
Abstract
Transglutaminases (TGs) are multifunctional, calcium-dependent enzymes that have been recently implicated in stroke pathophysiology. Classically, these enzymes are thought to participate in cell injury and death in chronic neurodegenerative conditions via their ability to catalyze covalent, nondegradable crosslinks between proteins or to incorporate polyamines into protein substrates. Accumulating lines of inquiry indicate that specific TG isoforms can shuttle into the nucleus when they sense pathologic changes in calcium or oxidative stress, bind to chromatin and thereby transduce these changes into transcriptional repression of genes involved in metabolic or oxidant adaptation. Here, we review the evidence that supports principally a role for one isoform of this family, TG2, in cell injury and death associated with hemorrhagic or ischemic stroke. We also outline an evolving model in which TG2 is a critical mediator between pathologic signaling and epigenetic modifications that lead to gene repression. Accordingly, the salutary effects of TG inhibitors in stroke may derive from their ability to restore homeostasis by removing inappropriate deactivation of adaptive genetic programs by oxidative stress or extrasynaptic glutamate receptor signaling.
INTRODUCTION
Stroke is a prevalent disorder in which the central nervous system (CNS), the vascular system, and the environment fail to dynamically interact and adapt to an imposed external stress. Adaptation is the ability of a cell or an entire organism to homeostase to a physiologic or pathologic stress (e.g., energy and oxygen depletion or free radicals) leading ultimately to survival. Classically, adaptive responses are characterized by a short, early phase dominated by the activation of preexisting proteins, such as the stress kinase, AMPK; and by a later, more durable phase involving de novo gene expression. The latter responses are mediated via the activation of transcriptional activators and co-activators that initiate the transcription of a battery of genes important in cell protection. In stroke, transcriptional repression has been widely reported, and this repression can be adaptive or maladaptive. 1 Accordingly, an investigation of how to modulate the activation of the genetic adaptive response in injured neurons—and thereby enhance neuronal survival and maintain brain plasticity—is of extreme interest to the stroke community. To attain this important goal will not be easy, and will require the identification of targets that are (1) able to regulate not one single gene, but many genes induced by stress (adaptive response), (2) induced only by toxic signals and not by physiologic ones, and (3) readily modulated by pharmacologic and biologic intervention.
Epigenetic Modulators as Targets to Activate Broad and Deep Neuroprotective and Restorative Programs
Recently, a number of laboratories have observed protection from stroke with drugs that modulate gene transcription. Some of these drugs inhibit epigenetic enzymes involved directly in chromatin remodeling including DNA methylation (e.g., DNA methyl transferases), 2 and histone acetylation (e.g., Histone Deacetylase inhibitors, HDACi). 3 They are termed as epigenetic because they lie ‘above the genome’, but have the ability to modulate gene expression independent of changes in DNA coding. Epigenetic modulation of chromatin appears to affect the expression of many prosurvival and prodeath genes in favor of survival in the nervous system, but cell death in cancer cells, providing an unexpected benefit as therapeutic agents. Additional laboratories including ours are trying to determine whether these epigenetic modulators can not only arrest cell death in the CNS but also facilitate regeneration and plasticity. 4 During these investigations, another family of enzymes has emerged as novel epigenetic modulators, transglutaminases (TGs).
TRANSGLUTAMINASES: NEW EPIGENETIC KIDS ON THE CNS BLOCK
Transglutaminases are enzymes involved primarily in crosslinking. They appear to be induced by pathologic stimuli, such as oxidative stress or intracellular calcium dyshomeostasis. We have recently shown that these enzymes also modulate transcription. 5 Inhibition of their activity leads to normalization of genes repressed in neurodegenerative conditions and subsequently neuroprotection. There is an increase in TG transamidating activity in different models of stroke 6 – 9 and inhibition of TG with an FDA approved, nonselective inhibitor, cystamine, showed beneficial effects. 10 Despite these promising outcomes, the precise isoforms required and the mechanisms by which TGs function have not been thoroughly delineated. Here, we will review TG's role in stroke pathophysiology and its potential contribution to maladaptive transcriptional repression after ischemia.
Transglutaminase Family Members and Their Functional Domains
Transglutaminases are a family of proteins that consist of eight enzymes encoded by closely related genes 11 called TGM 1-7, Factor XIIIA (F13A1), and protein 4.2 (a structural protein that lacks the catalytic site). Alignment of these gene products reveals a high degree of sequence similarity and a well-conserved gene organization with similar intron and exon distribution. Though the enzymes are differentially expressed in the organs, they have a common ancestor related to cysteine proteases 12 and they appear to be close evolutionarily, as shown in their phylogenetic tree (Figure 1A). These similarities may explain the redundancy in the TG genome. The enzymes comprises four structurally distinct domains, an N-terminal β-sandwich, a catalytic core, and two β-barrel domains (Figure 1B). The catalytic core possess three critical amino acids (cysteine, histidine, and aspartic acid) for the transamidating activity, through which TGs crosslink proteins via an N(epsilon)-(gamma)-glutamyl lysine isopeptide bond or incorporate polyamines to protein substrates 13 (Figure 1C). For many years, the field believed that the isoforms expressed intracellularly were silent and could be activated only by dramatic increase in calcium in neurons such as that observed with excitotoxic levels of glutamate (100 μmol/L). New data have recently presented 14 that identifies several calcium-sensitive sites in the protein structure of TG2 (Kd ~ 0.1 to 0.15 μmol/L). 15 The clustering of these sites allows the enzyme to be quickly activated by local release of calcium from a specific subcellular compartment where TG can mediate signaling or trigger proteosomal degradation. 16 By contrast to high calcium, TG activity is inhibited by the nucleotides ATP and GTP. Indeed, TG2, 17 TG3 18 TG5, 19 and TG6 20 all possess a GTP-binding site in their β-barrel domain.
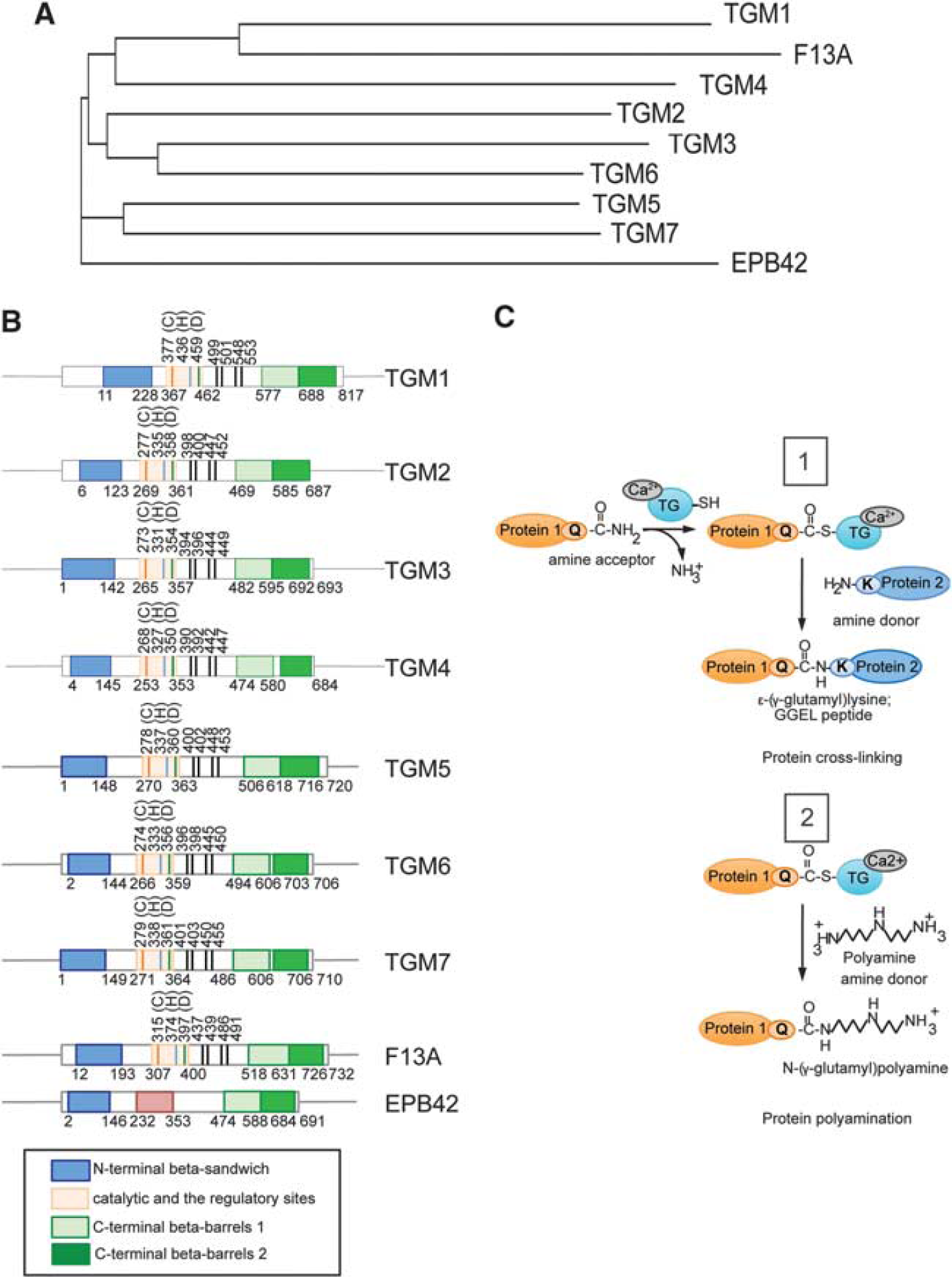
The transglutaminase (TG) family. (
Transglutaminases Are Believed to Crosslink Aggregated Proteins in Chronic Neurodegeneration, But What Role Do They Play in Acute Injuries such as Stroke Where Protein Aggregation Has Not Been a Dominant Theme?
Transglutaminases were initially invoked as mediators of neurodegeneration because of their ability to crosslink proteins by Dennis Selkoe in the early 80s: they have been implicated in the formation of insoluble aggregates in Huntington's disease (HD) and neurofibrillary tangles in Alzheimer's disease. 21 In this scheme, protein crosslinking occurs in ‘trans’ between a glutamate residue on one protein and a lysine residue on another protein. This type of crosslinking is necessary extracellularly to create a protective barrier between the skin and the external environment; 22 however, a physiologic role for TG acting intracellularly or extracellularly, in the brain, is unclear. Indeed, germ line deletion of TG2, the most abundant and best-studied isoform in the brain, resulted in increased life span and, surprisingly, increased number of intranuclear inclusion in HD mice, 23 , 24 which suggests that TG2's major role in the brain is not related to protein crosslinking.
Polyamination of Proteins by Transglutaminase
Polyamines, small cationic molecules present in millimolar concentration in the brain, 25 are potentially relevant TG substrates in CNS disorders. Transglutaminases can catalyze addition of polyamines to proteins, a process defined as polyamination. 13 This modification correlates with the alteration of normal protein functions, since the addition of positive charges on a protein surface can alter protein–protein interactions. Additionally, TG2 can polyaminate retinoblastoma protein and prevent its degradation by caspases. 26 A yeast two-hybrid screen using the C-terminal portion of TG2 as bait showed that this protein interacts with and polyaminates the viral oncoprotein E7. 27 Polyamination prevents E7 from binding retinoblastoma in the nucleus and thereby prevents E2F from being released and activating the proliferation program. In this way, TG2 acts as a tumor suppressor gene. Due to technical challenges, few proteins have been definitively shown to be polyaminated, but there are several validated targets: phospholipase A2 activity is increased by polyamination; 28 RhoA, responsible for the deleterious activation of the Rho-associated protein kinase pathway in ischemic-reperfusion in cardiomyocyte cells and the negative regulation of axon outgrowth; 29 the microtubule-associated protein tau, where polyamination inhibits calpain-mediated proteolysis 30 – 32 and contributes to cytoskeletal dysorganization; and actin, the main endogenous glutamyl substrate in cells undergoing apoptosis. 33 A more complete understanding of the ‘polyaminome’, the set of polyaminated proteins in the cell, will help to refine models of how polyamination changes cell functions and fates and whether this posttranslational modification is exclusively catalyzed by TGs. Polyamines have been implicated not only in cell death after ischemia, but also in regeneration, 34 , 35 so understanding the relative contributions of polyamination to cell death and regeneration will affect potential dose and timing of putative TG inhibitors.
Transglutaminase Levels and the Transamidating Activity Are Induced in Ischemia, Excitotoxicity, and Oxidative Stress
The best-studied TG isoform in the brain is TG2 due to its abundance and ubiquitous expression. It is expressed at low levels in neurons, astrocytes, and oligodendrocytes in normal conditions, but it is highly induced by the stress and injury that follows permanent 9 or transient ischemic stroke, 36 hemorrhagic stroke, 37 and other neurodegenerative conditions. Induction of TG appears to be, in part, transcriptional and deletion studies have identified several competent regulatory elements within the proximal promoter that allow precise regulation of TG message levels. Retinoids, 38 glucocorticoids, and cytokines (IL-6) 39 appear to induce TG message via transcription factors (TFs), such as NF-κB, 40 Sp1, 41 and Hypoxia Inducible Factor (HIF1α). 42 Jun and Fos, members of the AP-1 family, have been shown to induce TG2 transcription in neurons or glia 41 and this upregulation usually correlates with cell death. Building on prior work that showed that TG2 could be induced by a host of extracellular factors in glial cells, 43 , 44 we have recently showed that lethal, glutathione depletion-induced oxidative stress induces TG2 upregulation in cortical neurons 36 and that inhibition of TG in neurons prevents oxidative death. By contrast, protective concentrations of peroxide generated in astrocytes leads to downregulation of TG2 message. Accordingly, pharmacological TG inhibition in astrocytes leads to protection from oxidative death in adjacent neurons. 36 Thus, TG appears to act as a death mediator in both astrocytes and neurons.
In addition to TG2, TG1 was also dramatically upregulated in vitro and in an in vivo model of stroke. The increase in TG1 expression and TG1 and 2 activities was redox activated (Figures 2A and 2B) and necessary as well as sufficient for cell death (Figure 2D). 36 In this context, TG transamidating activity resides downstream of chronic ERK activation suggesting a potential role for ERK or its downstream transcriptional targets, in the regulation of TG1 and TG2 levels in the brain. In fact, Actinomycin D, an inhibitor of transcription, directly controls TG2 expression (Figure 2C). Another study supports NF-κB as a primary activator of TG2 transcription in astrocytes and showed nuclear translocation of p65 and p50, canonical NF-κB subunits, and their binding to the TG2 promoter. 45 Interestingly, TG1 and 2 can be upregulated by different groups of cytokines (e.g., TNFα, IL-6, and TGFβ) or toxins (lipopolysachharide) in mixed astrocyte-neuronal cultures. 46 By contrast to our in vitro data that shows that both TG1 and TG2 are required for cell death from oxidative death, TG2 ablation reduced the infarct volume in a model of permanent ischemia 47 and its overexpression in neurons via a Prp promoter, showed higher number of apoptotic cells and greater susceptibility to kainate stimuli. 48 Future studies will clarify why TG2 is necessary and sufficient in some paradigms, while TG2 and TG1 are necessary in other paradigms. Of course, the precise mechanism by which TG1 and TG2 mediate cell injury and death is still under investigation.
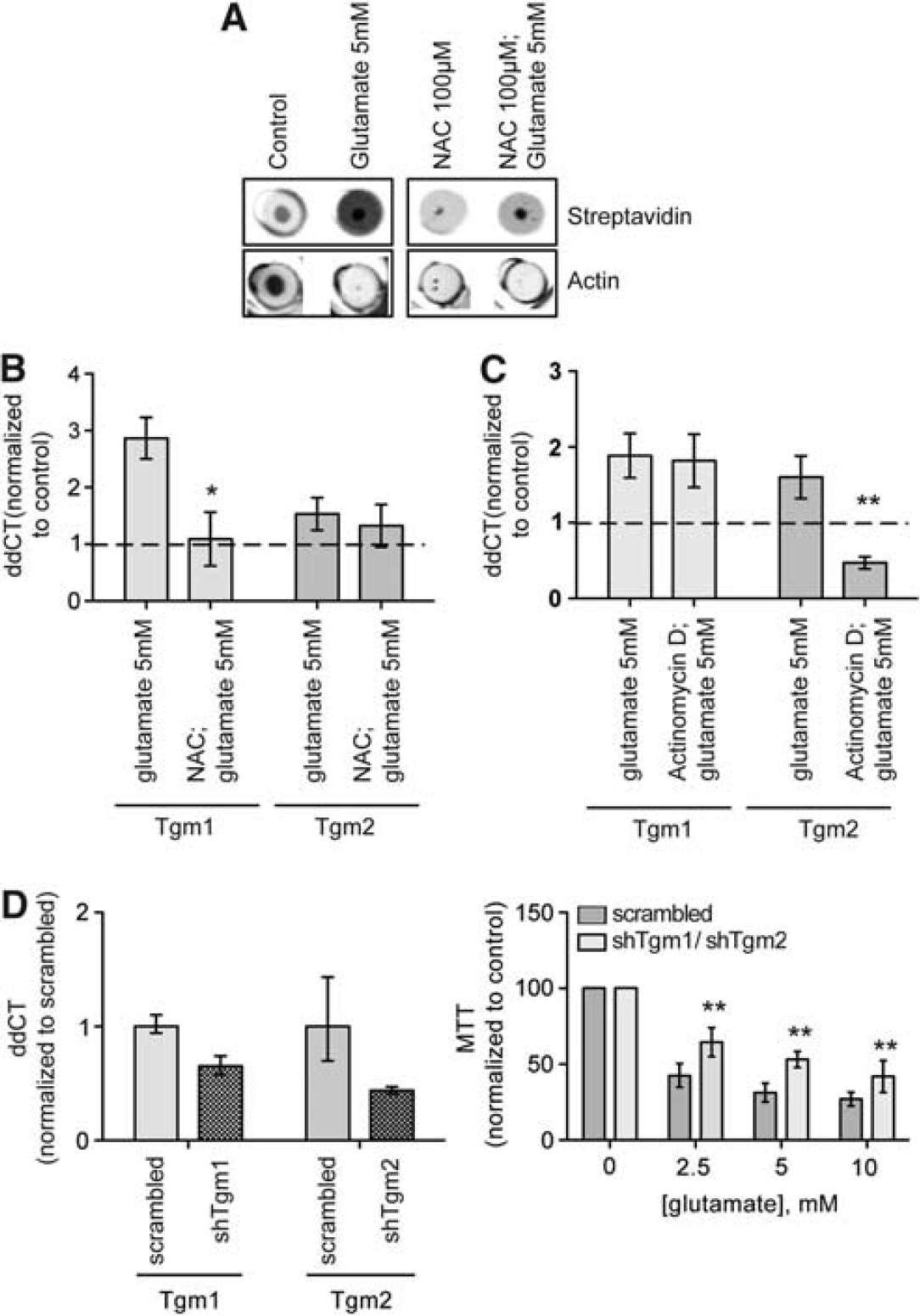
Transglutaminase (TG) activity and message levels are increased in neurons undergoing oxidative stress and the antioxidant N-acetyl cysteine (NAC), or the repressor of transcription actinomycin D or silencing two TG isoforms can protect neurons from oxidative stress-mediated cell death. (
Transglutaminase 2 possesses two nuclear localization sequences (NLS) and one putative nuclear export sequence that allows its translocation from the cytosol to the nuclear compartment during neuronal stress. 49 It has been estimated that 5% to 7% of TG2 resides usually in the nucleus; after 5 hours of ischemia, its nuclear levels are at least 10 times higher. 50 Following the observations that TG2 accumulates in the nuclear compartment of neurons after insult, and it can modify TFs (Sp1 and retinoblastoma, 51 and histone proteins 52 ), we showed an active role for TG2 as a transcriptional modulator in models of HD. Pharmacological and molecular TG2 inhibition and the expression of its cytosolic form were sufficient to normalize the expression levels of two genes implicated in the mitochondrial adaptive response, peroxisome proliferator-activated receptor-γ coactivator-1d and its downstream target, cytochrome c, normally repressed in HD. 53 Moreover, TG2 is bound to chromatin at promoters and intragenic regions, and it interacts directly with histone H3, suggesting a direct epigenetic modulation of the opening of the nucleosome and interfering with RNA polymerase II transcription. Conversely, TG1 does not present a putative NLS and has never been reported in the nuclear compartment. Of note, only few papers have been described TG1 in the brain, 54 – 56 , 36 therefore, a careful characterization of its cellular distribution and activation needs to be provided.
A Unifying Model for Transglutaminase in Ischemia
In spite of growing support for TG in neuronal injury and degeneration, a unifying model for its role in stroke has yet to be presented. We favor a model in which TG is activated by concomitant decrease in ATP/GTP levels and high intracellular calcium via extrasynaptic glutamate receptors after ischemia. ATP/GTP levels are significantly decreased in the ischemic core, 57 leading to the inability of the neuron to maintain ionic gradients that support membrane polarization, followed by calcium influx and mitochondrial damage. 58 High micromolar concentration of calcium induces a persistent activation of TG2 that results in increased polyamination of histones, leading to a more compact conformation of chromatin and repression of prosurvival and proplasticity genes (Figure 4B-3). In this scheme, TG is a sensor for calcium dyshomeostasis that signals to the cell nucleus to repress genes for survival and repair, thereby ensuring death. In the rest of the review, we will summarize evidence that supports this model, and clarify remaining gaps that need to be evaluated experimentally.
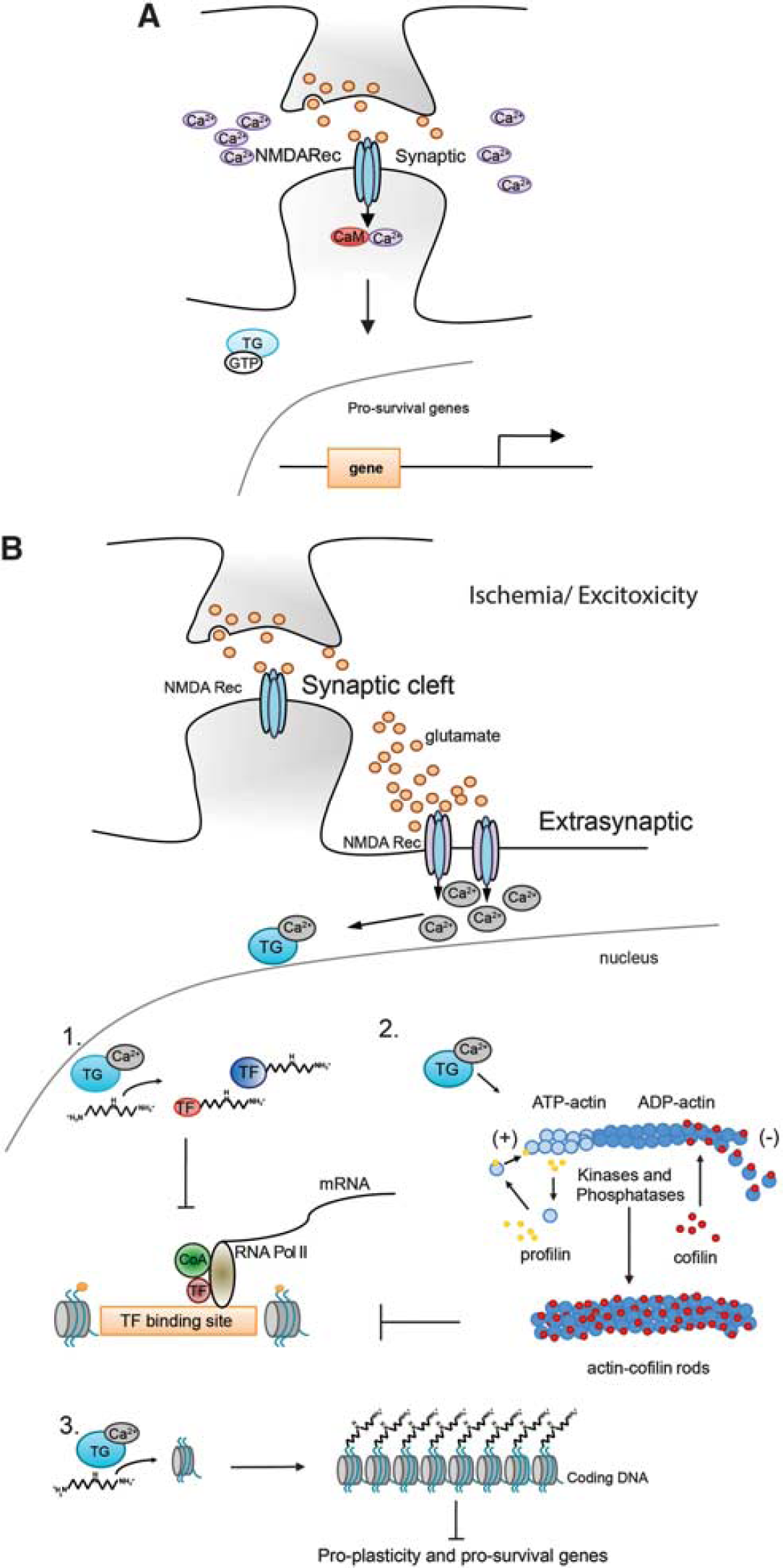
Potential mechanisms by which transglutaminase (TG) modulates gene transcription during ischemia/exicitotoxicity. (
PATHOLOGIC GENE REPRESSION AFTER STROKE: EXCITOTOXICITY, CYTOSKELETAL REARRANGEMENT, AND TRANSCRIPTIONAL REPRESSION AND THE INVOLVEMENT OF TGS IN STROKE PATHOBIOLOGY
Activation of Extrasynaptic Versus Synaptic Glutamate Receptors in Ischemia: Not All Roads Lead to Transglutaminase Activation
Over the past several decades, the clinical neuroscience community has come to understand the Janus faces of NMDA receptors and their divergent effects on gene expression. On the one hand, NMDA receptor blockade can prevent ‘excitotoxicity’ and provide remarkable histologic protection after experimental stroke; on the other hand, NMDA receptor activity is necessary not only for many eloquent brain functions, but it also appears critical for survival of neurons in development and postinjury. Initial models to explain the NMDA receptor paradox invoked differing levels of calcium entry based on the level and duration of NMDA receptor agonism. 59 Indeed, early on, Choi et al 60 showed that calcium was necessary for NMDA receptor-mediated neuronal death. Subsequent elegant studies by Tymianski and colleagues showed that calcium channeled via NMDA receptors leads to recruitment and activation of neuronal nitric oxide synthase and downstream deleterious consequences including peroxynitrite formation. 61 A refined model has emerged to explain the NMDA receptor paradox. Specifically, this model, developed by Bading and Hardingham, posits that the location of NMDA receptors determines whether they mediate prodeath or prosurvival signals. Accordingly, activation of synaptic glutamate receptors leads to 10-fold increases in calcium levels that are promptly bound to calmodulin and other calcium-binding proteins able to activate prosurvival signals (Figures 3 and 4A), whereas activation of nonsynaptic glutamate receptors (e.g., on perisynaptic sites, dendrites, and axons) induces a persistent release of calcium that leads to activation of prodeath signals; 59 (Figure 3).
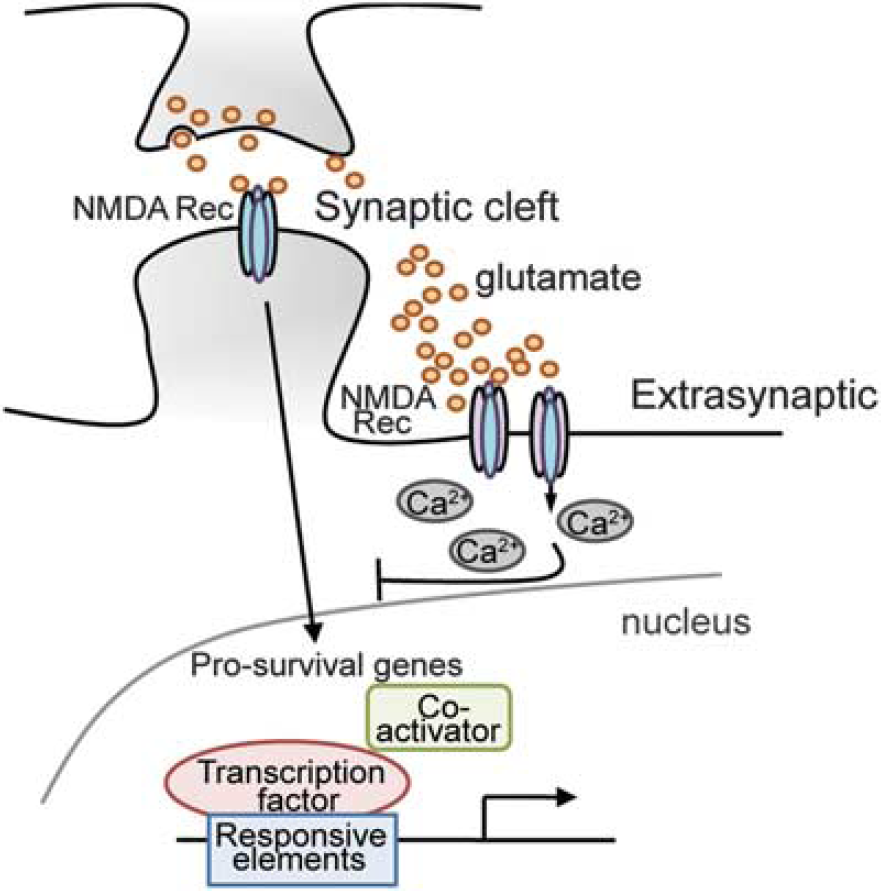
Excitotoxicity and the role of the NMDA receptors in gene regulation.
And while synaptic activation of glutamate receptors appears to trigger prosurvival genes via activation of CREB, peroxisome proliferator-activated receptor-γ coactivator-1d (a coactivator of mitochondrial biogenesis), NFAT (a calcium responsive TF), or the nuclear factor I subtype A, nonsynaptic glutamate receptor activation appears to counteract these prosurvival and proplasticity signals via the shut off of these selective pathways. For instance, nonsynaptic glutamate receptor activation leads to CREB dephosphorylation and reduced recruitment of the coactivator and histone acetyl transferase, CBP/p300 and thus diminished recruitment of RNA polymerase II and decreased histone acetylation. Recent studies suggest that CREB ‘shut off’ downstream of extrasynaptic glutamate receptors is determined by local elevations of calcium. Synaptic glutamate receptor activation leads to calcium saturation of caldendrin, a calmodulin like protein with EF hand calcium-binding motifs (calcium-binding motifs composed of two helixes (E and F) joined by a loop). Calcium saturated caldendrin binds to the NLS of a protein called Jacob and prevents it from moving to the nucleus to dysregulate transcription. By contrast, extrasynaptic glutamate receptor activation leads to calcium increases in the somatodendritic compartment where Jacob's NLS interfaces with Importin α to drive Jacob into the nucleus. The presence of nuclear Jacob correlates with dephosphorylated CREB and diminished neuronal viability and plasticity. 62 While the scheme is compelling, an understanding of how subplasmalemmal calcium changes at extrasynaptic sites are transduced into transcriptional repression remains obscure. Here, we propose a model in which the multifunctional calcium-dependent enzyme TG acts as a transducer of changes in extrasynaptic glutamate receptor calcium into repression of gene expression. Interestingly, both TG2 63 and Jacob enter the nucleus via Importin α, where they may interact and contribute to CREB shut off. Accordingly, TG2 is upregulated in cerebellar granule cells exposed to NMDA toxicity 64 and its inhibition in medium-sized spiny neurons derived from HD mice (YAC128) or wild-type controls is sufficient to protect neurons from NMDA-mediated toxicity. 5 A putative mechanism by which the inhibition of TG activity halts excitotoxic death could involve the derepression of peroxisome proliferator-activated receptor-γ coactivator-1d expression. 5 Indeed, it has been recently reported that peroxisome proliferator-activated receptor-γ coactivator-1d blocks extrasynaptic NMDA receptor activity and excitotoxicity in models of HD. 65
MULTIPLE TARGETS FOR GENE REPRESSION IN THE NUCLEUS
Transglutaminase 2 and Transcription Factors: Glutamine-Rich Activation Domains as Fertile Substrates for Transglutaminase-Mediated Posttranslational Modification
One of the mechanisms by which TG may induce gene repression involves direct modification of TFs. Transcription factors bind to specific DNA sequences and allow the transcription of the DNA in mRNA by promoting the recruitment of RNA polymerase to distinct regions of the chromatin. They usually contain a DNA binding domain that determine the selectivity of the TF for specific DNA sequences and a TF activation domain (TAD), which enables transcription in distinct regions of DNA. The TADs can present acidic domains, or glutamine-rich domains, or proline-rich domains. Since activation domains of many TFs, including prosurvival TFs such as Sp1, Sp3, and CREB are glutamine rich, and TG can add polyamines to glutamine residues, this model proposed that polyamination of glutamine-rich activation domains would inhibit interactions of TFs with coactivator proteins, necessary for the recruitment of the basal transcriptional machinery. Of note, among many other functions, Sp1 has been shown to mediate neuronal survival in response to oxidative stress 66 and CREB is a TF that has been implicated in cell survival, memory, and neuronal regeneration. Furthermore, the TATA binding protein TBP 67 and the coactivator, CREB Binding protein, CBP, 68 have long glutamine stretches that can be target of TG crosslinking and/or polyamination (Figure 4B-1). Another TFs presenting glutamine-rich domains are Oct1, Oct2, TAFII130, CBF-B and CBF-C, HAP1 and 2 and the multiprotein complex Mediator, which associates with general TFs and RNA Polymerase II to initiate transcription. Currently, the only empirical observations of a direct interactions between TG2 and TFs involve Sp1 that has been shown to be crosslinked in a model of alcoholic steatohepatitis; 69 ERK1, which has been detected in neuronal insoluble nuclear complexes with 14-3-3 and other components of the ERK pathway. 70 Finally, TG2 interacts with the HIF1β during in vitro ischemia, where TG2 limits HIF1α-mediated gene activation in a transamidating activity-independent way. 50 , 71 , 72 Better tools and a careful analysis of the nuclear ‘polyaminome’ will likely help defining novel candidates.
Targeting Chromatin to Mediate Gene Repression: Transglutaminase 2 and Histone Proteins
Other potential targets for TG in the nucleus after injury are histone proteins. Briefly, to fit the entire genome into the nucleus of a cell, DNA is compacted into higher order structures of which the basic unit is the nucleosome. The nucleosome is composed of DNA tightly wrapped around histone proteins. These histones are divided up into classical histones (H2A, H2B, H3, and H4), linker histones (H1), and variant histones (H2A.Z). The interaction of DNA (negatively charged) with histone proteins (positively charged) is regulated via posttranslational modifications at a series of highly conserved residues in the histone N-terminal tail. The best studied of these modifications is acetylation. Histone acetylation neutralizes positive charges and thus diminishes the interactions of core histone proteins with DNA, leading to increased transcription. The enzymes catalyzing this modification are histone acetyl transferases, the local ‘accelerators’ of gene expression. By contrast, HDACs are a family of enzymes involved in deacetylating histones and other proteins. Simply, they can be considered as local ‘brakes’ on gene expression. Inhibitors of HDACs have been widely studied in stroke models and appear to be protective in both pretreatment and posttreatment paradigms via their effects on salutary gene expression. 73 – 76 Among the genes implicated in the salutary effects of HDACi include the chaperone HSP 70, 77 , 78 the actin depolymerizing protein, gelsolin, 76 the tumor suppressor p21 waf1/cip1 75 and the glutamate transporter, Glt-1. 79 Interestingly, the expression level of class I histone proteins itself is modulated in a model of transient focal ischemia. 80 While HDACs 1, 2, and 3 expression is decreased in the ischemic core, the same proteins are induced in the penumbra, explaining the beneficial effects provided by the use of HDACi. Despite these exciting findings, it remains unclear how ischemic stress is transduced into changes in chromatin that lead ultimately to injury and lack of spontaneous repair. A possible model for transducing changes in calcium or oxidative stress into repression of gene expression invokes histone proteins as the major targets of TG2 crosslinking activity in the nucleus. Histone proteins have been shown to act as TG2 substrates in vitro by several groups. They have the propensity to crosslink 81 or to incorporate fluorescent or tagged polyamines at specific glutamines. 52 We have previously reported that in a model of HD, TG2 binds to histone H3, where it participates to transcriptional silencing of genes subserving many functions including mitochondrial biogenesis, chromatin structure, protein folding, and DNA repair. 5 Its selective inhibition was able to normalize the expression of 40% of the genes dysregulated by mutant huntingtin. To explain this profound effect, we hypothesize that TG2 and possibly other isoforms directly modify histones inducing chromatin repression. In this scheme, TGs add positively charged polyamines to histone tails thus increasing histone DNA electrostatic interaction and favoring a closed, repressed conformation of nucleosomes (Figure 4B-3).
Targeting Subnuclear Geography to Affect Gene Expression: Transglutaminase 2 and Actin
The cytoskeleton constitutes the framework of the cell on which many neuronal functions rely, including dendritic spine organization, intracellular transport of molecules from the nucleus to the axon, and growth cone formation. Actin, one of the major components of the cytoskeleton, has the ability to remodel its structure continuously by an ATP-dependent mechanism called treadmilling. Globular actin (G-actin), the free molecule, readily polymerizes into polarized where ATP-G-actin can bind from one end (the positive end) and ADP-G-actin is released by the other end (the negative end). This dynamic mechanism is enabled by an array of actin binding proteins, kinases, and phosphatases. Among them, the actin depolymerizing factor, cofilin, was shown to bind ADP actin and accelerate the filament severing and actin depolymerization at the negative pole. Cofilin activity is controlled by kinases able to inhibit its association with ADP actin and by phosphatases inducing its activation. During stress, a sharp decline in ATP concentration and the inactivation of the kinase-dependent cofilin phosphorylation leads to a rapid rise in the concentration of ADP actin and activated cofilin. This homeostatic pathway is engaged to halt ATP consumption by reducing ATP actin addition to actin polymer. When ATP levels are not promptly restored, cofilin and ADP actin bind together and form filaments that associate into cylindrical aggregates, called rods. Rods have been observed in the nucleus and cell body of neurons on oxidative stress, 82 glutamate excitotoxicity, 83 and ischemia, 84 and they slowly contribute to neuronal dysfunction and death. Interestingly, the formation of actin rods implies not only an alteration of the synapses but it also contributes to an impairment of gene transcription. It has been reported that both nuclear actin and cofilin are part of the preinitiation complex and are implicated in the transcription of most RNA polymerase II genes, 85 – 87 directly connecting actin filaments to transcriptional regulation. When the actin-cofilin equilibrium is perturbed by oxidative stress and excitotoxicity, transcription is affected leading to gene repression. One of the factors associated with nuclear stress is calcium release from the neighboring endoplasmic reticulum that can lead to the activation of the calcium-sensor proteins and TGs. Accordingly, nuclear rods have been observed in a cell model of HD downstream calcium dysregulation and upstream of transcriptional repression. In this scheme, calcium dysregulation leads to TG2 activation, crosslinking of cofilin and actin, and rod formation. 88 The findings suggest that TG2 could induce transcriptional repression associated with extrasynaptic glutamate signaling via effects on cofilin and actin, changes in subnuclear geography, and inhibition of RNA polymerase directly (Figure 4B-2).
THERAPIES AND STATE OF ART FOR INHIBITORS
Several low molecular weight inhibitors have been developed to counteract aberrant TG-transamidating activity in a host of disease states. The most widely used TG inhibitor in vivo is cystamine, a low molecular weight diamine that is protective in several rodent models of neurologic disease. 89 – 91 It is a reversible inhibitor, acting as a competitive amine with an IC50 in cells of 180 μmol/L 92 and it inhibits TG activity when delivered at a dose of 100 mg/kg dose in mice via intraperitoneal injection. 93 While the specificity of cystamine for TG2 has been questioned because of its ability to inhibit caspases, 24 , 94 numerous studies have shown that cystamine can influence activities via TG-specific actions. For example, cystamine can enhance message of a large number of transcripts, including DNAJb10, an HSP40 chaperone that is decreased in postmortem brain extracts from HD patients; 90 , 95 it can also posttranscriptionally activate Nrf2 in a 3-Nitropropionic Acid lesioning; and it can enhance trafficking of brain-derived neurotrophic factor. 95 Thus, while cystamine has many off target effects from TG2 inhibition, its established effects on TG2 inhibition likely contribute in important ways to its therapeutic effects on neurodegeneration. Additionally, cystamine partially protects from ischemic damage in the CA1 hippocampal region in gerbils 96 and it attenuates intracerebral hemorrhage-induced brain swelling and intracerebral hemorrhage-induced functional deficits. 37 It is currently in a Phase II clinical trial for HD (Raptor Pharmaceutical Corp., Novato, CA, USA). Interestingly, the administration of cystamine in combination with drugs able to modulate transcription, such as the Sp1 selective inhibitor, Mithramycin, or the HDACi SAHA (Suberoylanilidehydroxamic acid) showed a synergistic effect in extending mice life span by 40% 97 or ameliorating the eye morphology in a Drosophila model of HD. 98 The mechanisms involved in this synergism are still undefined.
In addition to cystamine, irreversible, specific TG2 inhibitors are a topic of active investigation.
99
Irreversible inhibitors or suicide inhibitors act by covalently modifying the cysteine in the catalytic group of TG2. Many possess a nucleophilic group able to form a stable chemical bond after reacting. Among them, dihydroisoxazoles have been shown to have low toxicity and good bioavailability
100
but no investigation has been performed in the CNS. Another class of inhibitors was developed using the TG2 substrate carbobenzyloxy-
CHALLENGES
Despite the fascinating role of the TG family of enzymes in neuronal injury and degeneration and the evolving roles of TG isoforms as transcriptional regulators, the mechanisms of action in the brain are only beginning to be fully understood. The reasons are protean, and reflect poor biochemical tools to follow TG levels; the absence of in situ biomarkers of local TG activity; and poor attention dedicated to the role of the other seven isoforms of the family in the brain. Another issue resides in the attempt to understand if TG2 has a prosurvival or proapoptotic role in the cell. It is possible that, as has been previously proposed, TG2 functions as a sensor of calcium increase and oxidative stress and it mediates prosurvival messages when its transamidating activity is silent, but it turns to a ‘natural born killer’ when the cell cannot homeostase to high levels of stress. 105 Furthermore, it has been suggested that TG2 is normally degraded by the proteasome at steady-state calcium levels; only in the presence of calcium increase, TG2 is stabilized and activated in its transamidating activity. To monitor TG2 stability in the cell, novel reporters for TG protein levels should be synthesized. Examples of sensitive reporters with a high dynamic range are the ones developed to monitor stability of the TFs HIF1α 106 or Nrf2. 107 In both cases, the enzyme luciferase has been fused to degrons that target these proteins for proteasomal degradation. When the TFs are stabilized intracellularly, it is possible to monitor their stabilization through luciferase activity. The detection is performed using a luminometer or the in vivo imaging system, an instrument that allows also in vivo detection of the luciferase activity and it would provide a way to monitor TG2 levels in cells undergoing different kinds of stress. Another crucial tool would be the one able to measure TG2 endogenous activity. A brilliant new system has been developed in the laboratory of Dr Truant, where TG2 has been fused to two different fluorescent proteins at its amino and carboxyterminal domains and, according to TG2 conformation, they will provide different fluorescent spectra intensity via FRET. When the two proteins are in proximity, it means that TG2 is in a close conformation, inactive and bound to GDP, and an increase in the FRET signal will be registered. Conversely, when TG2 is in an open conformation (active and bound to calcium), the detected signal will be significantly decreased. 102 The importance of the system resides in the ability to monitor the subcellular localization of TG2, its active or inactive state and can help in defining how inhibitors or specific cellular stresses change its conformation and how the change in conformation relates to TG2 toxicity in the cell. Based on this idea, it would be interesting to design constructs containing two known substrates for TG2 that when crosslinked by the transamidating activity, can provide a specific fluorescent spectrum to detect in live-cell imaging the timing and intensity of TG2 activation.
Another important challenge involves the development of new mouse models. The germline knockout for TG2 has been useful to define its initial contribution to HD but over time, compensation by other TG isoforms 108 has been shown, creating some suspect on the reliance of the results reported. Furthermore, the over-expressor mice provided results that are completely controversial with the once observed in the constitutive knockout. For instance, in an middle cerebral artery occlusion stroke model, the neuronal-specific overexpression of TG2 suggested a protective effect of this enzyme in the brain. 50 Interestingly, a similar global protection was observed in TG2 knockout animals. 109 It has been argued that this apparently mutually exclusive effect is caused by a protective role of TG2 in neurons and a detrimental role in astrocytes. Promoter-specific and inducible knock-out mice might clarify the role(s) of the TGs in neuronal injury and repair.
Finally, a novel and exciting discovery describes the ability of TG2 to crosslink neurotransmitters such serotonin to proteins. 110 More attention should be devoted in understanding how this may affect dynamics of neurotransmitter function when TG2 is activated in neurons. It would be important to understand how frequently TG2 uses neurotransmitters as substrates and what this modification induces in the target proteins. A similar enthusiasm was seen when polyamination was described. 13 Unfortunately, only few examples of targets have been discovered and adequate tools to study polyamination are still missing.
CONCLUSIONS AND FUTURE PERSPECTIVE
In conclusion, data from several laboratories indicate that TG transamidating activity is aberrantly upregulated after calcium dyshomeostasis and oxidative stress. In cellular and rodent models of stroke and neurodegenerative diseases, TG inhibition has been shown to be effective at halting cell death even when begun many hours after the onset of a death stimulus. The wide therapeutic window of TG inhibitors in vitro suggests it can be considered as a valid therapeutic strategy. Nevertheless, only one TG inhibitor available at present, cystamine and its reduced form cystamine, whose specificities have been questioned, can effectively cross the blood–brain barrier. As such, novel and more specific inhibitors are needed to prove the importance of this family of enzymes in ischemic damage. To develop better therapeutic strategies, a deeper understanding of physiologic and pathologic roles of TG transamidating functions, the role of TG-mediated posttranslational modifications, the degradation pathways controlling TGs levels in the CNS, and their targets in damaged neurons need to be investigated. Historically, TG's role in cell death has been primarily focused on its ability to crosslink proteins into aggregates, and to promote protein dyshomeostasis. A new era is emerging for TGs. In fact, converging lines of inquiry from several groups, including our own, suggest an important nuclear regulatory function. Future studies will determine whether the beneficial effects of TG inhibitors in stroke models accrues from derepression of adaptive transcriptional pathways via modulation of epigenetic proteins, the nuclear cytoskeleton, or TFs directly, and whether inhibition of TG can counteract some if not all of the deleterious effects of extrasynaptic glutamate signaling on neuronal survival and possibly repair.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Footnotes
ACKNOWLEDGEMENTS
A special thank to Dr Sama Sleiman, Dr Thong Ma, Dr Hossein Aleyasin, and Dr Saravanan Karuppagounder for the stimulating discussions on chromatin modifiers and therapeutic interventions. David Brand for his editorial help.