
Abstract
In recent years, several studies have unequivocally shown the occurrence of cortical spreading depressions (CSDs) after stroke and traumatic brain injury (TBI) in humans. The fundamental question, however, is whether CSDs cause or result from secondary brain damage. The aim of the current study was, therefore, to investigate the role of CSDs for secondary brain damage in an experimental model of TBI. C57/BL6 mice were traumatized by controlled cortical impact. Immediately after trauma, each animal showed one heterogeneous direct current (DC) potential shift accompanied by a profound depression of electroencephalogram (EEG) amplitude, and a temporary decrease of ipsi- and contralateral regional cerebral blood flow (rCBF) suggesting bilateral CSDs. Within the next 3 h after TBI, CSDs occurred at a low frequency (0.38 CSD/h per animal, n = 7) and were accompanied by rCBF changes confined to the ipsilateral hemisphere. No significant relationship between the number of SDs and lesion size or intracranial pressure (ICP) could be detected. Even increasing the number of posttraumatic CSDs by application of KCl by more than six times did not increase ICP or contusion volume. We therefore conclude that CSDs may not contribute to posttraumatic secondary brain damage in the normally perfused and oxygenated brain.
Keywords
Introduction
After traumatic brain injury (TBI), the initial—or primary—mechanical impact on the brain results in almost immediate, nonreversible damage of cerebral tissue. This sets in motion a cascade of secondary events finally leading to a delayed and progressive destruction of the adjacent brain parenchyma. The pathophysiology of this process, however, remains poorly understood. Brain edema formation with subsequent increase of intracranial pressure (ICP), reduced cerebral blood flow, exitotoxicity, inflammation, and apoptosis are some of the most frequently discussed mechanisms (Nortje and Menon, 2004).
An increasing amount of data generated in the past few years suggest a key role for cortical spreading depressions (CSDs) for secondary brain damage. A CSD is a simultaneous depolarization wave traveling over the cerebral cortex with a velocity of 2 to 3 mm/mins followed by a depression of neuronal activity for a couple of minutes. A shift in the direct current (DC) potential by 5 to 30 mV, which may also be accompanied by silencing of electroencephalogram (EEG) activity, is a robust and frequently used method to detect CSD waves in experimental settings.
It has become clear from experimental and more recent clinical work that CSDs are involved in the pathophysiology of several neurological disorders such as migraine (Milner, 1958; Lauritzen, 2001), cerebral ischemia (Mies and Paschen, 1984; Nedergaard and Astrup, 1986; Back et al, 1996), trauma (Mayevsky et al, 1996; Strong et al, 2002; Rogatsky et al, 2003), and intracerebral hemorrhage (Mun-Bryce et al, 2001; Dreier et al, 2006). In the intact brain, CSDs cause hypoxia and marked neuronal swelling leading to changes in dendritic structures and loss of spines (Takano et al, 2007). These changes, however, last only for few minutes and do not cause longlasting neuronal damage (Nedergaard and Hansen, 1988). Brain tissue that is metabolically compromised, however, is obviously not able to compensate the additional metabolic and excitotoxic strain imposed by CSDs (Obeidat and Andrew, 1998), for example, after focal cerebral ischemia, there is a correlation between the number of peri-infarct DC shifts and ischemic neuronal damage (Mies et al, 1993). Definite proof for the detrimental role of CSDs for neuronal survival in compromised brain tissue came from experiments where additional CSDs were induced with KCl in the ischemic hemisphere after focal cerebral ischemia (Back et al, 1996; Busch et al, 1996). Assessment of infarct volume by magnetic resonance imaging showed a stepwise increase in the total volume of the ischemic injury after each CSD.
So far, it is unclear if spreading depression-like events may contribute to secondary lesion expansion after TBI in a similar way. There are different studies demonstrating that spreading depressions occur after experimental TBI. Cortical spreading depression-like DC shifts could be detected after penetrating ballistic brain injury (Williams et al, 2005), fluid percussion injury (Rogatsky et al, 2003), and cortical cold injury (Hermann et al, 1999; Trabold et al, 2006). Gradually, it has become clear that posttraumatic spreading depressions occur also in humans (Mayevsky et al, 1996; Strong et al, 2002; Parkin et al, 2005; Fabricius et al, 2006). However, it remains to be clarified whether spreading depressions after TBI only reflect or indeed promote the progression of secondary brain injury. Therefore, the present experimental study aims to determine whether CSDs are associated with and contribute to posttraumatic brain damage, that is, if there is a cause—effect relationship between CSDs and secondary contusion expansion after TBI.
Materials and methods
Experimental Groups
Experiments were performed on a total of 29 male C57Bl6 mice (25 to 28 g b.w.; Charles River, Kisslegg, Germany). In a first study (n = 7), EEG, DC potential, laser Doppler flow (LDF), and ICP were measured up to 180 mins after TBI. In a second study, the influence of KCl-induced spreading depressions on posttraumatic ICP (n = 5) and contusion volume (n = 17) was assessed. In one group, additional CSDs were induced with KCl and ICP was measured for 120 mins (n = 5). In this group, one animal was excluded from analysis because of an intraoperative blood pressure below the autoregulation threshold of 50 mm Hg. In a second group, animals (n = 5) were killed 15 mins after trauma for histological assessment of the primary lesion volume. In a third group (n = 6), the spontaneous occurrence of CSDs together with EEG and regional cerebral blood flow (rCBF) was recorded for 120 mins and animals were killed 24 h after trauma for histomorphometric quantification of contusion volume. In a forth group (n = 6), additional CSDs were induced by the application of KCl to the cerebral cortex of the traumatized hemisphere and contusion volume was also assessed 24 h after trauma.
Anesthesia and Trauma Application
Anesthesia and noninvasive intraoperative monitoring was performed as previously described (Thal and Plesnila, 2007). Briefly, animals were anesthetized in a halothane chamber (4%) and anesthesia was maintained by intraperitoneally injected medetomidin (0.5 mg/kg), midazolam (5 mg/kg), and fentanyl (0.05 mg/kg). Animals were endotracheally intubated and mechanically ventilated using 30% O2 (MiniVent 845; Hugo Sachs Elektronik, March-Hungstetten, Germany). Anesthesia was maintained up to 4 h by hourly injections of one third of the dose necessary for initial anesthesia induction. Rectal temperature was kept constant at 37.0°C throughout the experiment using a feedback-controlled heating pad connected to a rectal probe (Heater Control Module; FHC, Bowdoinham, ME, USA). Arterial blood pressure was measured with a noninvasive blood measuring system (RTBP 2000; Kent scientific, Torrington, CT, USA). Ventilation was adjusted based on the continuous measurement of ETCO2 (endtidal CO2) by microcapnography (CI240; Columbus Instruments, Columbus, Ohio, USA).
The right parietal cortex was injured as previously described (Zweckberger et al, 2003, 2006; Plesnila et al, 2007). Briefly, the animal's head was fixed in a stereotactic frame and the skull was exposed by a midline incision. A large craniectomy was performed above the right parietal cortex with a high-speed drill under continuous cooling with saline (Figure 1). Controlled cortical impact (CCI) was performed perpendicular to the intact dura mater with a velocity of 6 m/sec, an impact duration of 150 ms, and a displacement of the brain of 0.75 mm. Immediately after the impact, the craniotomy was closed with the initially removed bone flap using conventional tissue glue (Histoacryl®; Braun-Melsungen, Melsungen, Germany). At the end of all experiments, animals were put into an incubator heated to 35°C for 30 mins until recovery of spontaneous motor activity.
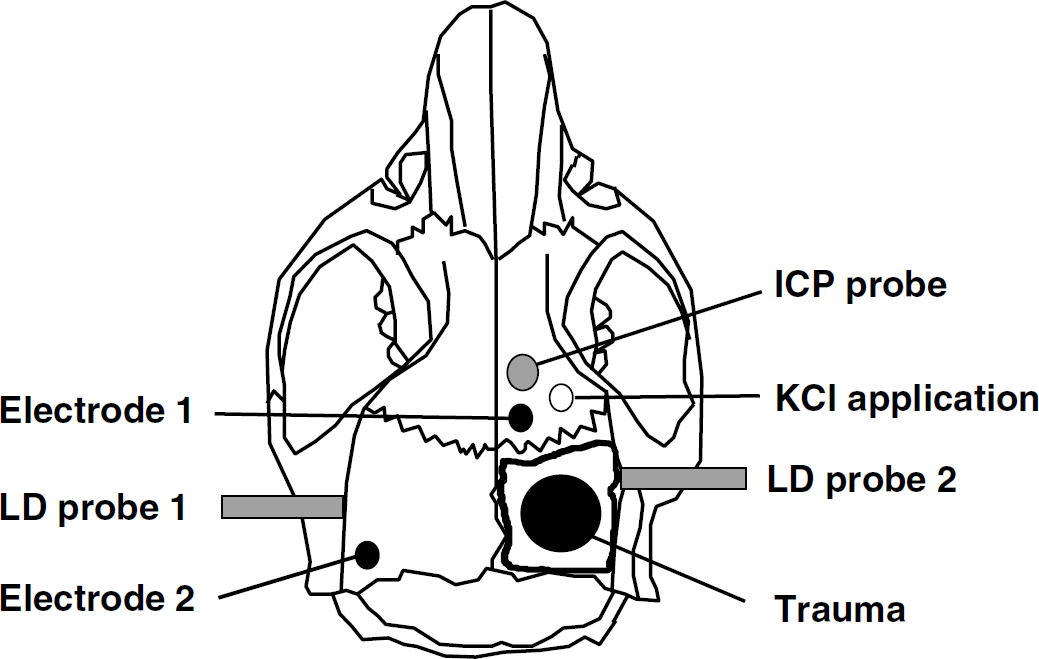
Schematic drawing of a mouse skull and the experimental setup describing the location of the following: craniectomy and trauma application, DC electrodes for the measurement of CSDs, ICP probe, laser Doppler probes for the measurement of CBF, and the burr hole for KCl application.
Electrophysiological Recordings
Two miniature calomel electrodes (mercury chloride, Hg2Cl2) were used for recording of CSDs and EEG through the intact skull as previously described (Back et al, 1996; Trabold et al, 2006). One electrode was placed over the right hemisphere, that is, ipsilateral to the trauma site, the other electrode over the left, contralateral hemisphere (Figure 1). The contact between skull and electrode was established by a saline-rinsed cotton wick. Animals were grounded by a silver-filled steel needle inserted into the neck muscles. The signal was amplified by a DC amplifier (B Radermacher, Köln, Germany) and recorded with a rate of 200 Hz using a computerized digital monitoring system (DaisyLab 5.0; Datalog GmbH, Mönchengladbach, Germany). Continuous measurements were performed from 30 mins before to 180 (study 1) or 120 mins (study 2) after trauma.
Measurement of Regional Cerebral Blood Flow
Regional cerebral blood flow was measured bilaterally with a flexible laser Doppler probe (Perimed 4001 Master; Perimed, Järfälla, Sweden) glued onto the exposed parietal skull over the middle cerebral artery territory as previously described (Plesnila et al, 2004). Because laser Doppler fluxmetry measures rCBF in arbitrary units results are expressed as percentage of baseline.
Measurement of Intracranial Pressure
Intracranial pressure was measured using a microprobe (Ø 0.9 mm; Mammendorfer Institut für Physik und Medizin, Mammendorf, Germany) as previously described (Zweckberger et al, 2003). The probe was inserted 3 mm into the brain parenchyma rostral to the craniotomy site (Figure 1) using a micromanipulator. Intracranial pressure was measured 10 mins before and up to 3 h after trauma.
Induction of Cortical Spreading Depression
Cortical spreading depression-like DC deflections were induced by epidural application of KCl. A burr hole was made 2 mm rostral of the craniotomy site and 2 mm lateral of the sagittal suture (Figure 1), and 2 μL of a 1-mol/L KCl solution were applied with a micropipette on the intact dura. Thirty seconds thereafter, the KCl was washed out with 5 μL physiological saline solution. Beginning 15 mins after trauma, one single CSD was generated every 10 mins. Altogether 10 to 14 CSDs were generated per animal in more than 100 mins. In control animals, 2 μL saline (1 mol/L) instead of KCl were used.
Quantification of Contusion Volume
Contusion volume was quantified as previously described (Zweckberger et al, 2003, 2006; Plesnila et al, 2007). Briefly, 24 h after CCI, animals were deeply anesthetized with 4% halothane and killed by cervical dislocation. Brains were removed, frozen in powdered dry ice, and stored at −80°C until further use. Ten-micrometer thick coronal sections were taken from 15 levels 500 μm apart throughout the contusion using a cryostat (CryoStar HM 560; Microm, Walldorf, Germany). One section from each level was stained with cresyl violet and digitized using a camera connected to a microscope. The contusion area on each section was quantified using an imaging analysis system (Olympus DP-soft, Munich, Germany) by an investigator blinded to the treatment of the animals. Contusion volume was calculated as follows: A1*d + 0.5*d*(A2 + A3 + ··· + A15). The variable d represents the distance between neighboring sections, that is, 0.5 mm.
All procedures described above are in accordance with the Institutional and National Guidelines for the Conduct of Animal Experiments and were approved by the Government of Upper Bavaria.
Statistical Analysis
All results are presented as mean ± s.d. Differences between groups were evaluated using the Mann—Whitney Rank Sum test with Bonferroni correction. Measurements over time (EEG amplitude, LDF) were tested versus baseline with Friedman repeated measures analysis of variance on Ranks followed by Dunn's comparison procedure as post hoc test. Differences with a P > 0.05 were considered significant. Calculations were performed with a standard statistical software package (SigmaStat 3.0; Systat Software, Erkrath, Germany).
Results
Mean arterial blood pressure and ETCO2 were within the expected range for long-term anesthesia in mechanically ventilated and traumatized mice throughout the study, that is, 60 to 80 mm Hg and 35 to 40 mm Hg, respectively (Supplementary Tables 1 and 2). Controlled cortical impact caused a marked increase in ICP from 3 ± 1 mm Hg before trauma to 15 ± 7 mm Hg 180 mins after trauma (Supplementary Table 1; P < 0.02).
CSDs after TBI
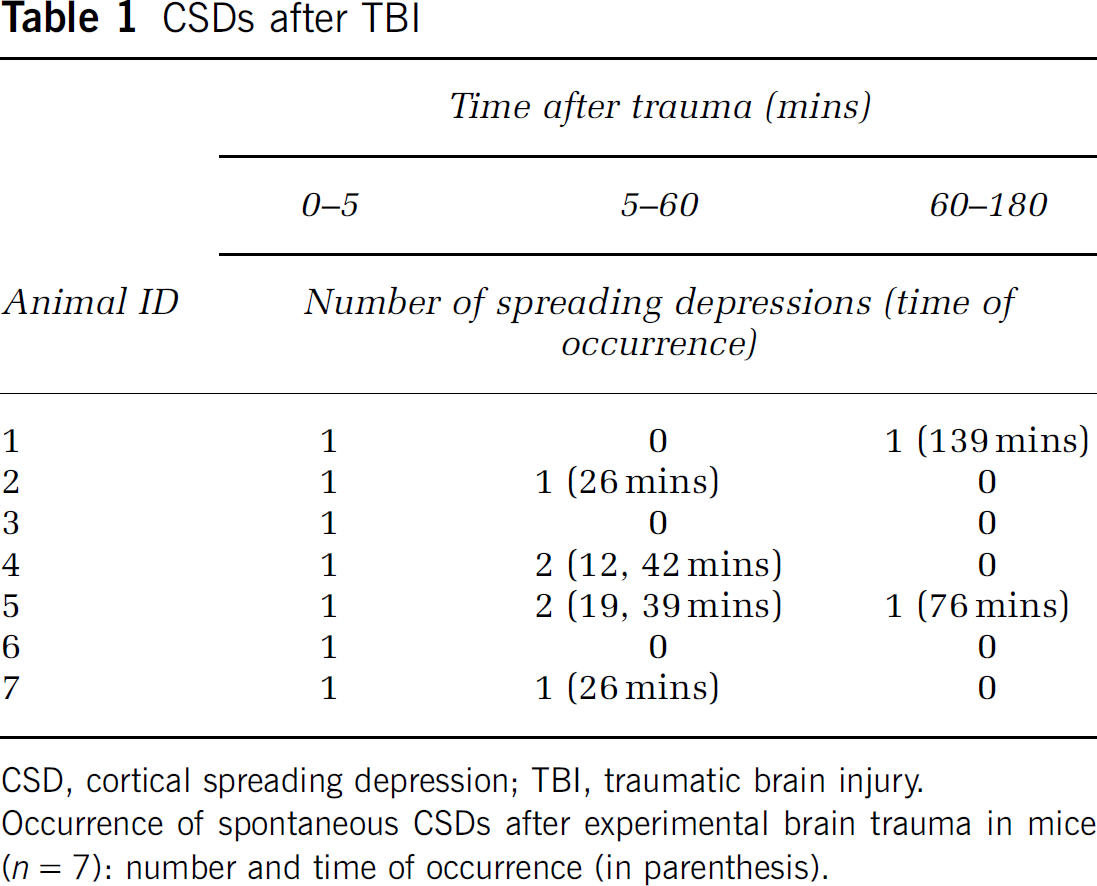
CSD, cortical spreading depression; TBI, traumatic brain injury.
Occurrence of spontaneous CSDs after experimental brain trauma in mice (n = 7): number and time of occurrence (in parenthesis).
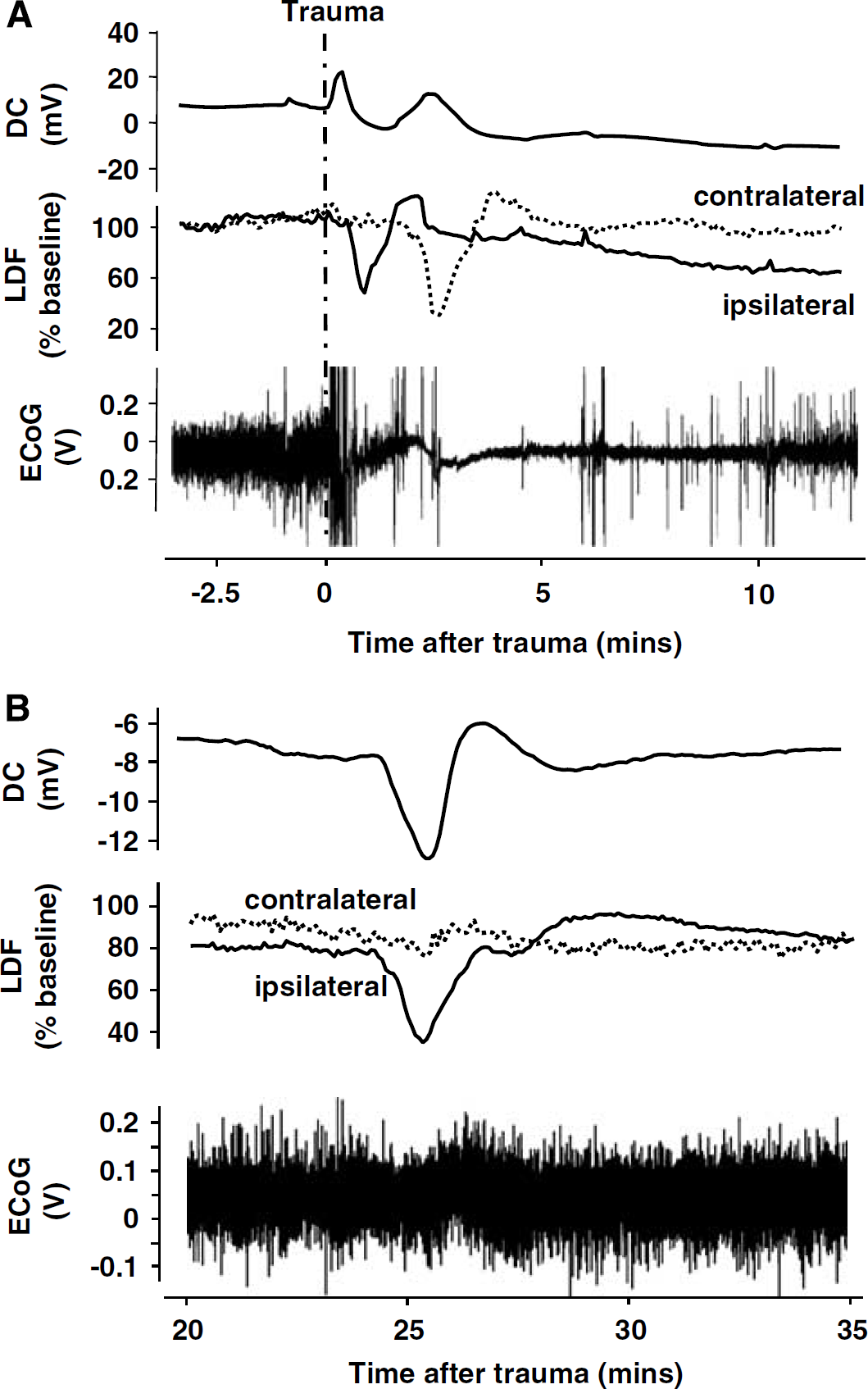
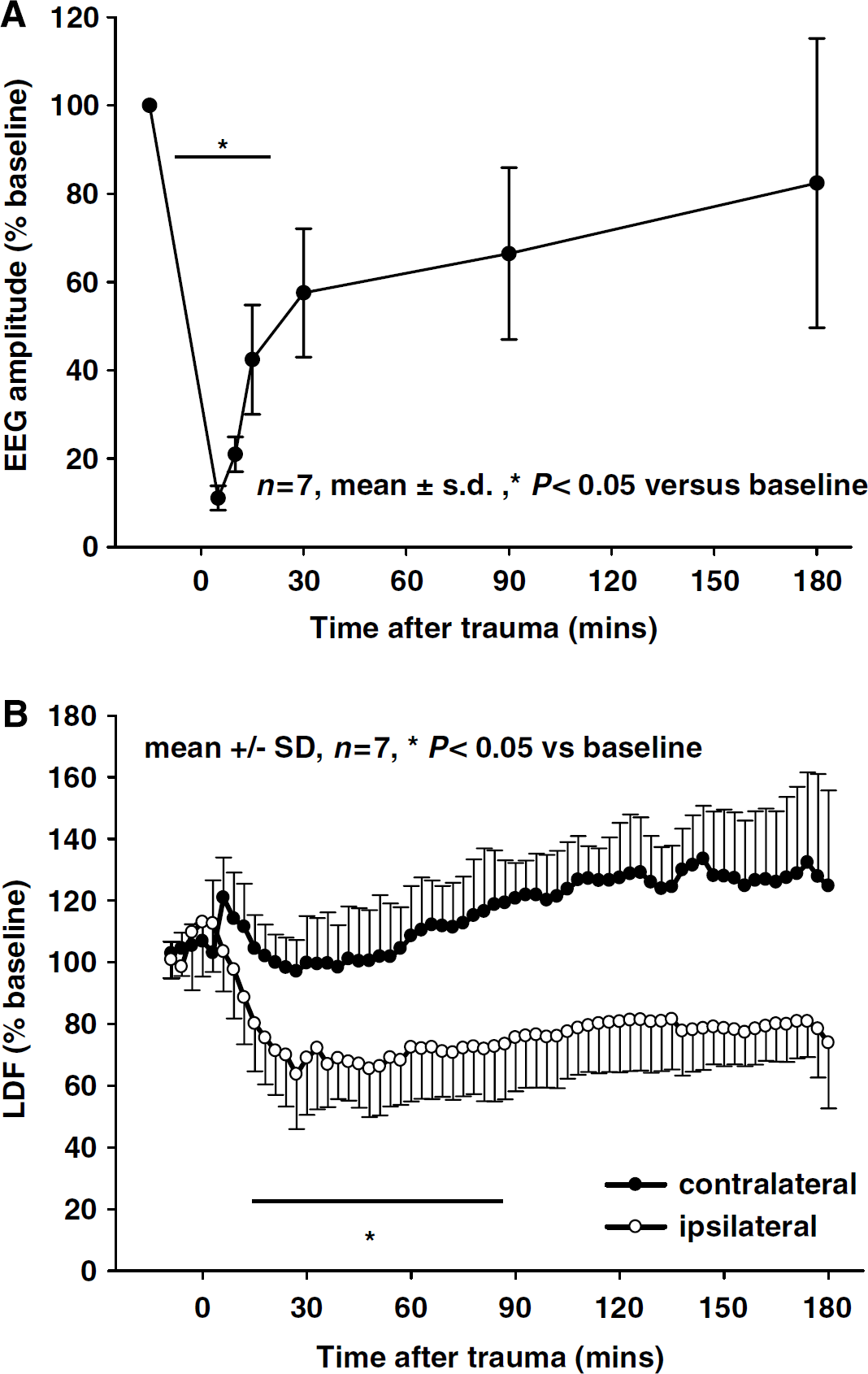
No changes in DC potential, EEG, and rCBF were recorded in healthy animals, that is, before trauma. Immediately after CCI, all animals generated one large CSD accompanied by a sequential temporary decrease of ipsi- and contralateral LDF (Figure 2A, upper two recordings). These changes were paralleled by a profound depression of EEG amplitude down to 14% ± 6% of baseline, which remained on this level for several minutes (Figure 2A, bottom). The EEG depression recovered only slowly to 82% ± 32% of baseline within the next 3 h. Recovery occurred relatively rapid within the first 30 mins and slowed down thereafter (Figure 3A). Similar, although not parallel changes were observed in rCBF. Cerebral blood flow in the traumatized hemisphere dropped within the first 24 mins after TBI to a minimum of 63% ± 14% of baseline (P < 0.05) and did not show any significant recovery until the end of the observation period (71% ± 9% at 3 h; Figure 3B, open circles). Except a tendency toward mild hyperperfusion, rCBF in the contralateral hemisphere did not show any significant trauma-related changes (Figure 3B, closed circles).
(
(
During the next 3 h after TBI, only eight additional spontaneous spreading depressions could be recorded in the seven investigated mice, that is, 0.38 CSD/h per animal. The majority of these CSDs (six out of eight) occurred in the first 60 mins after trauma (Table 1). In contrast to the CSD, which was observed in each animal immediately after TBI, the subsequent CSDs were not associated with any major changes in EEG amplitude and were only accompanied by ipsilateral changes of rCBF, indicating that these late CSDs were restricted to the injured hemisphere (Figure 2B).
Interestingly, the number of CSDs recorded during the first 2 to 3 h after TBI showed a trend toward an inverse correlation with ICP and contusion volume measured 2 and 24 h after TBI, respectively (Supplementary Figure 2). Because the assessment of these parameters was not the primary aim of the current study, further experiments with a higher number of animals are needed to clarify this observation.
In the second part of the study, we aimed to investigate the role of CSDs for secondary brain damage by inducing additional CSDs after TBI. Also, in this part of the study, all physiological parameters (arterial blood pressure and ETCO2) were within the normal range and no significant differences were observed between groups (Supplementary Table 2). Again, no CSDs were recorded before trauma and application of 1 mol/L NaCl in traumatized animals did not elicit any CSD. Application of 1 mol/L KCl, however, induced transient negative DC shifts followed by characteristic rCBF changes in a highly reproducible manner (Supplementary Figure 1).
Similar to the findings in the first part of our study, all 12 animals of the second study generated an early spontaneous depolarization in the first 10 mins after trauma (Supplementary Table 3) with the same characteristics as described above, that is, EEG depression and rCBF reduction in both hemispheres. Three out of six control animals exhibited further five spontaneous depolarizations during the next 2 h, values well in line to those obtained in the first part of the study (0.42 versus 0.38 CSD/h per animal, respectively). Altogether, that is, including the first CSD observed in each mouse, control animals generated 0.9 CSD/h. In the group where additional CSDs were induced, 5.7 CSD/h per animal were generated, that is, more than six times more as compared with the control group (Supplementary Table 3).
Induction of additional CSDs during the first 2 h after TBI had no effect on ICP, that is, no correlation between the number of CSDs and ICP was observed (Figure 4A). The same observation was also made for contusion volume. No correlation was observed between the number of CSDs and the contusion volume measured 24 h after TBI (Figure 4B).
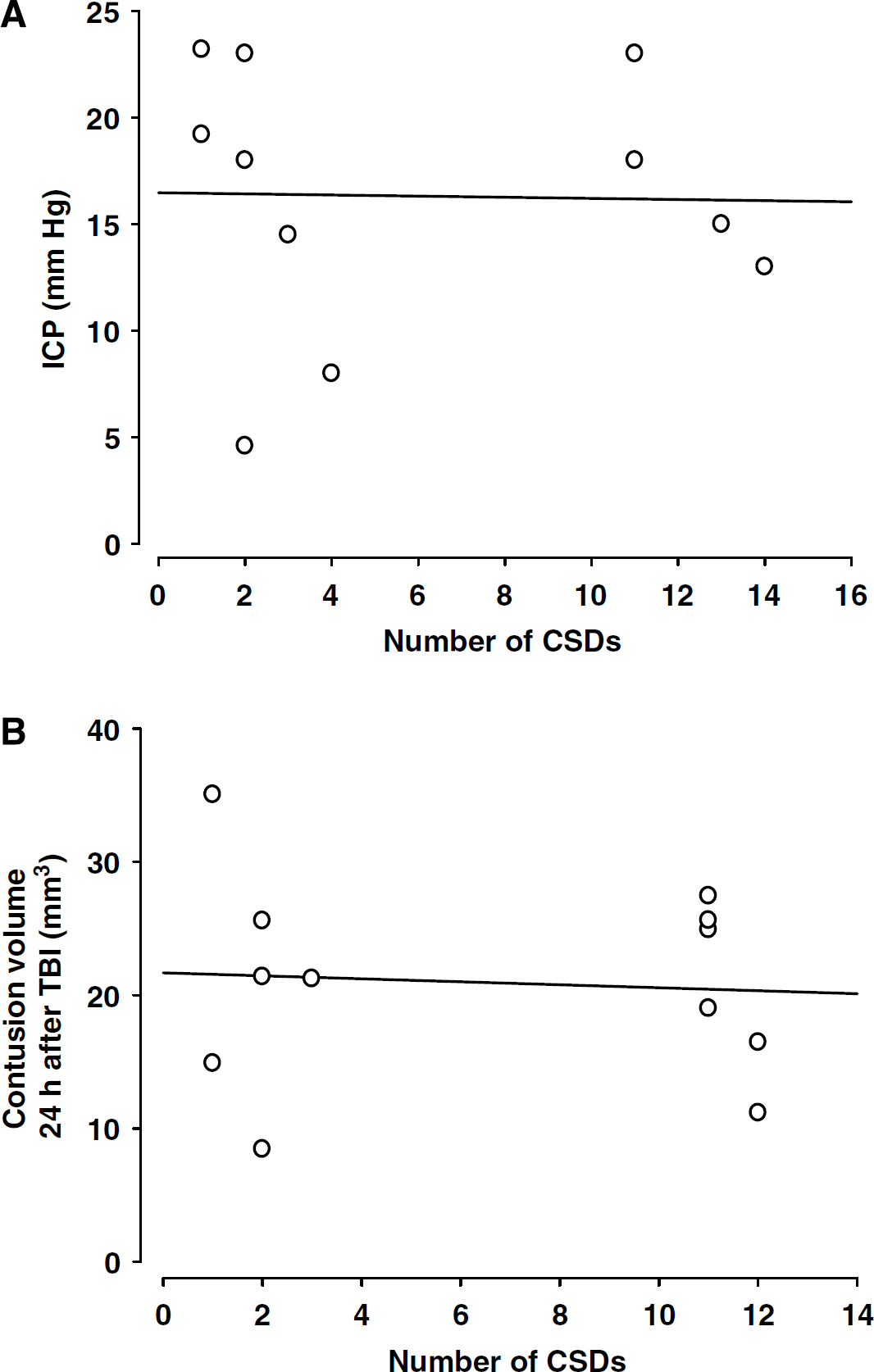
(
A similar effect of the induction of additional CSDs was also observed on secondary contusion expansion. Fifteen minutes after CCI, the mean contusion volume of control animals was 13.0 ± 1.6 mm3 (Figure 5; n = 6). Twenty-four hours after trauma when secondary contusion expansion reached its maximum, control animals had a contusion volume of 21.1 ± 3.7 mm3 (n = 6), that is, the initial contusion volume increased secondarily by 8.1 mm3 or 62%. Animals displaying six times more CSDs as compared with controls had a similar contusion volume, that is, 20.8 ± 2.6 mm3 (n = 6). These findings indicate that additional CSDs have no influence on the secondary expansion of the traumatic cortical contusion (Figure 5).
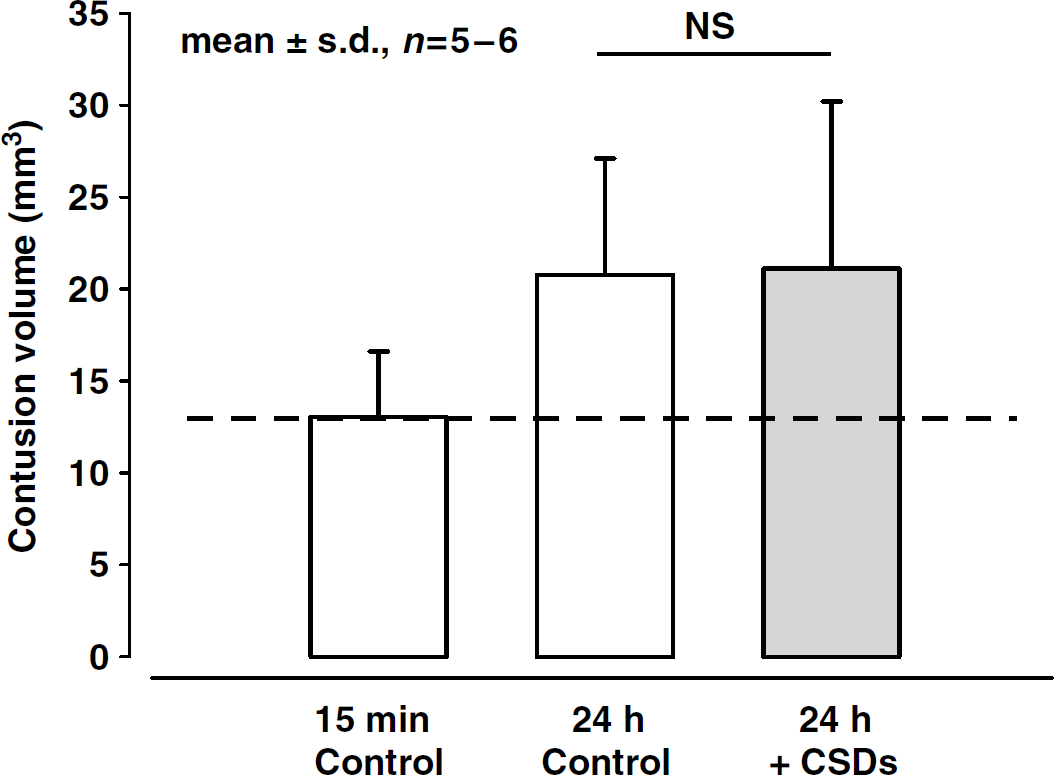
Volume of the traumatic contusion 15 mins (control group 1, n = 5) and 24 h after trauma with or without induction of additional CSDs (n = 6 each). Values are given as means ± s.d.
Discussion
The results of the current study show that CSDs occur at a very low frequency (0.7 to 0.9 CSD/h) and that inducing a sixfold higher number of CSDs does not lead to additional loss of brain tissue after experimental TBI in mice.
Controlled cortical impact, as used in the current study, is a widely used experimental brain trauma model. We used a CCI device especially developed for mice, thereby achieving a high intra- and interindividual reproducibility as reported previously (Zweckberger et al, 2003, 2006; Plesnila et al, 2007). Most importantly for the current study, our model allows the precise quantification of secondary brain tissue damage by histomorphometric comparison of contusion volumes 15 mins and 24 h after trauma. Contusion volume increases by ~60% during this time with the most pronounced lesion expansion within the initial 3 h after trauma (Zweckberger et al, 2006). Decompression craniectomy or inhibition of p53-mediated cell death signaling reduces contusion expansion significantly, thereby proving that secondary mechanisms are responsible for lesion expansion (Zweckberger et al, 2003, 2006; Plesnila et al, 2007). To ascertain optimal experimental conditions for the current study, we developed a protocol that allows noninvasive measurement of almost all relevant physiological parameters (Thal and Plesnila, 2007). Mice were intubated and mechanically ventilated to maintain physiological oxygenation during CSD measurements. Endtidal pCO2 was measured noninvasively by microcapnometry to allow adjustment of ventilation and, hence, physiological blood gas values during all experiments. Blood pressure was also recorded noninvasively with a tail cuff to avoid additional surgical procedures, thereby preventing fluid loss. Recordings show the lack of secondary systemic insults, that is, hypotension, during the experimental period (Supplementary Tables 1 and 3). Cerebral blood flow was measured with a flexible laser Doppler probe through the intact skull. The anesthesia protocol used for the current experiments is known not to affect physiological ICP (Zweckberger et al, 2003) and cerebral blood flow (Zornow et al, 1992), and does most likely also not affect the threshold for CSDs, as known for volatile anesthetics, for example, halothane, which are otherwise widely used in experimental research (Saito et al, 1995; Kitahara et al, 2001). Most importantly, the CSD measurement itself was also performed with noninvasive calomel electrodes, which allow stable and reproducible EEG and CSD measurements through the intact skull, thereby avoiding any additional mechanical alteration of the brain that may elicit additional spreading depression waves (Back et al, 1996; Trabold et al, 2006).
The phenomenon of secondary contusion expansion is mainly attributed to cell death in the traumatic penumbra, the damaged but still viable brain parenchyma adjacent to the contusion. This area contains scattered damaged neurons within an intact neuropil, is prone to perturbation of perfusion and concomitant secondary insult, and, most importantly, can be salvaged from progressive damage, because it is sensitive to therapeutic interventions (Zweckberger et al, 2006).
Although many different mechanisms are responsible for secondary lesion expansion, in recent years, many studies focused on the pathogenic potential of CSDs. In normal brain, CSDs induce tissue hypoxia and neuronal swelling (Takano et al, 2007), upregulate immediate early genes (Hermann et al, 1999; Urbach et al, 2006), and may precondition the brain toward subsequent ischemic events (Kobayashi et al, 1995; Horiguchi et al, 2005). Despite these changes, CSDs do not cause any longlasting structural damage to otherwise healthy brain tissue (Nedergaard and Hansen, 1988). In ischemic brain, however, repetitive waves of depolarization result in stepwise depletion of energy stores and functional as well as structural deterioration of the penumbra leading to an increase of infarcted tissue volume (Ohta et al, 2001; Selman et al, 2004; Hopwood et al, 2005; Umegaki et al, 2005). The deleterious effects of CSDs are believed to be mainly mediated by a lack of compensatory hyperperfusion usually occurring to match the metabolic strain caused by repolarization (Back et al, 1994). Other possible mechanisms of CSD-induced penumbral deterioration are the development of vasogenic edema by an increase in MMP-9 activity (Cunningham et al, 2005), or the activation of proinflammatory cytokine pathways (Jander et al, 2001), which, however, may well be secondary to changes in cerebral blood flow.
In the view of the pathophysiological similarities between the ischemic and the traumatic penumbra (Bramlett and Dietrich, 2004), it is conceivable that CSDs may also play a role for the secondary expansion of the traumatic penumbra. So far, however, it was unclear whether spreading depressions only accompany or indeed cause secondary brain damage after TBI. Hermann et al (1999), for example, observed no correlation between lesion size and the amount of spreading depressions after cortical cold injury, a model associated with secondary lesion expansion. It was concluded that in contrast to the metabolic penumbra in focal ischemia, depolarization may not aggravate the preexistent metabolic dysfunction in the vicinity of traumatic lesions. In addition, spreading depressions occurred at a much lower frequency than after experimental models of stroke, an observation well in line with the results of the current study performed in a clinically more relevant kinetic model of TBI. In a study using fluid percussion injury in rats, Rogatsky et al (2003) evaluated the relationship between trauma severity, posttraumatic ICP, and the number of spreading depressions. In animals with moderate or mild nonfatal trauma, CSDs occurred at a low frequency or even ceased and ICP did not correlate with the number of CSDs. In contrast, fatal TBI was often associated with multiple CSDs and with a close correlation of ICP versus the number of CSDs. These findings therefore suggest that CSDs may play only a role in the most severely damaged brain where secondary insults, for example, ICP-induced global reduction of cerebral blood flow, play a major pathophysiological role. These findings are in line with our current results showing that under conditions of a moderately increased ICP (15 ± 7 mm Hg) not reducing CBF to ischemic levels, CSDs appear at a low frequency (0.7 to 0.9 CSD/h), do not correlate with ICP, and are not associated with progressive loss of viable brain tissue in the traumatic penumbra. Support for the hypothesis that CSDs cause secondary brain damage only when associated with a secondary insult, for example, hypotension or pathologically increased ICP, comes from a previous study from our laboratory showing that postinjury lesion expansion after cold injury in rats was significantly worsened by hypotension (Trabold et al, 2006). These findings are also further supported by two clinical studies measuring CSDs in patients suffering from TBI (Mayevsky et al, 1996; Strong et al, 2002) or subarachnoid hemorrhage (Dreier et al, 2006).
Taken together, our results in mice suggest that after TBI, CSDs have only then a detrimental potential when they occur together with ischemia or hypoxia. Consequently, the prevention of cerebral or systemic secondary insults, for example, pathologically elevated ICP or hypoxia, may be the most efficient strategy to reduce or avoid CSD-induced brain damage.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.