
Abstract
Corticotropin releasing hormone (CRH) and its family of related peptides are involved in regulating physiologic responses to multiple stressors, including stroke. Although CRH has been implicated in the exacerbation of injury after stroke, the mechanism remains unclear. After ischemia, both excitotoxic damage and inflammation contribute to the pathology of stroke. CRH is known to potentiate excitotoxic damage in the brain and has been shown to modulate inflammatory responses in the periphery. Here the present authors examine the relative contribution of the two known CRH receptors, CRH-R1 and CRH-R2, to ischemic injury using CRH receptor knockout mice. These results implicate CRH-R1 as the primary mediator of ischemic injury in this mouse model of stroke. In addition, the authors examine a potential role for CRH in inflammatory injury after stroke by identifying functional CRH receptors on astrocytes and microglia, which are cells that are known to be involved in brain inflammation. By single cell PCR, the authors show that microglia and astrocytes express mRNA for both CRH-R1 and CRH-R2. However, CRH-R1 is the primary mediator of cAMP accumulation in response to CRH peptides in these cells. The authors suggest that astrocytes and microglia are cellular targets of CRH, which could serve as a link between CRH and inflammatory responses in ischemic injury via CRH-R1.
Corticotropin releasing hormone (CRH) and urocortin (Ucn) are peptides that regulate physiologic responses to stress, including behavioral and immunologic challenges (Fisher, 1989;Owens and Nemeroff, 1991;Venihaki et al., 2001). The two known classes of mammalian CRH receptors, CRH-R1 and CRH-R2, are G-protein coupled and signal through multiple intracellular pathways (Chalmers et al., 1995;Grammatopoulos et al., 2001;Heldwein et al., 1996). CRH and Ucn both bind CRH-R1 and CRH-R2, although CRH-R2 binds Ucn with 40-fold greater affinity than CRH (Vaughan et al., 1995). Recently, two new members of the CRH family have been identified, Ucn II and Ucn III. Both bind selectively to CRH-R2 with high affinity and have distinct localizations in the brain and periphery (Lewis et al., 2001;Reyes et al., 2001).
CRH has been implicated in the pathogenesis of ischemic injury (Lyons et al., 1991;Strijbos et al., 1994;Yatsushiro et al., 1997). CRH mRNA is upregulated acutely after stroke (Choi et al., 2001;Wong et al., 1995), and administration of CRH receptor antagonists before and immediately after ischemia reduces ischemic and excitotoxic brain damage (Loddick et al., 1998;Lyons et al., 1991;Mackay et al., 2001;Strijbos et al., 1994). The mechanism of CRH exacerbated injury in ischemia remains unclear. CRH may modulate ischemic injury through excitotoxic or inflammatory pathways. CRH potentiates the excitatory effects of glutamate (Bishop and King, 1992) and mediates limbic seizures in the developing rat brain (Baram et al., 1997). In addition, blockade of CRH receptors with a nonselective receptor antagonist reduces neuronal damage associated with excitotoxic seizures in the hippocampus (Maecker et al., 1997).
CRH may also influence stroke damage via inflammatory pathways given its known role as a proinflammatory mediator in peripheral immune responses (reviewed in Baigent, 2001). CRH and Ucn are upregulated in inflamed tissue (Crofford et al., 1992, 1993;Hargreaves et al., 1989;Kageyama et al., 1999;Karalis et al., 1991;Kohno et al., 2001;Mastorakos et al., 1995), and both peptides increase inflammatory cytokine production (Agelaki et al., 2002;Kohno et al., 2001;Leu and Singh, 1992;Pereda et al., 1995;Schulte et al., 1994). It is well established that stroke damage is mediated, in part, through inflammation, with the release of cytokines and leukocyte recruitment (Barone and Feuerstein, 1999;Beamer et al., 1995;DeGraba, 1998;Stevens et al., 2002). Glial cells (astrocytes and microglia) are early mediators of the inflammatory response in the brain through the production of proinflammatory cytokines (Etienne-Manneville et al., 1999;Gregersen et al., 2000;Lee et al., 2000b;McMillian et al., 1994). Trauma, tissue injury, and disease in the CNS lead to microglial activation and production of TNFα and IL-1, which may play a role in neurotoxicity (Giulian et al., 1993). Astrocytes, activated during CNS inflammation, produce inflammatory cytokines (TNFα, IL-1, and IL-6) that aid microglia in the initiation of an immune response, which may, in turn, exacerbate neuronal death (Etienne-Manneville et al., 1999;Lee et al., 2000b). The fact that CRH/Ucn modulates inflammation in the periphery suggests that CRH receptor pathways may play a similar role in cerebral ischemic injury via actions of CRH on astrocytes or microglia. This idea is strengthened by the observation that CRH stimulates changes in intracellular Ca2+ in rat astrocytes (Kapcala and Weng, 1992;Takuma et al., 1994) and that CRH-R1 immunostaining localizes on astrocytes in mouse brains (Bishop et al., 2000). Thus, nonneuronal cells in the CNS may be important cellular targets of CRH in ischemia.
Here the authors examine the relative contributions of the two known CRH receptors, CRH-R1 and CRH-R2, to ischemic injury using mice deficient in one or the other of these CRH receptor subtypes. The authors also investigate whether murine astrocytes and microglia serve as potential targets of CRH actions in the mouse brain. These results implicate CRH-R1 as the primary mediator of the injurious effects of CRH in cerebral ischemia, which is consistent with pharmacologic studies using CRH-R1 specific antagonists (Loddick et al., 1998;Mackay et al., 2001;Yatsushiro et al., 1997). The authors also show that astrocytes and microglia express functional CRH receptors that, after activation with CRH or Ucn, show marked increases in intracellular levels of cAMP. These results suggest that these non-neuronal cells are potential cellular targets of CRH that may indeed play a role in the exacerbation of neuronal injury following stroke.
MATERIALS AND METHODS
Peptides
Rat urocortin and r/h CRH (Bachem, Torrace, CA, U.S.A.) were dissolved in water. The CRH receptor antagonist CP154,526 (provided by R. Mansbach, Pfizer Central Research, Groton, CT, U.S.A.) was dissolved in DMSO (final DMSO concentration for experiments was <0.1%). The CRH receptor antagonist αhelCRH9-41 (Peninsula Laboratories, San Carlos, CA, U.S.A.) was dissolved in NH4CO3 (0.2M). UcnII (a gift from R. Isfort, Proctor & Gamble, Cinnicinati, OH, U.S.A.) was dissolved in water.
Mice
Male C57Bl/6J mice were obtained from Jackson Laboratories. CRH-R1 deficient mice (Crhr1−/−) were obtained from Dr. Wolfgang Wurst, Max Planck Institute of Psychiatry, Munich, Germany (Timpl et al., 1998). Crhr1−/− mice used in these experiments were of mixed genetic background C57Bl6J/129Ola/CD-1. Mice lacking CRH-R2 receptor (Crhr2−/−) were generated and maintained as previously described (Coste et al., 2000). Crhr2−/− mice were backcrossed four generations onto C57Bl/6J. Littermates from each of the knockout lines were used as controls. All procedures met NIH guidelines with the approval of the Oregon Health & Science University Institutional Animal Care and Use Committee.
Focal ischemia
Cerebral focal ischemia was induced by middle cerebral artery occlusion (MCAO) (Clark et al., 1997) in adult Crhr1−/−, Crhr2−/− and littermate controls. Mice were anesthetized by halothane inhalation (4%/L O2) and maintained with 1.5%/L O2. The middle cerebral artery (MCA) was blocked by threading silicone-coated 7–0 monofilament nylon surgical sutures through the external carotid to the internal carotid and finally blocking the bifurcation into the MCA and anterior cerebral artery. The filament was maintained intraluminally for 60 minutes while the mice were under anesthesia. The filament was then removed, thereby restoring blood flow. Rectal temperature was monitored in each mouse during surgery to prevent artifacts induced by body temperature fluctuations. Cerebral blood flow (CBF) was monitored throughout the surgery by laser doppler flowmetry (Periflow 5000; Perimed, Sweden). Animals were killed 48 hours postischemia. Brains were removed, and a 1-mm section was taken at bregma for infarct measurements. To visualize the region of infarction, sections were placed in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in 0.9% phosphate-buffered saline and stained at 37°C for 30 minutes (Bederson et al., 1986). After staining, the sections were transferred to 4% paraformaldehyde. The percent of infarct was determined from the slice by comparing the area of infarct versus the total area of the ipsilateral hemisphere (Infarct Area/Ipsilateral Hemisphere Area)%. The present authors have shown previously that the infarct area of a coronal section taken near bregma correlates highly (r2 = 0.96) with the volume of infarct in this model (Hill et al., 1999).
Astrocyte and microglia isolation
Cells were isolated from 2 to 4 day old C57BL/6J, Crhr1−/−, and Crhr2−/− pups by a procedure modified from Giulan et al. (Giulian and Baker, 1986). Briefly, the cortices were separated from the meninges, triturated, trypsinized, passed through a 130 μm filter, and plated at approximately 10 cortices/75cm2 flask in DMEM with 5% FBS. Medium was exchanged 4 days later and then every 3 days for a total of 12 to 14 days. To obtain mixed glia cultures, flasks were trypsinized and replated for subsequent assays and used 48 to 72 hours later, depending upon the assay. To obtain cultures enriched for astrocytes or microglia, flasks were shaken overnight at 250 rpm to separate astrocytes and microglia. The nonadherent microglia were removed and were replated for 1 hour followed by manual agitation and a single wash to remove nonadherent cells. For the adherent microglia, the medium was exchanged every 3 days with 50% DMEM medium and 50% astrocyte conditioned medium and used 7 days later. The remaining astrocytes were trypsinized and replated for subsequent assays. Cultures were stained with antibodies to GFAP and CD11b (as described below) to determine the degree of enrichment; the cultures routinely contained greater than 95% of the enriched cell population (astrocyte or microglia).
Immunostaining
Enrichment analysis
Astrocytes and microglia were cultured in 4-well chamber slides (VWR, Brisbane, CA, U.S.A.) for 3 to 4 days and were approximately 70% confluent at time of staining. Cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature. Slides were then washed three times in phosphate buffered saline (PBS). Slides were stained overnight at 4°C with the primary antibody rabbit anti-mouse GFAP (Sigma, St. Louis, MO, U.S.A.) or with rat anti-mouse CD11b (PharMingen, San Diego, CA, U.S.A.). A biotinylated secondary antibody (goat anti-rabbit) was used to detect GFAP immunostaining, and a goat anti-rat antibody coupled to alkaline phosphatase was used to detect CD11b using a commercially available staining kit (Vector Laboratories, Burlingame, CA, U.S.A.). Cells were then analyzed under a microscope to determine the extent of enrichment.
Colocalization analysis
Mixed glial cells were cultured at low density in 4-well chamber slides for 2 days. Cells were fixed in methanol for 5 minutes at −10°C. Slides were washed as described above. Cells were incubated at 4°C overnight with the following: rabbit anti-mouse GFAP, rat anti-mouse CD11b (Pharmigen), and goat anti-CRH-R1 anti-sera (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), which reacts with the carboxy terminus of mouse CRH-R1 and CRH-R2 receptors. Goat anti-rabbit-Texas Red (Molecular Probes, Eugene, OR, U.S.A.) was used to detect GFAP staining; goat anti-rat-AP (Sigma) with the Vecta Red-AP substrate kit (Vector Laboratories) was used to detect the CD11b; and donkey antigoat-biotin (Santa Cruz Biotechnology) with avidin-FITC (PharMingen) was used to detect CRH-R1. Specificity of the CRH-R1 anti-sera was tested using a CRH-R1 control peptide (10-fold excess) (Santa Cruz Biotechnology) before incubation with cells. Slides were washed and coverslipped in Slow Fade (Molecular Probes). Images were acquired using a Bio-Rad MRC 1024 ES laser scanning confocal imaging system attached to an inverted Nikon Eclipse TE300 microscope. The acquisition system uses a krypton/argon laser with excitation lines at 488, 568, and 647 nm, and sequential detection using two 8-bit photomultiplier tubes (PMTs). Images were processed using the Lasersharp post-processing software (Biorad, Hercules, CA, U.S.A.).
CRH receptor stimulation and cAMP assay
Mixed glia and astrocytes were cultured in 12-well tissue culture plates for 3 to 4 days and were approximately 90% confluent at time of assay. Microglia were cultured in 24-well tissue culture plates for 7 days with two media changes (50% DMEM medium and 50% astrocyte conditioned medium) and were approximately 60% confluent at time of assay. Cells were preincubated for 90 minutes in serum-free DMEM. After this time, the medium was replaced with serum-free DMEM containing 1 mM 3-isobutyl-1-methyxanthine (IBMX; Sigma) to inhibit cAMP phosphodiesterase activity (Beavo et al., 1970). After 30 minutes, medium was replaced with fresh DMEM plus 1 mM IBMX containing appropriate concentrations of CRH, Ucn (Bachem, Torrance, CA, U.S.A.), or UcnII (a gift from R. Isfort, Proctor & Gamble, Cinnicinati OH, U.S.A.). After appropriate incubation times (5 minutes for astrocytes; 10 minutes for mixed glia and microglia) at 37°C, the medium was removed and cells were lysed overnight at −20°C in extraction medium (95% ethanol, 20 mM HCl). Samples were dried under vacuum, and cAMP was measured using a commercially available RIA kit (Biomedical Technologies, Stoughton, MA, U.S.A.). Inhibition experiments were performed in the previously mentioned medium with 10 nM Ucn and the CRH receptor antagonists αhelCRH9-41 or CP154,526.
Single cell RT-PCR
Mixed glial populations were plated in Terasaki plates (VWR) at dilutions that yield a single cell per well. Plates were then inverted to allow the cell(s) to collect in the apex of the hanging droplet of media. Confirmation of single cells was determined by visualization 2 hours after plates were inverted. Single cells were collected and pelleted in a 0.65-ml eppendorf tube. Single cells were suspended in 15 μl reverse transcriptase buffer (Life Technologies, Rockville, MD, U.S.A.) and stored at −80°C until they were used. Single cell samples were treated with DNase twice to remove DNA. To verify successful removal of amplifiable DNA, each sample was tested in the presence and absence of reverse transcriptase. Reverse transcription was performed at 42°C for 1 hour in the presence of 2 μl 0.1M DTT, 1 μl 10 mM dNTPs (Perkin Elmer, Branchburg, NJ, U.S.A.), 1 μl random hexamers (Pharmacia, Piscatatway, NJ, U.S.A.), 1 μl RNasin (Promega, Madison, WI, U.S.A.), and 1 μl Superscript reverse transcriptase II (Life Technologies). Reactions were stopped by heating at 90°C for 5 minutes and were stored at −20°C until PCR amplification. PCR reactions contained 1× PCR buffer (Perkin Elmer), 200 μM dNTPs, 1 mM MgCl2, 2.5 U Taq (Perkin Elmer), 400 nM antisense, and 400 nM sense primers. An initial denaturing of 5 minutes at 94°C was followed by 45 cycles of 1 minute 94°C, 1 minute 56°C, and 2 minutes 72°C, with a final elongation of 10 minutes 72°C. The quality of the cDNA samples was tested using primers for the housekeeping gene, L3 (Peckham et al., 1989). Samples containing the 150 bp band for L3 were used in further studies. In all other amplifications, a nested primer approach was performed using the PCR conditions described previously. External primer pairs were used for the initial round of PCR, resulting in fragment sizes as follows: GFAP 206 bp, CRH-R1 157 bp, and CRH-R2 190 bp. A second round of amplification was performed using 1 μl of first-round amplification products in the presence of internal primers. The resultant band sizes were as follows: GFAP 72 bp, CRH-R1 60 bp, and CRH-R2 68 bp. RNA from mouse NIH 3T3 cells was used as a negative control for the presence of CRH receptor expression.
Primers
L3 housekeeping gene: sense (5′-tgccaggtcatccgcatcattg-3′), antisense (5′-tgacatcaatcatctcatcctgcc-3′) GFAP external: sense (5′-accaaactggctgatgtctacc-3′), antisense (5′-tcatgtgcctcctgtctatacg-3′) GFAP internal: sense (5′-ggagagggacaactttgcac-3′), antisense (5′-cagcctcaggttggtttcat-3′) CRH-R1 external: sense (5′-atcctcatgaccaaactccg-3′), antisense (5′-tgaagacaaccctggagacc-3′) CRH-R1 internal: sense (5′-tacaggaaggctgtgaaggc-3′), antisense (5′-acatgtaggtgatgcccagg-3′) CRH-R2 external: sense (5′-ctacacctactgcaacacgacc-3′), antisense (5′-ttcgcagtgtgagtagttgacc-3′) CRH-R2 internal: sense (5′-acccggagccctagtagaga-3′), antisense (5′-ttccgggtcgtgttgtactt-3′)
Statistics
Results are presented as the mean ± standard deviation. Statistical comparisons were made using Student's t-test. Mean differences were considered statistically significant when P < 0.05.
RESULTS
Crhr1−/− mice but not Crhr2−/− mice show reduced infarcts
To test the relative role of CRH-R1 and CRH-R2 in ischemic injury, the present authors performed MCAO in mice with a targeted mutation in the CRH-R1 (Crhr1−/−) or CRH-R2 (Crhr2−/−) gene. Crhr1−/− mice that were subjected to 60 minutes of MCAO showed a significant reduction in infarct size compared with wildtype littermates (24.5 ± 10% vs. 52.2 ± 15%; P < 0.005) (Fig. 1). Crhr2−/− mice, however, did not show a difference when compared with wildtype littermates (48.2 ± 18% compared with 57.6 ± 14%) (Fig. 1). These results indicate a primary role for CRH-R1 in mediating ischemic damage in stroke and suggest that CRH-R2 is unlikely to exacerbate such injury.
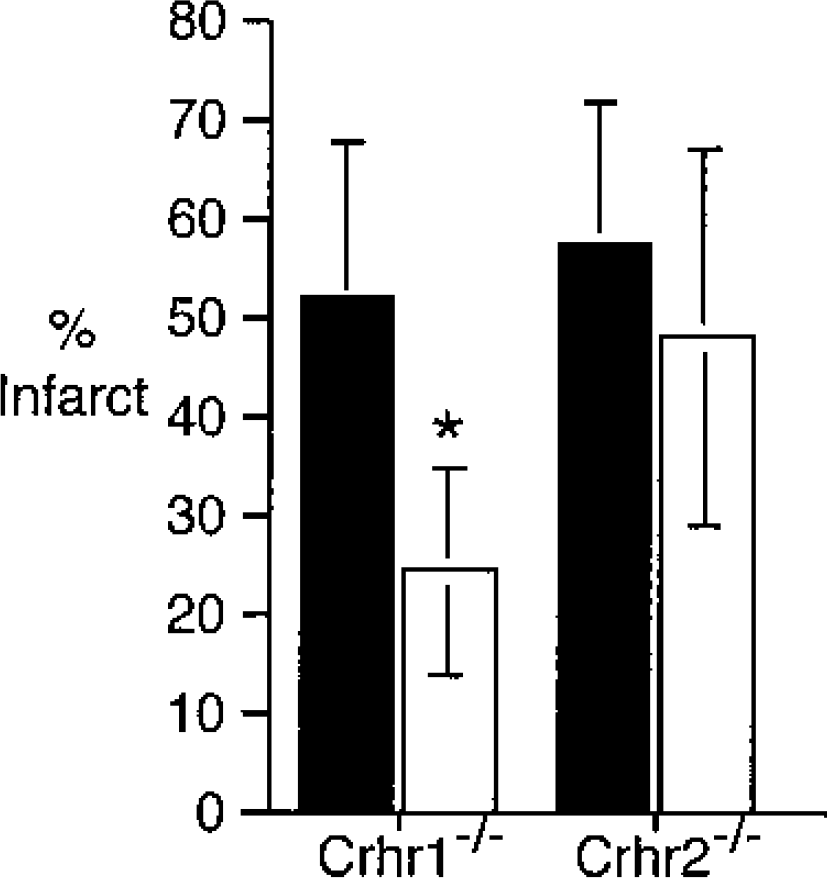
CRH-R1 but not CRH-R2 knockout mice show decreased infarct size. Crhr1−/− (white bars; n = 7), Crhr1−/− wildtype littermates (black bars; n = 7), Crhr2−/− (white bars; n = 9), and Crhr2−/− wildtype littermates (black bars; n = 7) were subjected to 60 minutes MCAO. Animals were killed 48 hours later, and percent infarct was determined by TTC staining and digital imaging. Percent infarct was calculated as described in Materials and Methods based upon the area measurements of one slice. Error bars represent SD. ∗ P < 0.005 receptor knockouts versus wildtype littermates using Student's t-test. CRH, corticotropin releasing hormone; MCAO, middle cerebral artery occlusion; SD, standard deviation.
CRH receptor expression in mouse glial cells
To explore the possibility that glial cells may be cellular targets of CRH actions in ischemia, the authors tested whether CRH receptors are expressed on mouse astrocytes or microglia. The authors stained cultured mouse astrocytes and microglia with an antibody that recognizes both CRH-R1 and CRH-R2. Astrocytes were distinguished from microglia based upon staining with the astrocyte-specific marker, GFAP. Microglia were identified with the microglial marker CD11b. Fig. 2A and Fig. 2D show positive staining (red) of astrocytes and microglia, respectively. These cells were tested for the presence of CRH receptors using a CRH-R1/CRH-R2 antibody. Strong staining is seen with this antibody (green; Fig. 2B and Fig. 2E). The specificity of the staining was confirmed using a 10-fold excess of CRH-R1 control peptide to block staining (data not shown). Fig. 2C and Fig. 2F show colocalization of the receptor staining with the cell specific markers (merge shown in yellow). These results indicate that CRH receptors are expressed on both mouse astrocytes and microglia. As a negative control, the authors tested the mouse fibroblast cell line NIH 3T3 for CRH receptor staining and found no such staining (data not shown).
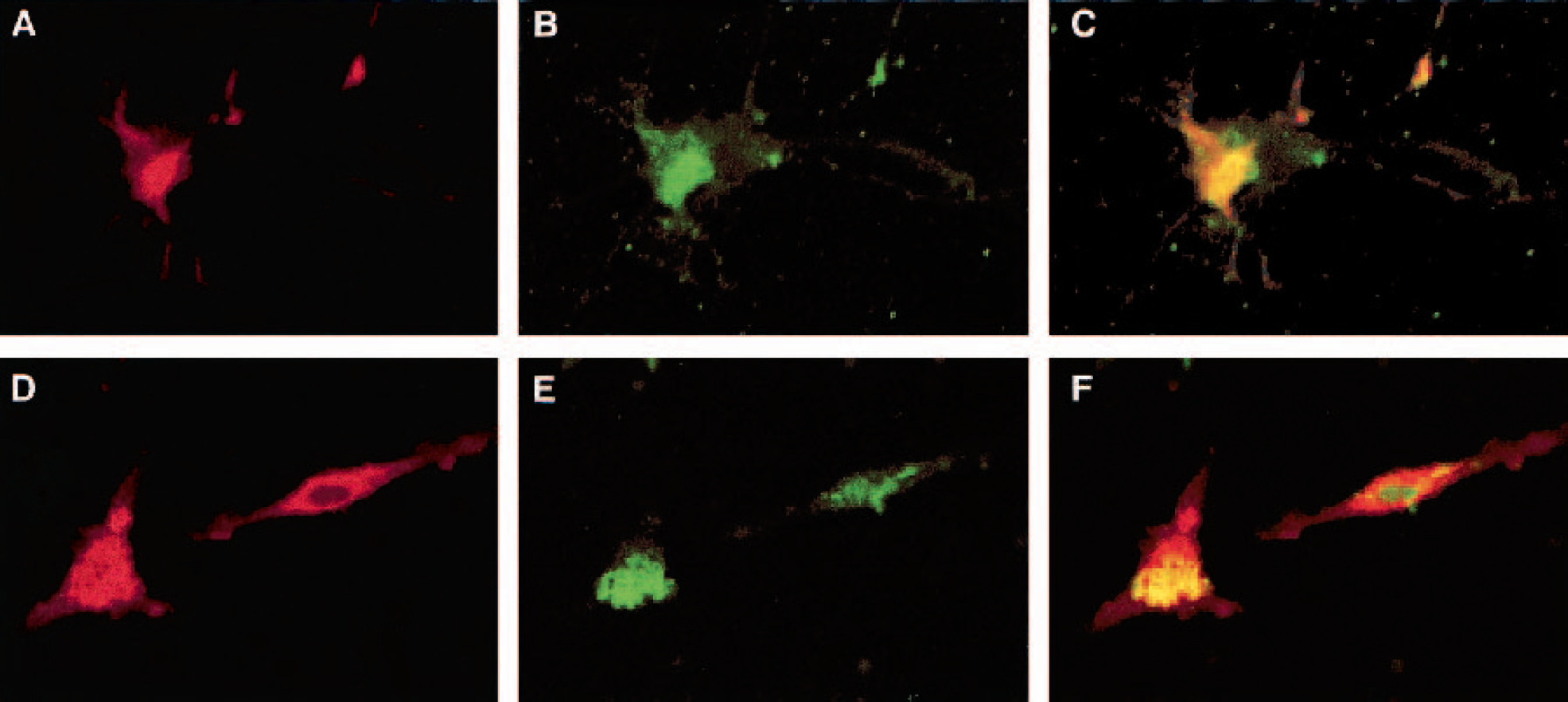
Colocalization of CRH receptors with GFAP and CD11b in vitro.
mRNA expression of both CRH-R1 and CRH-R2 in mouse astrocytes and microglia
The authors next sought the identity of CRH receptors expressed by astrocytes and microglia using single cell RT-PCR for CRH-R1 and CRH-R2 from mRNA obtained from each of the cell types. The authors used a nested primer approach to visualize gene products on an ethidium bromide gel. Single cells were obtained from mixed glial populations, as described in Materials and Methods. To distinguish astrocytes from microglia, primers for the astrocyte-specific marker GFAP were used. Figure 3 (left panel) shows expected band sizes for CRH-R1 (60 bp) and for CRH-R2 (68 bp), as well as a band corresponding to GFAP (72 bp) that identifies this cell as an astrocyte. In Figure 3 (middle panel), mRNA for both receptors is also seen in microglia, which are cells that do not express GFAP. Again, the negative control NIH 3T3 cDNA was used. Figure 3 (right panel) shows amplification of the housekeeping gene L3 (150 bp) but no amplification of CRH-R1 or CRH-R2 in the NIH 3T3 fibroblast cell line. All band identities were confirmed by sequence analysis (data not shown). Thus these data indicate that messages for both forms of CRH receptors are expressed in microglia and astrocytes.
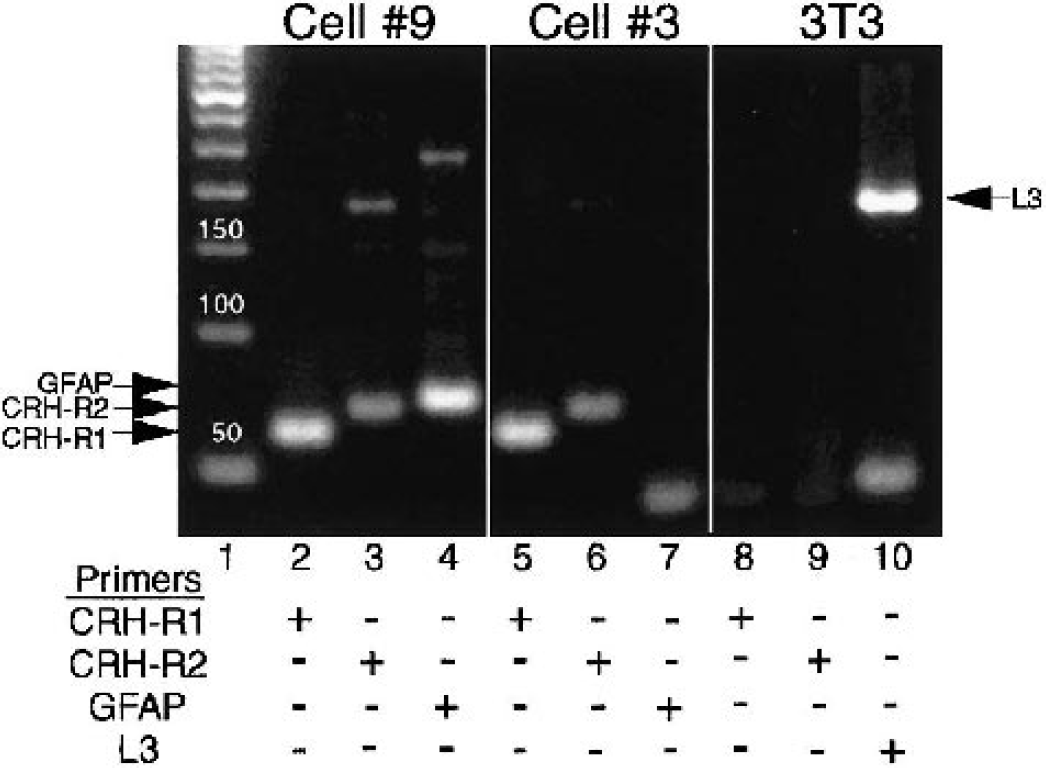
Nested RT-PCR showing amplified product for both CRH-R1 and CRH-R2 in individual cells either positive or negative for GFAP. Lane 1, 50 bp ladder. Lanes 2 and 5, positive for R1 product; 60 bp. Lanes 3 and 6, positive for R2 product; 68 bp. Lane 4, positive for GFAP product; 72 bp. 3T3 cells were included as a negative control. Lanes 8 and 9, negative for R1 and R2, respectively. Lane 10, positive for L3 housekeeping gene; 190 bp. Upper bands in lanes 3 and 4 are product from initial PCR. Lower bands in lanes 7 and 10 are bands resulting from primer dimer formation. CRH, corticotropin releasing hormone.
Evidence for CRH-R1 coupling to cAMP in mouse astrocytes and microglia
The authors stimulated neonatal cultures of mixed glia with various doses (10−11–10−6 M) of CRH and Ucn to test whether CRH receptors expressed on astrocytes and microglia are functionally coupled to adenylate cyclase. Stimulation with either peptide caused a robust dose-dependent accumulation of cAMP within 10 minutes (Fig. 4A). CRH and Ucn also induced a rapid dose-dependent increase in cAMP in cultures that were enriched for astrocytes (Fig. 4B) and microglia (Fig. 4C).
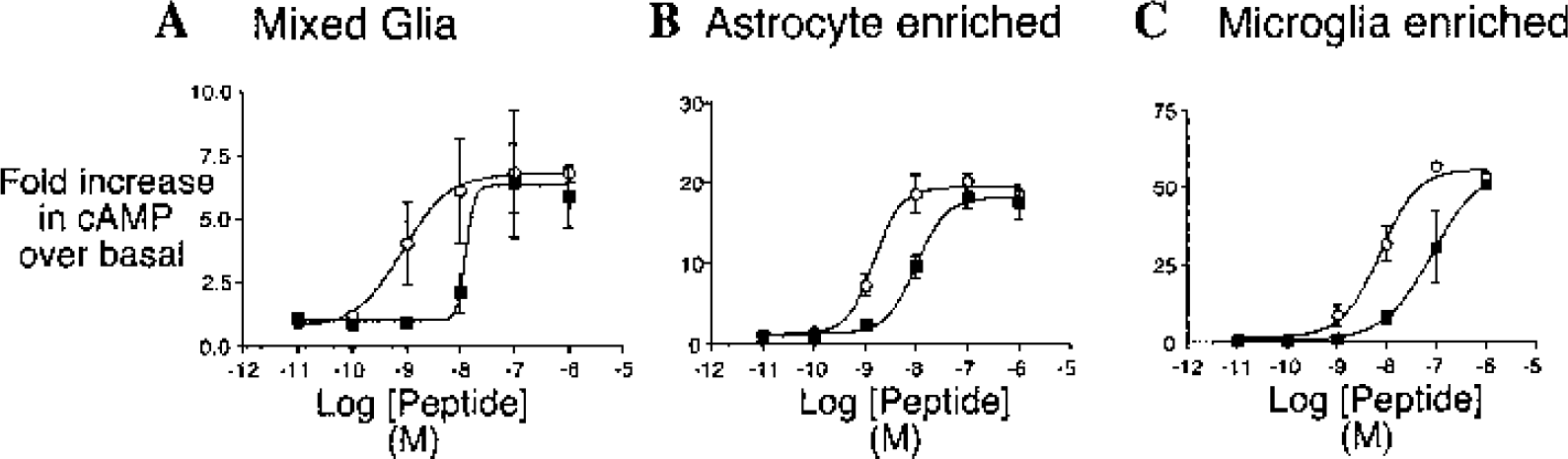
Dose-dependent accumulation of cAMP in glial cultures after stimulation with CRH and Ucn.
The authors used αhelCRH9-41, an antagonist that blocks both receptors, as well as the selective CRH-R1 antagonist CP154,526 to determine whether one or both receptors were responsible for CRH/Ucn-induced cAMP production. Cultures were incubated with 10 nM Ucn in the presence of 5 μM αhelCRH9-41or 5 μM CP154,526. Ucn-induced cAMP accumulation was inhibited by both antagonists to near-basal levels in mixed glia, astrocyte, and microglia cultures (Fig. 5A). In addition, Ucn II, a peptide specific for CRH-R2, did not increase cAMP production in astrocytes (Fig. 5B), further supporting CRH-R1 signaling as the primary route of cAMP induction. Further, the authors examined the effect of Ucn stimulation upon cAMP accumulation in astrocytes from Crhr1−/− and Crhr2−/− mice. As in cells from wildtype mice, Ucn induced a significant cAMP response in Crhr2−/− astrocytes (black bars; Fig. 5C), which was inhibited with CP154,526 (shaded bars). Crhr1−/− astrocytes showed no cAMP accumulation with Ucn treatment. Stimulation with forskolin, a nonreceptor agonist of cAMP production, induced robust increases in cAMP (open bars; Fig. 5C), demonstrating cAMP induction responses in these cells. These results demonstrate that functional coupling to adenylate cyclase in these cells requires CRH-R1.
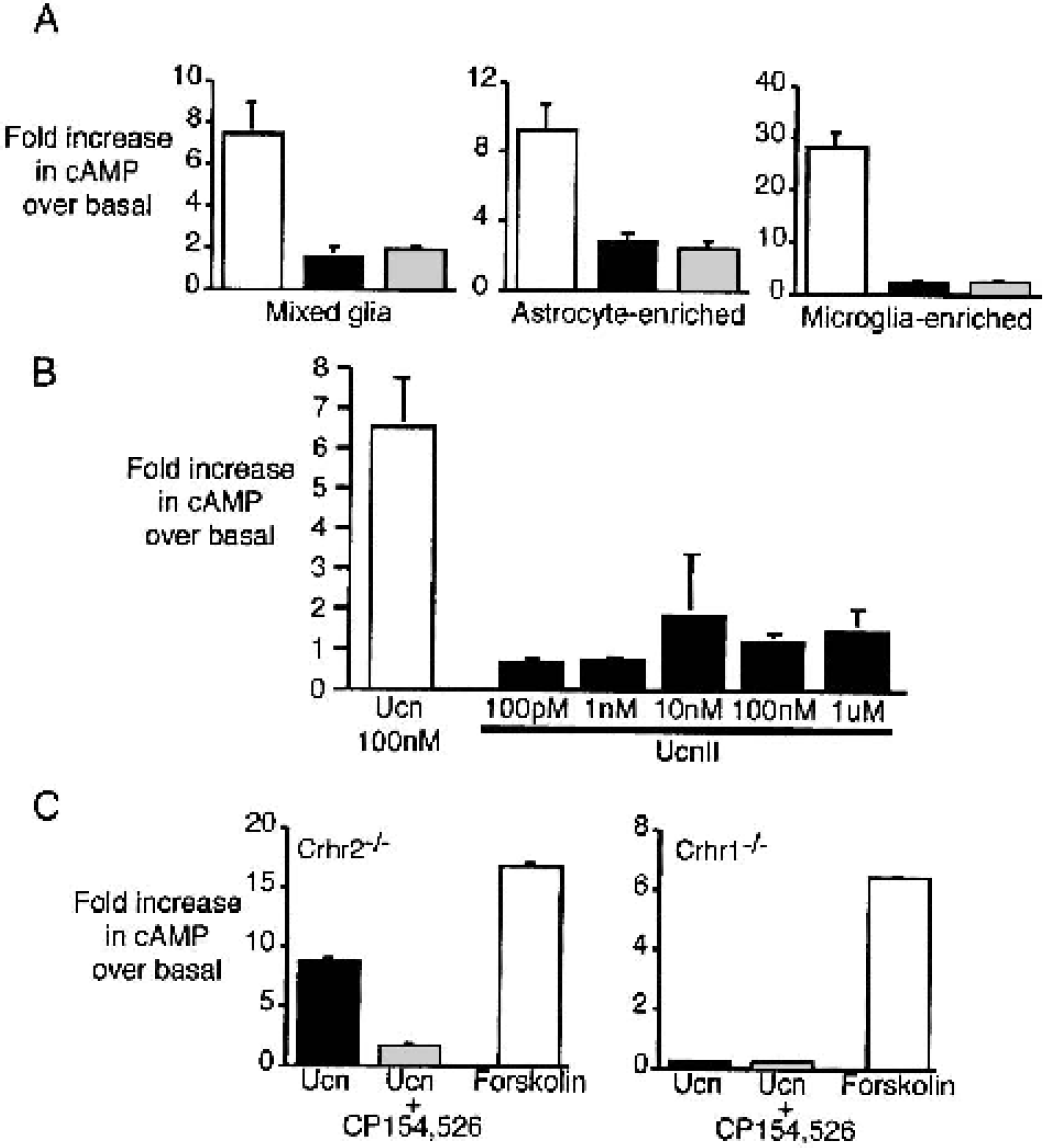
CRH-R1 mediated cAMP accumulation in glial cultures.
DISCUSSION
The authors demonstrate that CRH-mediated exacerbation of neuronal damage after cerebral ischemia is primarily via CRH-R1. Mice that lack CRH-R1 have a significantly reduced infarct size compared with wildtype mice. In contrast, CRH-R2 deficient mice show normal damage after ischemia. Thus CRH-R1 activation is dominant in mediating the deleterious effects of CRH/Ucn in ischemic injury, whereas CRH-R2 plays no significant injurious role in this process. These findings are consistent with rat models of ischemia, where CRH-R1 antagonists inhibited ischemic damage (Loddick et al., 1998;Mackay et al., 2001). The mechanism of CRH action during stroke is still unclear; however, the route of damage is most likely a result of direct actions of CRH in the cerebral cortex and not the result of increased glucocorticoids, since Strijbos et al. (1994) demonstrated that the glucocorticoid antagonist RU486 does not alter ischemic damage.
Neuronal damage from ischemia has been associated with direct excitotoxic effects as well as via inflammatory mediators (Barone and Feuerstein, 1999;Lee et al., 2000a). CRH may be involved in either mechanism of injury; it has been shown to be an exctiotoxic agent (Baram and Hatalski, 1998;Bishop and King, 1992;Maecker et al., 1997) as well as a mediator of inflammation (reviewed in Baigent, 2001). In the present study, the authors examined astrocytes and microglia as potential targets of CRH in the ischemic brain. The authors demonstrate that both astrocytes and microglia express CRH receptors by immunostaining and single cell PCR. Moreover, the authors report that CRH-R1 is functionally coupled to increases in cAMP after CRH/Ucn stimulation in both cell types. The authors' single cell PCR analysis revealed mRNA for CRH-R1 and CRH-R2 expression in both cell types, although the functional analysis using the CRH-R1 specific antagonist showed complete inhibition of the cAMP accumulation induced by CRH and Ucn, indicating activation of cAMP through CRH-R1. Further, Ucn failed to stimulate cAMP accumulation by CRH-R1 deficient astrocytes but induced a robust response in wildtype and CRH-R2 deficient glia. In addition, the CRH-R2 specific peptide Ucn II failed to stimulate cAMP accumulation in wildtype glial cultures. CRH-R2 transfected HEK293 cells were used as a positive control for Ucn II activity (data not shown). In other cell types, CRH-R2 can signal independent of cAMP (Karteris et al., 2000;Kiang, 1994;Kiang 1997); therefore, activation of CRH-R2 in glial cells may be coupled to other second messenger systems. However, because the authors' in vivo studies implicate only CRH-R1 as the mediator of stroke damage, the authors do not view CRH-R2 activation as a significant modulator in stroke outcome.
Both astrocytes and microglia become activated during CNS inflammation and produce proinflammatory cytokines (TNFα, IL-1, and IL-6), which initiate an immune response and exacerbate neuronal damage (Etienne-Manneville et al., 1999;Giulian et al., 1993;Lee et al., 2000b;McMillian et al., 1994). Although the present authors are unaware of studies demonstrating a direct inflammatory effect of CRH upon glial cells in the CNS, a number of studies indicate that CRH-related peptides regulate cytokine production from inflammatory cells in the periphery (Agelaki et al., 2002;Kohno et al., 2001;Leu and Singh, 1992;Pereda et al., 1995;Schulte et al., 1994;Venihaki et al., 2001). For example, CRH has been shown to augment the production of IL-1, TNFα and IL-6 in macrophages during endotoxin challenge both in vitro and in vivo (Agelaki et al., 2002). Microglia are considered to share a common lineage with monocytes; thus it is possible that CRH/Ucn modulate cytokine production in these cells during inflammation. Additionally, systemic immunoneutralization of CRH suppresses infiltration of inflammatory cells in an experimental model of aseptic inflammation (Karalis et al., 1991), and inhibition of cellular infiltration into the brain has been shown to reduce ischemic damage in several animal models (Clark et al., 1995;Grogaard et al., 1989). Thus, the present authors speculate that CRH may recruit inflammatory cells from the periphery into the brain and enhance the production of inflammatory cytokines.
A proposed role for CRH/Ucn in modulating inflammatory processes in ischemia does not preclude mediation of damage caused by excitoxicity, particularly in light of the fact that both CRH-R1 and R2 are expressed on neuronal cells (Chen et al., 2000;Lovenberg et al., 1995). Indeed, multiple studies have examined the role of CRH/Ucn in excitotoxic damage (Baram and Hatalski, 1998;Baram and Ribak, 1995;Maecker et al., 1997;Strijbos et al., 1994), and a neuroprotective effect of CRH receptor antagonists has been shown in a model of limbic seizures in the developing rat brain and in the hippocampus during kainate-induced seizures (Baram et al., 1997;Baram and Hatalski, 1998;Maecker et al., 1997). In addition, Strijbos et al. (1994) reported inhibition of NMDA-induced neuronal damage, using a CRH receptor antagonist.
In contrast to these latter studies that support an injurious role for CRH, several recent reports indicate a potential protective role for CRH and Ucn in excitotoxic and ischemic damage (Brar et al., 2000;Brar et al., 1999;Lezoualc'h et al., 2000;Pedersen et al., 2001, 2002). In addition, it is worth noting that work performed by Craighead and colleagues (2000) does not support a protective or injurious role; their studies show no effect of CRH on neuronal cell death directly or in combination with NMDA. These discrepancies may be caused, in part, by the fact that CRH receptors are now known to couple to several distinct second messenger systems. Recently, Grammatopoulos et al. (2001) reported that CRH receptors were capable of activating at least five different G-proteins. The authors speculate that preferential activation of specific G-proteins varies with the physiologic condition of the cell. This suggests a tightly regulated response that may depend upon various factors, such as peptide avidity, receptor density, availability of signaling molecules, and posttranslational receptor modification. For instance, Ucn protects hippocampal neurons from excitotoxic death upon signaling through two G proteins (Gsα and Gq/11). Inhibition of one or the other G proteins fails to provide protection (Pedersen et al., 2002). Thus neuroprotection versus neurotoxicity may depend upon the state of activation or differentiation of an individual cell. In addition, with at least four peptide family members in the mammalian CRH family (CRH, Ucn, Ucn II, and Ucn III), the varying affinity of receptors for each peptide may trigger different cascades of signaling. The controversy between a protective or damaging role for CRH may be reconciled as more is understood about the signaling events following CRH receptor activation.
In conclusion, the authors demonstrate that in the mouse, CRH-R1 appears to be the primary CRH receptor through which CRH exerts an injurious role in cerebral ischemia. The authors suggest that astrocytes and microglia may act as cellular targets of CRH and propose that these cells could serve as a link between CRH and inflammatory responses in ischemic injury via CRH-R1. Understanding the inflammatory component in CRH related stroke damage may lead to new targets of therapeutic drug design.
Footnotes
Acknowledgments
The authors thank Aurelie Snyder for her assistance with the confocal imaging.