adenine dinucleotide phosphate oxidase 2, NOX-2, 88 a major source of reactive oxygen species (ROS) in the brain vasculature. 89 Reactive oxygen species, in turn, react vividly with NO to attenuate vasodilation, and in this way, capillary dysfunction (elevated CTH) could attenuate microvascular flow responses via a hypoxia sensitive mechanism. Indeed, HIF-1 has been reported to upregulate NOX-2 levels as early as 1 hour after experimental TBI, and again 2 to 4 days after injury. 90
If the suppression of CBF responses is coupled to the metabolic needs of the tissue, then hypoxia would be predicted to cause further attenuation of vasodilator responses to improve oxygen extraction efficacy, while reductions in the brain's oxygen metabolism would be expected to relax the need for flow suppression. Therapeutic, moderate hypothermia is reported to reduce CMRO2 by 5% to 7% per degree Celsius reduction in core temperature. 91 The prediction that impaired microvascular flow responses after TBI represent adaptations to secure tissue oxygenation when CTH is high, is consistent with the finding that responses to standardized vasodilatory stimuli are reduced during hypoxia, but increased during hypothermia. 92 We discuss the therapeutic effects of hypothermia further below.
Severe Capillary Transit Time Heterogeneity Increase: Hypoxia and Tissue Damage
If CTH values become very high, then uncontrolled hyperemia (luxury perfusion) leads to even more severe tissue hypoxia than outlined above, and is highly likely to cause tissue damage (see Table 1). If vasodilation is suppressed, low CBF and low PtO2 may contribute to tissue damage in several ways—see Figure 4E. First, the reduction of PtO2 is likely to cause spreading depolarizations and neuronal injury. 93 Second, low PtO2 activates HIF-1α in TBI models. 87 Increased HIF-1α, in turn, is a powerful stimulus for BBB opening and edema formation, possibly via the upregulation of aquaporin expression.87,94 In addition to the detrimental effects of brain edema, pericapillary fluid is likely to further compress capillaries and thereby add to a vicious cycle of further CTH increase and hypoxia. Third, HIF-1 also upregulates nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX-2) levels, a prominent source of superoxide—a ROS—as early as 1hour after experimental TBI, and again 2 to 4 days after injury. 90 Reactive oxygen species reacts with NO to form the much more toxic peroxynitrite. 95 Although NO depletion and peroxynitrite both impair smooth muscle cell relaxation and thereby offer a putative mechanism for the crucial flow suppression in this stage, 96 peroxynitrite also inactivates tissue plasminogen activator, increasing thrombogenicity and thereby the risk of further tissue damage. Both platelet accumulation and widespread hypoperfusion have been observed as early as 30minutes after TBI in rats, but their spatial distributions did not suggest that hypoperfusion is secondary to thrombosis. 97 In biopsies from TBI patients, microvascular constrictions and microthrombosis have been demonstrated within hours of injury, 28 consistent with the notion that flow suppression, and possibly relative tissue hypoxia, may be present at the time of astrocytic endfeet swelling early after TBI. 28 The extent to which microthrombosis causes significant hypoperfusion after TBI, however, remains unclear. 21 Finally, hypoxia and peroxynitrite are also known to damage mitochondria. 96 Mitochondrial dysfunction, in turn, exacerbates the energy crisis by reducing the amount of ATP that can be obtained from the available oxygen, and amplifies the production of ROS. 96 The generation of ROS has also been shown to alter mitochondrial dynamics and functions. Accordingly, ROS induces accelerated mitochondrial fission, increased permeability of the mitochondrial membrane, accumulation of Ca2+, and the production of even more ROS. 98 Mitochondrial dysfunction is therefore likely to increase the risk of cellular damage and apoptosis in the already compromised neuronal tissue.
The hallmark of hypoxic tissue injury caused by severely disturbed capillary flow patterns is that CBF remains above the ‘classic’ ischemic threshold—and even above normal CBF in cases of luxury perfusion. Another key feature of this condition is that a large proportion of blood passes through the capillary bed too fast to permit proper extraction of oxygen by the tissue. As a result, the oxygen tension of mixed venular blood is predicted to be significantly higher than that of tissue. Therefore, conditions of high CTH mimic both a diffusion hindrance of oxygen, 20 and partial shunting of blood through anatomic shunts. 54
IS THE EVOLUTION OF CEREBRAL BLOOD FLOW AND OXYGEN EXTRACTION FRACTION AFTER TRAUMATIC BRAIN INJURY CONSISTENT WITH FLOW-METABOLISM COUPLING DURING VARIOUS DEGREES OF CAPILLARY DYSFUNCTION?
Regional brain metabolism after TBI is affected by the patient's medication and level of consciousness, and by the extent of neuronal death in relation to both the initial injury and subsequent hypoxic episodes. With this caveat, we will briefly discuss whether the evolution of CBF, OEF, and their relation to outcome, can be understood in terms of changes in capillary perfusion patterns, accompanied by either successful (although perhaps futile) coupling of CBF to the metabolic needs of the tissue, or by insufficient flow suppression (luxury perfusion).
Based on the time course of the capillary changes reviewed above, we hypothesize that CTH is high during the initial hours after injury owing to increased ICP and glial swelling—see Figure 5. We hypothesize that regional CBF is suppressed to near-ischemic levels in the affected tissue, securing optimal tissue oxygenation under these conditions. In this state, the metabolic reserve is predicted to be either exhausted or low, and accompanied by low PtO2. This is consistent with findings that low tissue oxygen is a strong, independent predictor of poor outcome. 99 The prediction that the early stage of hypoperfusion represents a stage of severe metabolic impairment is supported by the finding that during this stage, CBF is correlated with motor score and outcome, whereas no such correlation can be found beyond 12h. 6 Overgaard et al 10 confirmed that outcome in patients who proceed to develop hyperemia (consistent with a milder CTH increase and either compensatory hyperemia, or luxury perfusion) is better than in those who display continued hypoperfusion (consistent with moderate or severe CTH increase and tissue hypoxia). Also, in a fluid percussion injury model of TBI in rats, contusive injury 3 occurs in regions that show severe reductions in CBF as early as 30 minutes after the injury. 100
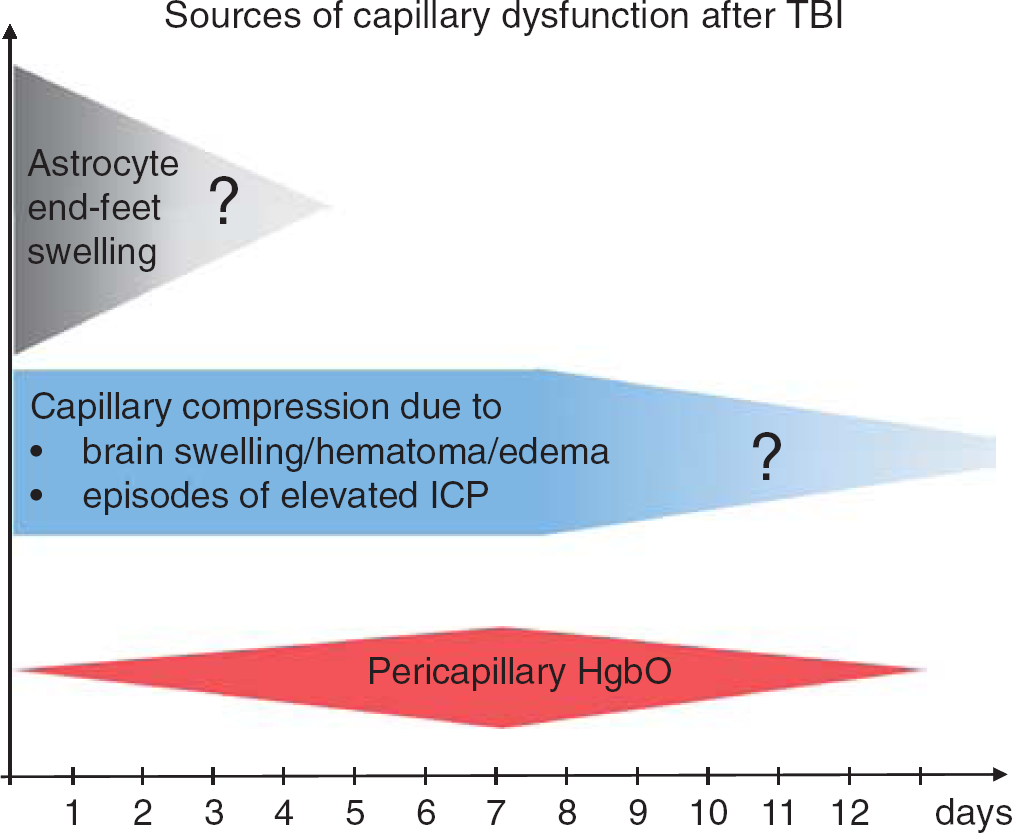
Sources of capillary dysfunction after traumatic brain injury (TBI). The colored panels summarize the sources of capillary flow disturbances as identified in this review, and their estimated duration (x-axis). They include astrocyte endfeet swelling observed 3 hours to 3 days after contusions in humans (Figure 1), capillary compressions due to local swelling and increased intracranial pressure (ICP) throughout the critical period after the injury, and oxidative stress and nitric oxide depletion caused by pericapillary oxyhemoglobin. The extent to which structural damage to the capillary wall is permanent is crucial because permanent changes in capillary transit time heterogeneity (CTH) are potential sources of long-term, neurodegenerative changes. 150 Reproduced and modified from ref. 116.
The prediction that the ‘optimal CBF’ for brain oxygenation in the injured brain may be lower than that of normal brain is supported by studies that have suggested unwanted effects of augmenting CPP and thereby CBF: Coles et al 101 demonstrated that an increase in CPP from 70 to 90mm Hg increased both CBF and cerebral blood volume, while reducing OEF, CMRO2, and neuronal electrical function. The prediction that CBF-increases may result in paradox reductions in brain oxygenation due to malignant CTH is consistent with findings by Budohoski et al 102 , who recorded spontaneous changes in MAP, arterial flow velocities, PtO2, and blood oxygenation in TBI patients. In 46% of their recorded cases, PtO2 changed in proportion to blood flow, as one would expect if oxygen delivery were limited by CBF alone. In the remaining 54 %, however, PtO2 was anti-correlated with MAP and flow, i.e., increases in MAP and blood flow reduced PtO2, or vice versa, 102 consistent with a state of elevated CTH in which increases in CBF no longer improves tissue oxygenation as it would be predicted by the classic Crone–Renkin equation. In these cases, some degree of flow suppression may be present, and the observed reductions in PtO2 may increase OEF sufficiently to meet the metabolic needs of the tissue, in which case, these events would be accompanied by reductions in blood oxygenation at the site of PtO2 measurement. This correlation was observed in only 60% of cases, suggesting that malignant CTH and insufficient suppression of flow may be a frequent finding after TBI. 102 The proposed, conceptual algorithm for interpreting dynamic 1 autoregulation data in terms of degree of capillary dysfunction, luxury perfusion, and metabolic impairment, is summarized in Table 1.
GLUCOSE EXTRACTION AND METABOLISM AFTER TRAUMATIC BRAIN INJURY
Under aerobic conditions, oxidative phosphorylation generates 29 to 30 ATP molecules per glucose molecule, whereas lactate production under anaerobic conditions generates only two. Knudsen et al 103 used indicator dilution data and mathematical analysis to show that the extraction of glucose and glucose analogs in the brain is limited by CTH, and that efficient glucose extraction during hyperemia depends on homogenization of capillary transit times, similar to the properties of oxygen extraction. 22 The extended flow-diffusion equation predicts how CTH affects the extraction efficacy of any freely diffusible solute. 22 Although we have yet to refine the model to take into account the kinetics of glucose transporters and glucose metabolism, our preliminary analysis suggests that elevated CTH tends to favor the extraction of glucose over oxygen, and hence aerobic glycolysis. 104 Because of this differential extraction, increases in CTH would be expected to cause the conversion of some glucose into lactate, even though CBF remains at or above standard ischemic threshold. This is consistent with animal studies by Ginsberg et al, 105 , who showed unusually high levels of glucose analog extraction in areas of only moderately altered CBF, and with reports of elevated lactate levels in the CSF of patients who show no signs of elevated OEF. 106 The prediction that glucose extraction is better preserved than oxygen extraction at high CTH—where OEF is expected to be high and the energy crisis most severe—is consistent with findings by Abate et al, 107 who found the net clearance of glucose analogs, but not CMRO2, to be preserved at high OEF in TBI patients, by Robertson et al 108 , who found that the combination of elevated lactate production and reduced CMRO2 predicted subsequent infarction in TBI patients with no signs of ischemia, and by Ginsberg et al 105 , who showed that neuronal necrosis in a rat TBI model occur in areas that show the aforementioned acute CBF–glucose metabolism uncoupling. A thorough analysis of the differential effects of CTH and tissue metabolism on the net extraction of glucose and glucose analogs must be undertaken to understand how capillary flow disturbances affect the so-called Lumped Constant, which relates glucose analog uptake to actual glucose metabolism,109,110 and thereby the conclusions of tracer-based glucose metabolism studies after TBI.
DISCUSSION
This review identifies four major sources of capillary flow disturbances early after TBI. First, animal studies and biopsies from TBI victims show that astrocytic endfeet swelling is a source of capillary compression immediately after injury, lasting for up to 2 to 3 days in humans. Second, animal studies suggest that elevated ICP is the source of capillary compression and considerable, functional shunting of oxygenated blood through the capillary bed, although CPP and CBF is maintained. Third, widespread pericyte constriction and dislocation are likely to disturb capillary flow patterns early (hours) after brain injury. Finally, blood from hematoma, contusions, and early exudation of erythrocytes is likely to act as a sink for NO, counteracting local vasodilation in a pattern that resembles the course of subarachnoid hemorrhage over the first 2 weeks after injury—see Figure 5. The long-term effects of permanent capillary damage and flow disturbances are discussed in ref. 111.
The most important effect of elevated CTH is that it causes some blood to pass through the capillary bed at transit times too short to permit efficient extraction of its oxygen. Therefore, tissue survival becomes contingent on the attenuation of CBF and CBF responses to limit functional ‘shunting’ under conditions of high CTH. Studies of dynamic autoregulation and pressure reactivity in TBI patients show that ‘passive’ changes in ICP and cerebral flow velocities in response to changes in MAP are associated with high mortality and poor functional outcome.112–115 We speculate that such vasoparalysis reflect the failure of metabolic vasomotor control mechanisms—including those who block ‘futile’ vasodilation and luxury perfusion in states of high CTH. Note that hyperemia that is coupled to brain metabolism (Figure 4B) carries a much better prognosis than luxury perfusion (Figure 4D), which represent an important exception from the flow-metabolism coupling otherwise assumed in Figure 5. See Table 1.
The temporal course of capillary changes observed after TBI suggests that successive stages of capillary flow disturbances may exist—see Figure 5. First, an initial, dramatic increase in regional CTH, during which CBF is suppressed, PtO2 is low, and OEF is high. Second, when CTH improves as a result of reduced capillary compression, the less severe reduction of OEFmax can ultimately be compensated by elevated CBF alone, giving rise to a hyperemic state. Meanwhile, areas in which CTH remains elevated are predicted to remain hypoperfused, and show little benefits from augmented CBF. Finally, the clearance of blood breakdown products through the perivascular space is predicted to cause widespread capillary and arteriolar vasoconstrictions for a period of time, coinciding with the observation of radiographic vasospasms. 116
Our review suggests that invasive monitoring of PtO2 and blood oxygenation responses to spontaneous fluctuations in MAP and flow velocities may provide means of identifying malignant CTH and the presence neurovascular coupling, albeit locally. We speculate that dynamic autoregulation data and patient outcome data can provide means of testing our interpretation of such physiologic data and possibly inform their use in individualized TBI therapy. For global measurements of brain oxygenation by the formalism presented here, we recently demonstrated that CTH can be estimated as part of routine computerized tomography perfusion imaging, 117 providing a means of noninvasively assessing the extent of capillary dysfunction across the whole brain as part of the initial patient assessment.
Which Cerebral Perfusion Pressure, Cerebral Blood Flow, and Intracranial Pressure Provide Optimal Tissue Oxygenation after Traumatic Brain Injury?
When portions of the closed cranial cavity become occupied by brain edema and hematoma, even small changes in the volume of intracranial blood vessels, brain parenchyma, or of CSF, can lead to dramatic changes in ICP that may interfere not only with CTH, but with the brain's blood supply.118,119 The CPP is the difference between the mean arterial blood pressure and ICP, and therefore the pressure difference that drives CBF. Under the assumption that CBF determines cerebral oxygenation, it is therefore rational to maintain CPP within the normal range in patients with elevated ICP to maintain brain oxygenation. Accordingly, current guidelines recommend a CPP between 50 and 70mm Hg and further suggest that patients with intact autoregulation may tolerate higher CPP. 120 In clinical practice, CPP is managed with infusion of fluids and vasopressors.
Although elevated ICP certainly interferes with cerebral oxygenation if CBF becomes hindered, this review posits that oxygen extraction may be impaired by distortions of capillary flow patterns at CPP and ICP thresholds lower than those at which CBF becomes impaired—to an extent that exceeds the benefits of maintaining normal CBF in TBI patients. Below, we first discuss whether the maintenance of CPP within the range of normal autoregulation is a necessary and sufficient condition for avoiding brain damage, and then whether therapies that target both a lower ICP and a slightly lower CBF in fact optimize cerebral oxygenation.
In normal brain, cerebral autoregulation adjusts arterial and arteriolar tone to maintain relatively constant CBF across a wide range of MAP values.121–123 It is well known, however, that brain function is preserved at CPP values far below the autoregulatory range (MAP as low as 30 mm Hg) in resting, normal subjects 124 and in animal models of hypotension. 125 Homogenization of capillary flow patterns may indeed facilitate oxygen extraction (normal brain OEF is a modest 30%) under these conditions: in animal models of experimental hypotension, it has been reported that capillary flow heterogeneity is reduced as CPP falls.25,126 We have previously reanalyzed flow heterogeneity data from a study in which CPP was reduced below the normal autoregulatory range of 70 to 115 mmHg in rats:22,126 Using estimates of CBF, CTH, and PtO2, we found that tissue oxygenation was in fact similar to that of normotensive animals at a CPP of 50 mm Hg, and reduced by a modest 13% at a CPP as low as 30 mm Hg. 22
Similarly, the maintenance of CPP within the normal autoregulatory range does not appear to be a sufficient condition for the maintenance of cerebral oxygenation after TBI if ICP is elevated. A classic study of experimental reductions in CPP by either hypotension or elevated ICP showed that, whereas autoregulation is lost at a CPP of 80 mmHg in systemic hypotension, autoregulation and cerebral oxygen metabolism was maintained for CPP as low as 30mmHg in conditions of elevated ICP. 125 Bragin et al 25 studied the microvascular circulation under similar conditions and found that elevated ICP is associated with a high degree of microvascular shunting, and that the ‘preservation’ of CBF at low CPP is in fact associated with severe tissue hypoxia, tissue damage, and blood-brain barrier breakdown., 54
Managing Capillary Dysfunction
Taken together, reduction in CPP caused by reduced outward pressure (reduced MAP) and increased inward pressure (elevated ICP) on the microvasculature may have very different effects on CTH, and thus on brain oxygenation. Indeed, the use of ICP targeted 127 or (mainly) CPP targeted approaches 120 to manage patients with elevated ICP remain controversial, 128 particularly in view of the reductions in CBF that accompany therapeutic vasoconstriction to reduce ICP.
Below, we briefly discuss outcomes of therapeutic strategies that would be expected to reduce CTH or to ameliorate the metabolic effects of capillary dysfunction by reducing CBF or the metabolic needs of the tissue.
Hyperventilation (HV) is a powerful means of reducing intracranial vessel diameter, and thereby ICP. The accompanying reduction in CBF, we predict, provides efficient oxygen extraction for both normal and elevated CTH. This is consistent with studies by Diringer et al 7 , who examined nine patients 11.2 hours after TBI, before and after 30minutes of HV to a PaCO2 of 30mmHg. Although HV reduced global CBF and cerebral blood volume, OEF increased from 31% to 45%, and CMRO2 remained unchanged, suggesting that the metabolic needs of the tissue were met. 7 In a follow-up study, they performed regional analysis to assess the effects of 30 minutes of moderate and severe HV on regions with low CBF values: 8 Even in regions with CBF less than 10, 15, and 20mL/100mL/minute, OEF increased in proportion to the reduction in CBF, maintaining unaltered CMRO2 in what is likely to have been partly injured tissue.
Although brief periods of HV are considered safe, prolonged HV is currently discouraged on grounds that low PaCO2 levels may cause critical vasoconstriction and reduce CSF bicarbonate levels, reducing its pH buffering capacity. In the normal brain, the pH of CSF is tightly regulated via secretion of HCO3~ by the epithelium of the choroid plexus, but the capacity of this mechanism is poorly understood. 129 The lack of buffering capacity is thought to cause large changes in CBF and ICP in response to small changes in pH, for example in response to the PaCO2 changes during HV. 130 To examine these potentially harmful side effects, Muizelaar et al 130 conducted a randomized trial in which 36 patients received HV, 36 received HV and THAM (a substance that prevent the loss of pH buffer capacity in the CSF), and 41 controls. The number of patients with favorable outcome was significantly lower in the HV group compared with the HV + THAM group after 3 and 6, but not after 12 months. Notably, CBF values were significantly lower in the HV + THAM group (who showed alkalization of the CSF) than in the HV group, and their arteriovenous oxygen concentration difference values did not suggest ischemia. After this study, prolonged HV was abandoned, although several studies appear to prove its benefits. 131 One observation that has been taken to argue against the safety of HV in TBI is the accompanying reduction in PtO2. It should be kept in mind that such reductions improve oxygen extraction by creating higher blood–tissue oxygen concentrations differences. So while low PtO2 per se indicates impaired tissue oxygenation, we propose that HV-induced reductions in PtO2 within certain boundaries may not be contraindicative as suggested by some. 131 In addition to the benefits of reduced ICP, HV is known to reduce CTH in normal brain. 132
Indomethacin is a cyclooxygenase inhibitor that acts as a powerful vasoconstrictor, reducing CBF uniformly by one-third in both awake and anesthetized normal brain while maintaining CMRO2.133,134 Several studies in TBI patients have shown that an indomethacin bolus followed by continuous infusion can cause prompt and prolonged reduction of ICP and CBF,135,136 without causing reductions in CMRO2 135 or subsequent radiographic signs of ischemic damage. 137 See detailed discussions in refs. 138, 139. Subsequent studies have demonstrated beneficial effects of indomethacin in the treatment of TBI patients,140,141 and we speculate that these may be attributed in part to reductions in both ICP (and hence CTH) and CBF.
Mannitol is frequently used in the management of elevated ICP—allegedly because of its ability to reduce the degree of brain edema. Interestingly, mannitol also causes an acute reduction in blood viscosity, and thereby, one would expect, in CTH. In normal brain, the accompanying increase in OEFmax would be expected to cause rapid vasoconstriction when neurovascular coupling compensates for the more efficient oxygen extraction by reducing CBF. Indeed, mannitol (1g/kg) acutely reduces blood viscosity by 23%, vessel diameter by 12%, and ICP by 28% in the intact rat brain. 142 For comparison, HV to achieve a similar reduction in vessel diameter generates a similar reduction in ICP, 142 suggesting that the acute effects of mannitol may be explained in part to improved oxygenation and compensatory vasoconstriction in tissue with intact vascular regulation—rather than reductions in edema.
In animal models of TBI, mild to moderate hypothermia has proven neuroprotective in a large number of studies—See Dietrich et al 143 for a review of experimental considerations and putative protective mechanisms. Although the effects of mild hypothermia on clinical outcome in human TBI remain unclear,144,145 the treatment is considered superior in the management of refractory intracranial hypertension. 146 In addition to the neuroprotection conferred by reduced oxygen metabolism (above) and mechanisms described elsewhere,91,143 we speculate that reductions in CBF and thereby more efficient oxygen extraction under conditions of elevated CTH, may represent an important protective mechanism. Position emission tomography studies in normal pig brain showed reductions of CBF to 20 mL/100 mL/minute, and an increase in OEF to almost 90%, during gradual cooling to 32°C. 147 Although some might fear that such CBF reduction would cause ischemic damage, we note that biophysically, net oxygen extraction appears to be at its maximum for any CTH at this CBF value, 80 whereas the concurrent reduction in CMRO2 would be expected to protect the brain from hypoxic damage in the event of fluctuations in CBF, blood viscosity, or blood oxygenation (saturation and hemoglobin concentration).
CONCLUSION
Future studies should examine whether capillary flow disturbances are prevalent after TBI, and whether they, in combination with CBF and PtO2, predict tissue oxygenation. We propose that studies of pericyte and astrocyte endfeet function, of the effects of elevated ICP and blood viscosity on capillary flow patterns, and of edema formation, in relation to TBI may improve our understanding of hypoxic tissue injury after TBI, and lead to the identification of novel strategies to improve patient outcome. Our analysis suggest that maintenance of low CBF (just above the ischemic threshold) as well as low ICP, in combination with suppression (pharmacological and by hypothermia) of cerebral oxygen metabolism optimizes the oxygen supply–demand balance in brain tissue. Reports of acute changes in capillary morphology after TBI suggest that early intervention is critical. Dynamic autoregulation measurements may allow indirect clinical evaluation of the degree of CTH increase, and guide both the therapies above, and patient prognosis. Studies should address whether permanent capillary damage persist after both mild and severe injury, given the potential for elevated CTH to cause long-term neurodegenerative changes. 111 Neuroimaging based methods to study CTH, 148 or routine evaluation of CBF responses to standardized vasodilatory stimuli, may prove useful in this effort.
Footnotes
The authors declare no conflicts of interest.