
Abstract
Adult hippocampal neurogenesis is important for learning and memory, especially after a brain injury such as ischemia. Newborn hippocampal neurons contribute to memory performance by establishing functional synapses with target cells. This study demonstrated that the maturation of hippocampal neurons is enhanced by postischemia intermittent hypoxia (IH) intervention. The effects of IH intervention in cultured neurons were mediated by increased synaptogenesis, which was primarily regulated by brain-derived neurotrophic factor (BDNF)/PI3K/AKT. Hippocampal neo-neurons expressed BDNF and exhibited enhanced presynaptic function as indicated by increases in the pSynapsin expression, synaptophysin intensity, and postsynapse density following IH intervention after ischemia. Postischemia IH-induced hippocampal neo-neurons were affected by presynaptic activity, which reflected the dynamic plasticity of the glutamatergic receptors. These alterations were also associated with the alleviation of ischemia-induced long-term memory impairment. Our results suggest that postischemia IH intervention rescued ischemia-induced spatial learning and memory impairment by inducing hippocampal neurogenesis and functional synaptogenesis via BDNF expression.
INTRODUCTION
Neurogenesis refers to the birth of new neurons through proliferation, migration, differentiation, survival, and functional integration, particularly in the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus (DG) of the hippocampus. 1 The genetic ablation of neo-neurons in the hippocampus is associated with learning and memory impairment and the induction of neo-neuron proliferation in the hippocampus alleviates such lesion-induced impairment.2,3 The production and differentiation of hippocampal neo-neurons generates complex synapses that deeply integrate into the existing hippocampal circuit.1,4 Therefore, neo-neurons play an essential role in hippocampus-related behaviors.
Brain-derived neurotrophic factor (BDNF) is an important neurotrophin that is widely expressed in the hippocampus, and BDNF may be necessary for brain function. For example, BDNF-mutant mice exhibit learning deficits and BDNF expression is rapidly induced in the rat hippocampus after learning.5,6 Longterm potentiation (LTP), which is a memory formation mechanism, may enhance presynaptic function and remodel postsynaptic plasticity through the stabilization of existing synapses, the formation of new synapses, or the induction of receptor trafficking or diffusion. Brain-derived neurotrophic factor regulates the expression of PKA and CREB, which are required for LTP. 7 Brain-derived neurotrophic factor also regulates synaptic function by stabilizing and maintaining the structural complexity of synapses. 8 Neurogenesis is also regulated by BDNF. 9
Brain ischemia causes neurological disturbances, particularly with regard to memory. 3 Brain-derived neurotrophic factor expression is markedly reduced following chronic ischemia in rats and a rapid loss of synapse formation and accompanying impairments in learning and memory have been observed in ischemia models.10–12 We previously reported that an intermittent hypoxia (IH) intervention following stroke rescued ischemiainduced spatial learning and memory impairments, likely through the induction of hippocampal neurogenesis. 3 The present study investigated whether postischemia IH intervention altered the function of hippocampal neo-neurons in learning and memory.
MATERIALS AND METHODS
Animals
Adult (8-week-old) male Sprague-Dawley (SD) rats were used. The rats were housed in pairs in a temperature-controlled (22°C ± 1°C) vivarium on a 12-hour light:dark cycle (lights on at 0700 hours) with free access to food and water. The Institutional Animal Care and Use Committee of the National Yang-Ming University approved all of the experimental protocols.
Primary Neuron Culture
Whole brains were dissected from postnatal day 0 (P0) SD pups. The tissue was diced into small pieces and digested in a papain solution containing 1% 50 mM EDTA, 150 mM CaCl2, 10% cysteine (2 mg/mL), and 2% papain (510 units/mL) for 20 minutes followed by DNaseI treatment for 5 minutes at 37°C. The solution was centrifuged, and the pellets were diluted in Dulbecco's modified Eagle's medium supplemented with 10% horse serum and 1% penicillin–streptomycin (PS). The solution was plated in 60-mm tissue culture dishes or 12-well plates. The medium was replaced with NeuroBasal medium containing 2% B27 supplement, 1% PS, 50 mM glutamine, and 2.5 mM glutamate (NB/B27) on the following day. Ara-c (5 μM) was added 2 days later to reduce the overgrowth of glial cells. The medium was replaced with NB/B27 medium without glutamate after an additional 2 days. Half of the medium was replaced every 3 days for 2 weeks.
Focal Cerebral Ischemia
Right middle cerebral artery occlusion (MCAO) was induced using standard microsurgical techniques. The right middle cerebral artery trunk was ligated above the rhinal fissure with 10-0 suture thread. The complete interruption of blood flow was confirmed using an operating microscope. Both common carotid arteries were then occluded with nontraumatic aneurysm clips for 1 hour as described previously. 3
Intermittent Hypoxia
Rats were randomly assigned to the following groups: sham-MCAO with sham IH (Sham; n = 15), MCAO with sham IH (MCAO; n = 15), MCAO with IH (MCAOIH; n = 15), and MCAO with IH and zidovudine (AZT; n = 10). Rats receiving MCAO were allowed to recover for 7 days before receiving the IH or sham IH treatment. Intermittent hypoxia (12% oxygen) was administered for 4 hours per day for 7 days in a hypoxia chamber (70 × 40 × 35 cm3). Rats receiving sham IH sessions (i.e., Sham and MCAO groups) were placed in the hypoxia chamber and exposed to room air for 4 hours per day for 7 days (Figure 1A).
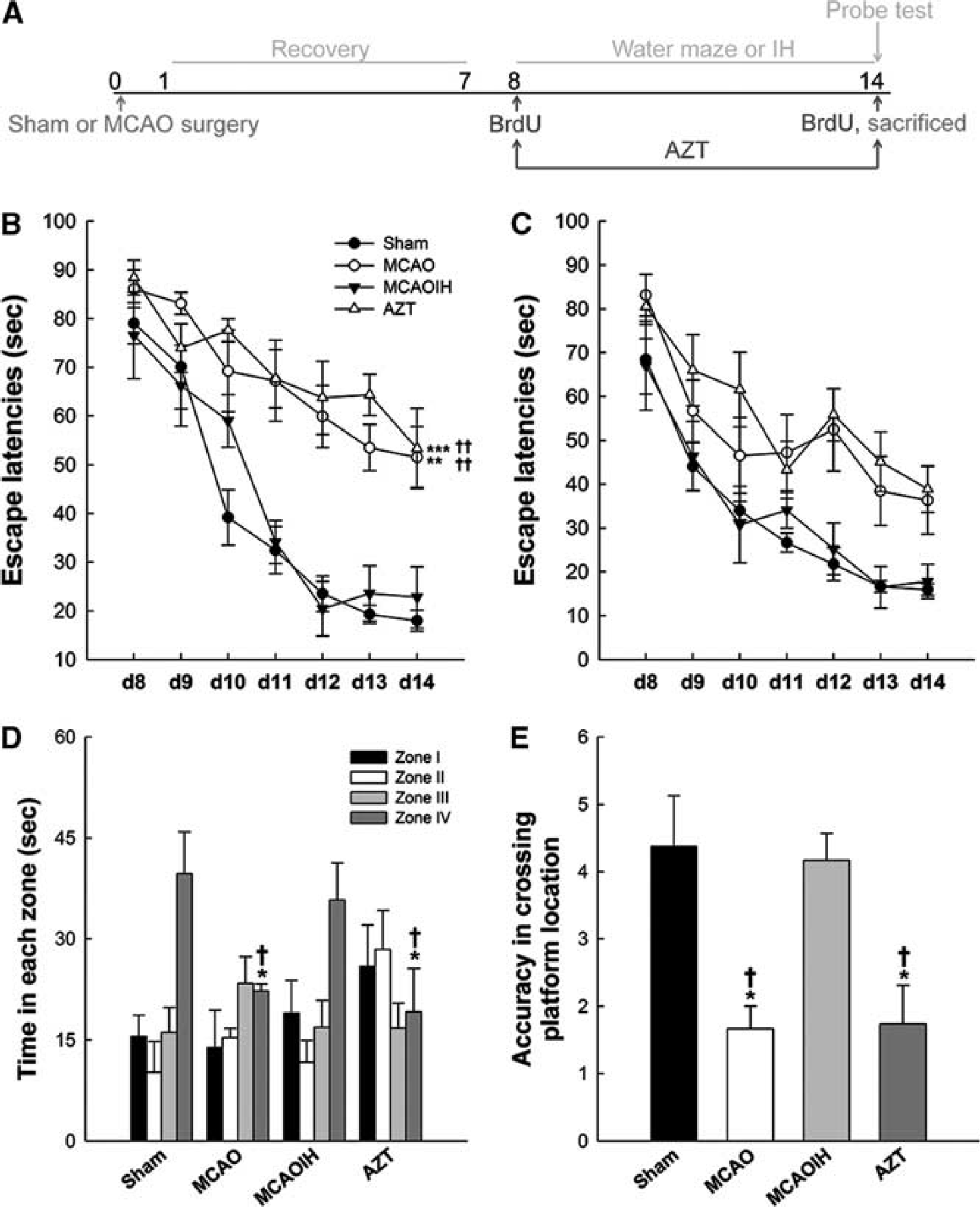
Postischemia intermittent hypoxia (IH) intervention alleviates long-term memory (LTM), but not short-term memory (STM), impairments. (
Primary neuron cultures were placed in a hypoxia chamber (Billups-Rothenberg, Del Mar, CA, USA) with 2% oxygen once after 12 days in vitro (div) (Hypoxia ×1) or three times between 12 and 14 div (Hypoxia ×3, all n = 5 for immunoblotting; n = 30, three cultures for immunostaining; n = 30 for Sholl analysis). The cultures were removed after hypoxic exposure and processed for subsequent analyses (Figure 3A).
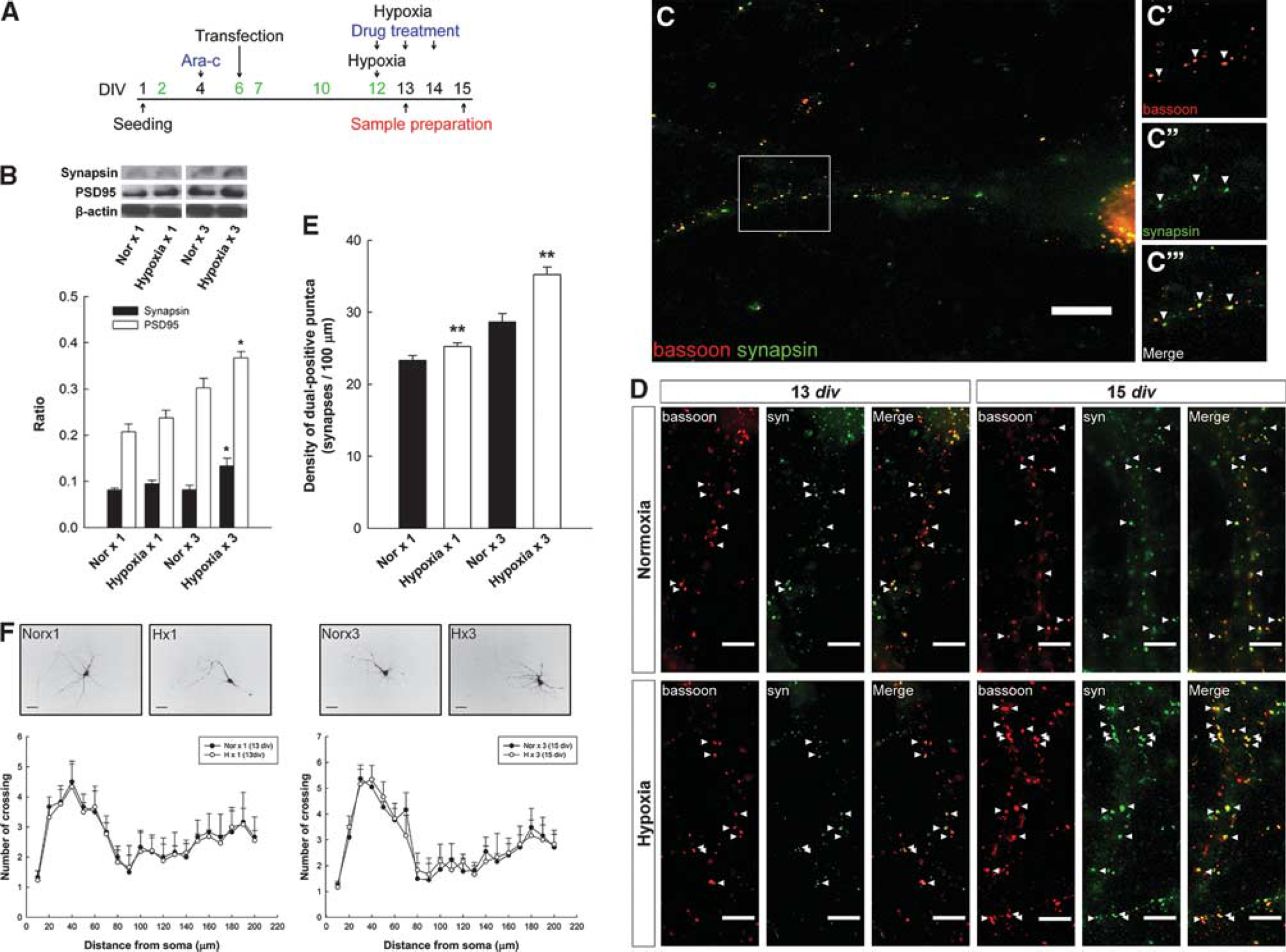
The repeated hypoxia increases the synaptic density and does not alter dendritic neurite branching in primary neuron cultures. (
Morris Water Maze
All rats were trained on the MWM (Morris Water Maze) on the same 7 days as the administration of the IH or sham IH treatments (days 8 to 14; Figure 1A). The rats were exposed to three training trials per day. The rats swam freely until they located the hidden platform and remained on it for 30 seconds. Rats that did not locate the platform within 90 seconds were gently guided to it. The rats rested in their home cages for 1 hour, followed by a probe test in which the platform was removed from the pool. The rats swam for 90 seconds during the probe test, and their behavior was recorded as in a previous study. 3
Pharmacological Treatments
The rats received intraperitoneal injections of BrdU (50 mg/kg; Sigma, St Louis, MO, USA) on days 8 and 14. The rats were killed 1 hour after the MWM probe test on day 14 (Figure 1A). Rats in the AZT group also received intraperitoneal injections of zidovudine (100 mg/kg; Sigma) on days 8 to 14 1 hour before IH intervention (Figure 1A).
The pharmacological treatments of the neuronal cultures were as follows: human BDNF (50 ng/mL), the TrkB inhibitor (2 μM K252a), the MEK inhibitor (50 μM PD98059), and the PI3K/AKT inhibitor (50 μM LY294002) were purchased from Sigma-Aldrich and added to the NB/B27 medium 12 to 14 div 1 hour before IH intervention (Figure 3A).
Transfection
Primary neuron cultures were transfected on 6 div using the expression vector pLenti–GFP Control Vector (a gift from Dr Sun). The culture medium was changed to a medium without antibiotics 1 to 2 hours before transfection in T-Pro NTRII transfection reagent (T-Pro Biotechnology, New Taipei, Taiwan).
Immunostaining
The animals were killed after the last behavioral test. Free-floating sections (20 μM) were processed using standard immunohistochemical procedures as described previously. 3 Immunocytochemical analyses were performed on primary neuron cultures on 13 or 15 div. Neurons were fixed in 4% paraformaldehyde in PBS (phosphate-buffered saline) and permeabilized with 0.2% Triton X-100 in PBS. The cells were blocked with 10% normal serum in PBS and incubated in primary antibodies at 4°C overnight. Species-specific secondary antibodies were used at a 1:500 dilution. The following antibodies were used: BrdU (Accurate, Westbury, NY, USA), BDNF (Santa Cruz, Dallas, TX, USA), bassoon (Stressgen, Farmingdale, NY, USA), DCX (Santa Cruz), GluA2 (Chemicon, Billerica, MA, USA), GluN2B (Chemicon), PSD95 (Cell Signaling, Boston, MA, USA), synapsin (Cell Signaling), and synaptophysin (Chemicon).
Immunoblotting
For in vivo analyses, the right hippocampus was collected and lysed in RIPA buffer as described previously. 3 For in vitro analyses, neuronal cultures were rinsed in PBS, scraped, and homogenized in the RIPA buffer. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA). The samples were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to the polyvinylidene difluoride membranes. The membranes were probed with antibodies at appropriate dilutions, visualized with ECL-plus, and analyzed using ImageQuant (GE Healthcare Biosciences, Pittsburgh, PA, USA). The following antibodies were used: AKT (Cell Signaling), pAKT (Cell Signaling), β-actin (Sigma-Aldrich), GluA2 (Chemicon), GluN2B (Chemicon), MAPK (Cell Signaling), pMAPK (Cell Signaling), PSD95 (Cell Signaling), synapsin (Cell Signaling), pSynapsin (Cell Signaling), and synaptophysin (Chemicon).
Enzyme-Linked Immunosorbent Assay
ELISA (enzyme-linked immunosorbent assay) was performed using the BDNF Emax immunoassay system (Promega, Madison, WI, USA). The right hippocampus (n = 5 for each group) was dissected and homogenized on ice in a lysis buffer (137 mM NaCl, 20 mM Tris-HCl (pH 8.0), 1% NP40, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 1 μg/mL leupeptin, and 0.5 mM sodium vanadate). The samples were processed by acidification and subsequent neutralization to increase the numbers of detectable proteins in the lysates. The samples were centrifuged at 12 500 r.p.m. at 4°C for 30 minutes, and the supernatants were stored at −80°C for ELISA.
Flat-bottomed, 96-well plates (BD Falcon, Franklin Lakes, NJ, USA) were coated with a monoclonal anti-BDNF antibody (mAb) without shaking overnight at 4°C. After washing with 0.05% Tris-buffered saline with Tween 20, the 96-well plates were blocked in a 1× block and sample buffer for 1 hour followed by a 2-hour incubation of the BDNF standard and hippocampal lysates. The captured BDNF was allowed to bind the secondary BDNF-specific polyclonal antibody for 2 hours. After washing with TBST, the amount of specifically bound polyclonal antibody was detected using a species-specific anti-immunoglobulin Y, horseradish peroxidase-conjugated antibody as a tertiary reactant for 1 hour. The unbound conjugate was removed by washing. The plates were incubated with a chromogenic substrate, and color changes were measured at 450nm in a microplate reader (Infinite 200, TECAN, San Jose, CA, USA).
Sholl Analysis
Neuronal branching complexity was analyzed in GFP-transfected neurons on div 13 to 15. Low-magnification images of isolated neurons were digitally enhanced in ImageJ software (National Institutes of Health, Bethesda, MD, USA). A series of concentric circles with gradually increasing diameters (10-μm steps) was centered on the cell body. Two-dimensional analysis was performed to count the number of branches that intersected successive concentric circles.
Image Analysis
To determine the number of immunoreactive (BrdU+/DCX+, BrdU+/ BDNF+, BrdU+/GluA2+, BrdU+/GluN2B+) cells throughout the entire DG of hippocampus and the number of synapsin+/bassoon+ puncta from primary neuron culture, the fluorescence images were obtained using an Olympus BX-61 microscope (Yuanyu Industry Co., Ltd., Taiwan). Besides, the synaptic puncta were analyzed starting from neuronal cell soma within 100-μm to reduce the variations of each cultures and treatments. To verify the intensity of immunorecative (synapsin+, synaptophysin+, or PSD95+) cells and the number of synaptophysin+ cells, the single plane confocal images from hippocampal sections were obtained using Leica SP2 confocal microscopy (Major Instruments Co., Ltd., Taiwan). The brain sections with the strongest intensity were scanned first with the same parameters setting. ImageJ analysis software was used for quantification.
Statistical Analysis
All data are expressed as the mean values ± s.e.m. Statistical significance was defined as P < 0.05. Repeated-measures analysis of variances (ANOVAs) followed by Tukey's post hoc tests were used to analyze the memory performance and Sholl analysis. One-way ANOVAs also followed by Tukey's post hoc test were used to assess the differences among groups. Student's t-test was used when appropriate.
RESULTS
Postischemia Intermittent Hypoxia Intervention Affects Long-Term, but not Short-Term, Memory
Intermittent hypoxia intervention following MCAO rescues ischemia-induced spatial learning and memory impairments likely via the induction of hippocampal neurogenesis. 3 The experiments presented here examined the effects of postischemia IH-induced neurogenesis on long- and short-term memory (STM). Performance on the first daily MWM trial reflected long-term memory (LTM), and the average performance on the other daily trials reflected STM. A repeated-measure ANOVA on escape latencies of LTM noted significant differences for group × time interaction (F = 1.79, P < 0.05), groups (F = 10.73, P < 0.001), and time (F = 37.596, P < 0.001). The LTM performance of the MCAO group was markedly impaired following ischemia compared with the Sham group (P < 0.01; Figure 1B). Intermittent hypoxia after ischemia reversed this impairment, as indicated by the significant improvement in LTM performance in the MCAOIH group compared with the MCAO group (P < 0.01). No differences in the LTM performance of the MCAOIH and Sham groups were observed (P = 0.986). Zidovudine treatment, which disrupts neurogenesis, blocked the memory-sparing effect of IH, as indicated by the impairment in LTM performance in the AZT group compared with the Sham and MCAOIH groups (P < 0.001 and P < 0.01, respectively). The AZT group showed a performance similar to that of the MCAO group (P = 0.946). These results indicate that postischemia IH-induced neurogenesis was necessary for the reversal of ischemia-induced spatial LTM impairment. However, no group × time interaction differences in STM performance were observed (F = 0.943, P = 0.532; Figure 1C), which suggests that postischemia IH-induced neurogenesis is linked to spatial LTM but not STM.
The MCAO and AZT groups did not exhibit a preference for the target quadrant (zone IV), although a preference was observed in the sham group (P < 0.05 for both groups; Figure 1D). The MCAOIH group spent significantly more time in the target quadrant than did the MCAO and AZT groups (P < 0.05 for both groups); however, the performance of this group was similar to that of the Sham group (P = 0.362; Figure 1D). The accuracy of platform location crossings during the probe test was also analyzed. The MCAO and AZT groups exhibited significantly poorer performance compared with the Sham group (P < 0.05 for both groups; Figure 1E). The MCAOIH group exhibited better accuracy than did the MCAO and AZT groups (P < 0.05 for both groups), although their performance was similar to that of the Sham group (P = 0.347; Figure 1E).
Postischemia Intermittent Hypoxia Intervention Enhanced the Maturation of Hippocampal Neurons
Hypoxia induces the premature maturation of precursor cell 13 and thus, we speculated that IH enhances hippocampal neuron maturation. We performed double immunohistochemical evaluations for BrdU (new born cells marker) and DCX (neuron marker) and calculated the number of BrdU+/DCX+ cells in the hippocampus in each group (Figure 2B). There was a significant decrease in the DCX+ cells in the AZT group, suggesting that the zidovudine treatment is enough to disrupt the hippocampal progenitor cells proliferation (Figure 2A). Furthermore, a significant decrease in the BrdU+/DCX+ number in the MCAO group compared with Sham (P < 0.05) and MCAOIH groups (P < 0.05) was observed (Figures 2C and D). Similarly, a significant decrease in the BrdU+/DCX+ number in the AZT group compared with the Sham (P < 0.01) and MCAOIH groups (P < 0.01) was observed (). No difference in the BrdU+/DCX+ number between the MCAOIH and Sham groups (P = 0.532; Figures 2C and D) was observed. These results suggest that ischemia led to decreased newborn cell maturation, and postischemia IH intervention reversed this effect. These results indicate that postischemia IH induced the proliferation and maturation of hippocampal newborn neurons that attenuate ischemia-induced spatial learning and memory deficits in rats.
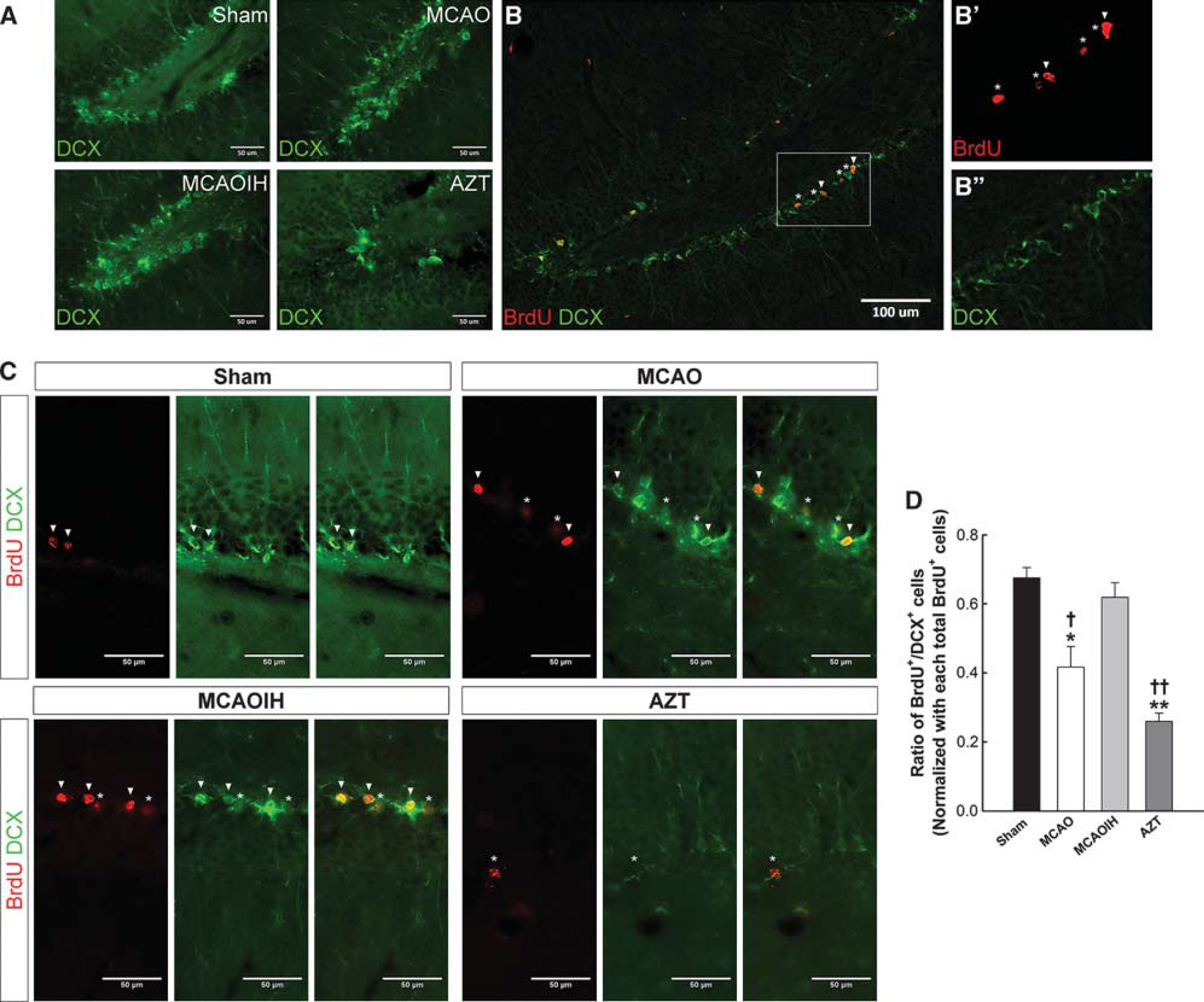
The maturation of newborn cells is reduced following ischemia and rescued following postischemia intermittent hypoxia (IH) intervention. (
The Effects of Intermittent Hypoxia Intervention are Mediated by Increased Synaptogenesis but not Dendritogenesis
Hippocampal neurogenesis is involved in memory formation2,3; however, the mechanism underlying this relationship remains unclear. Newborn neurons contribute to brain function by forming functional circuits with original neurons. 1 A rapid loss of synapse formation with memory impairment is observed in ischemia models. 10 Synaptogenesis is important for memory formation, although the effect of postischemia IH on synaptic density is not known. We developed an in vitro IH model in which primary neuron cultures were exposed to 2% oxygen. This oxygen concentration induces cellular proliferation, which is consistent with our previous finding that postischemia IH intervention induces the proliferation of newborn hippocampal cells. 14 We exposed neuron cultures to hypoxia either once (Hypoxia ×1 group) or three times (Hypoxia ×3 group) beginning on 12 div to mimic our in vivo IH intervention protocol. Control cultures were exposed to normoxia either once (Nor ×1) or three times (Nor ×3) in the incubator. Immunoblot assays were performed for synapsin, a presynaptic vesicle-associated protein, and PSD95, a scaffolding protein involved in the assembly of the postsynaptic density complex (Figure 3B). No significant differences in synapsin or PSD95 expression were observed in the Hypoxia ×1 group compared with the Nor ×1 group (P = 0.202 and P = 0.244, respectively; Figure 3B). However, a significant increase in the expression of synapsin and PSD95 was observed in the Hypoxia ×3 group compared with the Nor ×3 group (P < 0.05 for both groups; Figure 3B). To further verify the effects of IH intervention on mature synapses, we performed double immunocytochemical evaluation for synapsin and bassoon, which is a component of presynaptic cytoskeleton at the active zone (Figure 3C). The number of synapsin+/bassoon+ puncta presented as mature synapses 15 within 100-μm increments from the soma increased significantly in the Hypoxia ×1 group compared with the Nor ×1 group (P < 0.01) and in the Hypoxia ×3 group compared with the Nor ×3 group (P < 0.01; Figures 3D and 3E). These results suggest that repeated IH intervention increased the synaptic density. Therefore, we used repeated IH intervention for our in vitro protocol in subsequent experiments.
We transfected primary neuron cultures with the pLenti–GFP vector on 6 div to examine whether the IH-induced increase in the synaptic density was due to an increase in the number of dendritic neurite branches. The transfected neurons were exposed to hypoxia or normoxia on 12 to 14 div, and Sholl analysis 16 was performed on 13 or 15 div. The neuritic–dendritic arborization results for the normoxic and hypoxic cultures were similar (Figure 3F). The repeated-measure ANOVA results noted that no significant differences on the dendritic morphologies of the normoxic and hypoxic cultures were observed on 13 (F = 0.257, P = 0.619) or 15 div (F = 1.188, P = 0.324; Figure 3F), suggesting that IH intervention did not promote dendritic branching. These results indicate that IH intervention in primary neuron culture promoted synaptogenesis that was not dependent on dendritogenesis.
Brain-Derived Neurotrophic Factor/PI3K/AKT Primarily Regulated the Promotion of Synaptogenesis after Intermittent Hypoxia Intervention
Brain-derived neurotrophic factor is essential for synapse formation. 8 Therefore, we speculated that IH promoted synaptogenesis by inducing BDNF expression. We treated primary neuron cultures with BDNF under normoxic conditions (NB group) on 12 to 14 div and prepared samples on 15 div. Immunoblot analyses were used to measure synapsin and PSD95 expression (each n = 5 for immunoblotting; Figure 4A). We demonstrated a significant increase in synapsin and PSD95 expression in the NB group compared with the normoxia (N) group (P < 0.001; Figure 4A), suggesting that BDNF promoted synaptogenesis. Primary neuron cultures with hypoxia (H group) exhibited a significant increase in synapsin and PSD95 expression compared with the N group (P < 0.001 and P < 0.01, respectively), although no difference was observed compared with the NB group (P = 0.731 and P = 0.07, respectively; Figure 4A). Primary neuron cultures exposed to hypoxia and K252a treatment, used to abolish BDNF-induced increase of synapsin and PSD95 expression (HK group), exhibited significant reductions in synapsin and PSD95 expression compared with the N (P < 0.01 for both groups), NB (P < 0.001 for both groups) and H groups (P < 0.001 for both groups; Figure 4A). This result suggests that BDNF regulated IH-induced synaptogenesis. The number of synapsin+/bassoon+ puncta within 100-μm increments from the soma (n = 30, three cultures for immunostaining) significantly increased after BDNF and hypoxia treatment; however, K252a treatment markedly reduced these numbers (N versus NB: P < 0.01; N versus H: P < 0.01; N versus HK: P < 0.001; NB versus HK: P < 0.001; H versus HK: P < 0.001; H versus NB: P = 0.314; Figures 4B and C). The repeated-measure ANOVA exhibited that no difference in the number of dendritic neurite branches was observed (F = 1.354, P = 0.318, n = 30 for Sholl analysis; Figures 4D and E). Therefore, BDNF regulated IH-induced synaptogenesis.
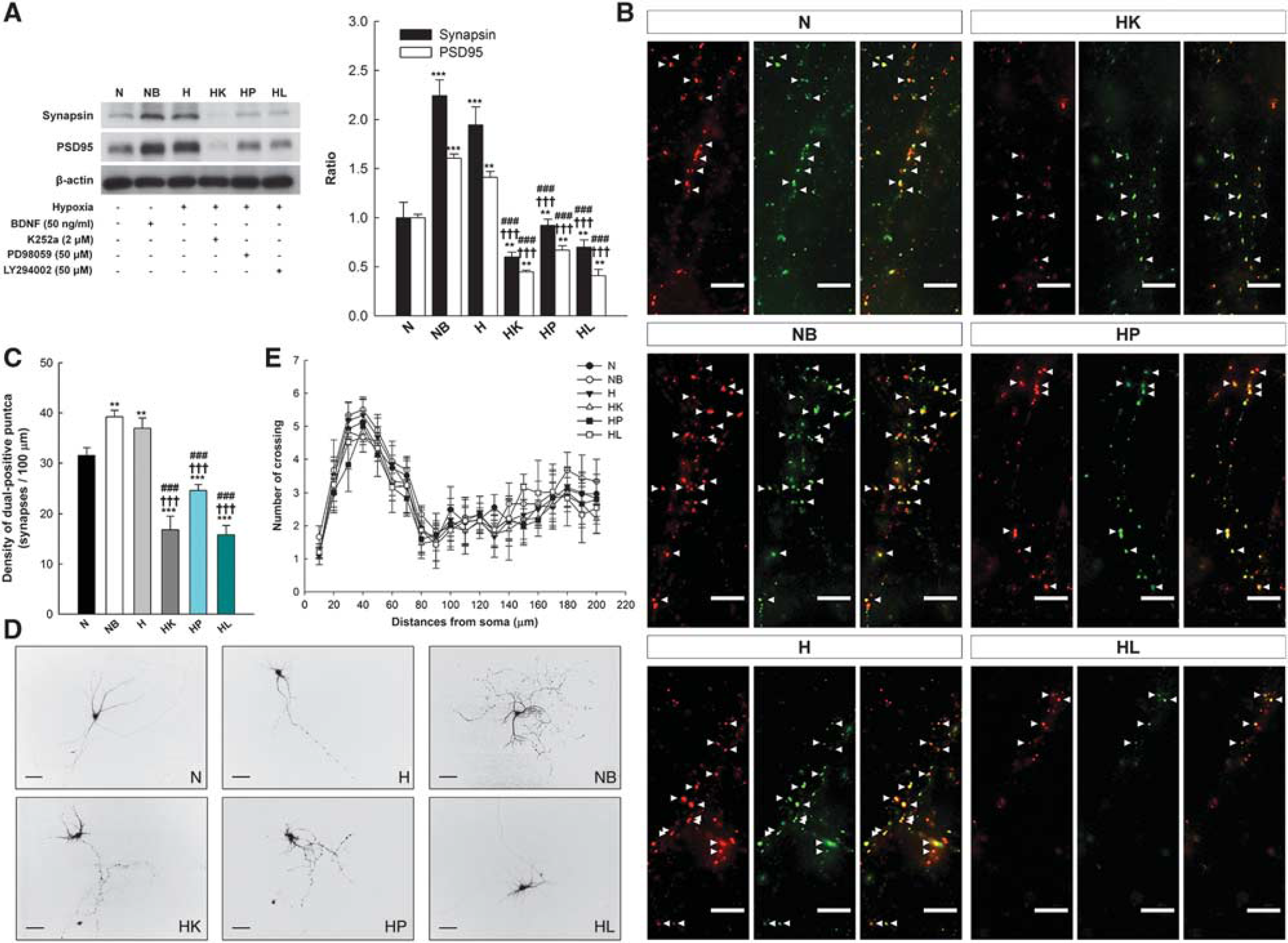
Changes in the synaptic density are not dependent on dendritic neurite branching. (
AKT and MAPK are important downstream proteins of BDNF that regulate synaptic plasticity. 17 We treated primary neuron cultures with the MEK 1/2 inhibitor, PD98059 (HP group), or the PI3K/AKT inhibitor, LY294002 (HL group), 1 hour before IH intervention on 12 to 14 div to investigate which downstream pathway mediated BDNF-regulated synaptic plasticity. Samples were prepared on 15 div. Lower levels of synapsin and PSD95 were expressed in the HP and HL groups compared with the N group (P < 0.01 for all groups), H group (P < 0.001 for all groups), and NB group (P < 0.001 for all groups; Figure 4A). The number of synapsin+/bassoon+ puncta in 100-μm increments from the soma was markedly decreased in the HP and HL groups compared with the N, H, and NB groups (P < 0.001 for all groups; Figures 4B and C). These decreases were independent of changes in the dendritic neurite branching (F = 1.354, P = 0.318; Figures D and E). A marked reduction in the ratios of synapsin (69.16% ± 3.78%) and PSD95 (68.02% ± 5.08%) were noted in the HL group compared with the HP group (synapsin: 47.43% ± 3.18%, P < 0.01; PSD95: 47.71% ± 3.582%, P < 0.01) but not the HK group (synapsin: 64.03% ± 2.57%, P = 0.515; PSD95: 65.08% ± 1.39%, P = 0.842). A significant reduction ratio of synapsin and PSD95 was observed in the HK group compared with the HP group (P < 0.01 and P < 0.05, respectively). These results suggest that pAKT expression is the primary mediator of BDNF-regulated synaptogenesis, and thus, IH intervention promotes synaptogenesis in primary neuron cultures primarily through BDNF/PI3K/AKT expression.
Postischemia Intermittent Hypoxia Intervention Increases Synapse Density and Enhances Synaptic Function in Hippocampal Dentate Gyrus Subregion
We proposed a possible mechanism for synaptogenesis after IH intervention in vitro. Whether synaptogenesis and postischemia IH in vivo share the same mechanism also requires further examination. Moreover, the role of the IH intervention-induced newborn neurons in these alterations remains unknown.
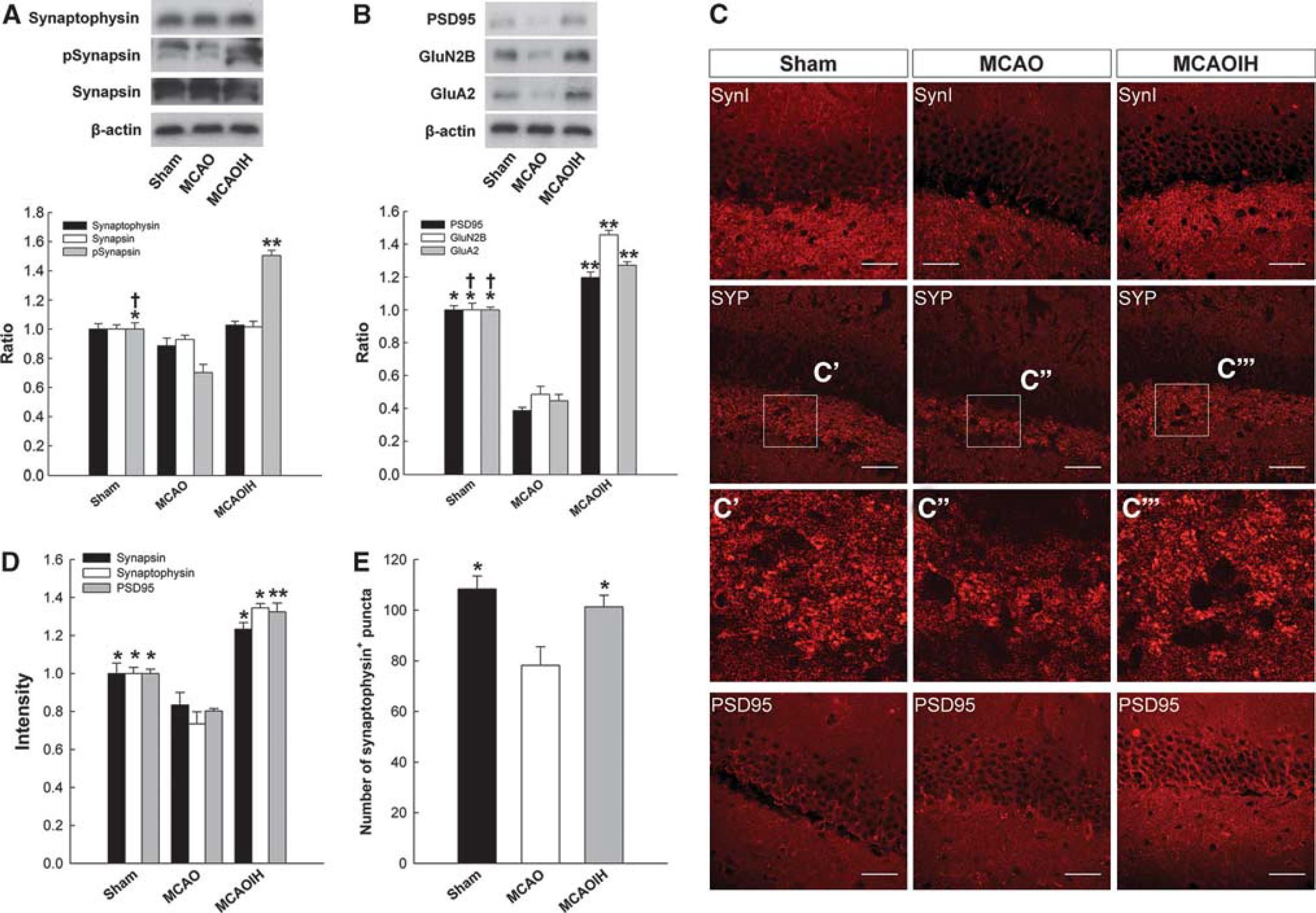
We investigated changes in synapse density to examine these issues. Immunoblotting was used to assess the expression of synapsin in the hippocampus. No differences in synapsin expression between the Sham, MCAO, and MCAOIH groups were observed (P = 0.267; Figure 5A). To confirm this result, we also measured another presynaptic marker, synaptophysin. However, IH did not increase synaptophysin expression among each group (P = 0.078; Figure 5A). To further verify the effects of postischemia IH intervention on hippocampal DG subregion, we used a Leica SP2 confocal microscope to examine the intensity of synapsin+ cells and the number of synaptophysin+ puncta, which is an index of the number of presynaptic axon boutons. 12 A marked reduction in the intensity of synapsin was noted in the MCAO group compared with the Sham (P < 0.05) and MCAOIH group (P < 0.05; Figures 5C and 5D). There was no difference between Sham and MCAOIH group (P = 0.093; Figures 5C and 5D). Similarly, MCAO group expressed less synaptophysin+ puncta than the Sham (P < 0.05) and MCAOIH group (P < 0.05; Figures 5C and 5E). There was no difference between Sham and MCAOIH group (P = 0.164; Figures 5C and 5E). Furthermore, the intensity of synaptophysin expression is an index of the number of presynaptic vesicles and relates to presynaptic terminal functional synapse.12,18 A marked reduction in the synaptophysin intensity was observed in the MCAO group compared with the Sham (P < 0.05) and MCAOIH groups (P < 0.05; Figures 5C and 5D). There was no difference between Sham and MCAOIH group (P = 0.062; Figures 5C and 5D). These results suggest that postischemia IH intervention rescue the loss of presynaptic density and may relate to the enhancing of presynaptic function in hippocampal DG subregion.
Postischemia intermittent hypoxia (IH) intervention induces synapse density and enhances synaptic function in the hippocampus. (
Based on our previous results, we next verified that whether post-IH intervention could enhance synaptic function. Synapsin is a phosphoprotein that enables vesicle release. Therefore, we measured hippocampal synapsin phosphorylation (Figure 5A). A marked reduction in pSynapsin expression was observed in the MCAO group compared with the Sham groups (P < 0.05) but rescued in MCAOIH rats (P < 0.01; Figure 5A). Besides, there was a marked increase of pSynapsin expression in MCAOIH rats as compared with Sham rats (P < 0.05; Figure 5A), suggesting that postischemia IH intervention altered the presynaptic density in vivo and enhanced the efficiency of presynaptic neurotransmitter release.
We used PSD95 as an index to examine whether the postsynaptic density is affected by postischemia IH intervention (Figure 5B). PSD95 expression was markedly reduced in the MCAO group compared with the Sham (P < 0.05) and MCAOIH groups (P < 0.01; Figure 5B). No difference in PSD95 expression between the MCAOIH and Sham groups was observed (P = 0.697; Figure 5B). Similarly, there was a significant decrease on the intensity of PSD95 in the MCAO group compared with the Sham (P < 0.05) and MCAOIH group (P < 0.05; Figures 5C and 5D). No difference on the intensity of PSD95 between MCAOIH and Sham groups was noted (P = 0.178; Figures 5C and 5D). These results suggest that IH intervention upregulated the postsynapse density complexes in the hippocampus. Collectively, these findings indicate that postischemia IH increased the synapse density and enhanced synaptic function in hippocampal DG subregion.
Postischemia Intermittent Hypoxia Intervention Induces Synaptogenesis via Brain-Derived Neurotrophic Factor Expression in Newborn Neurons
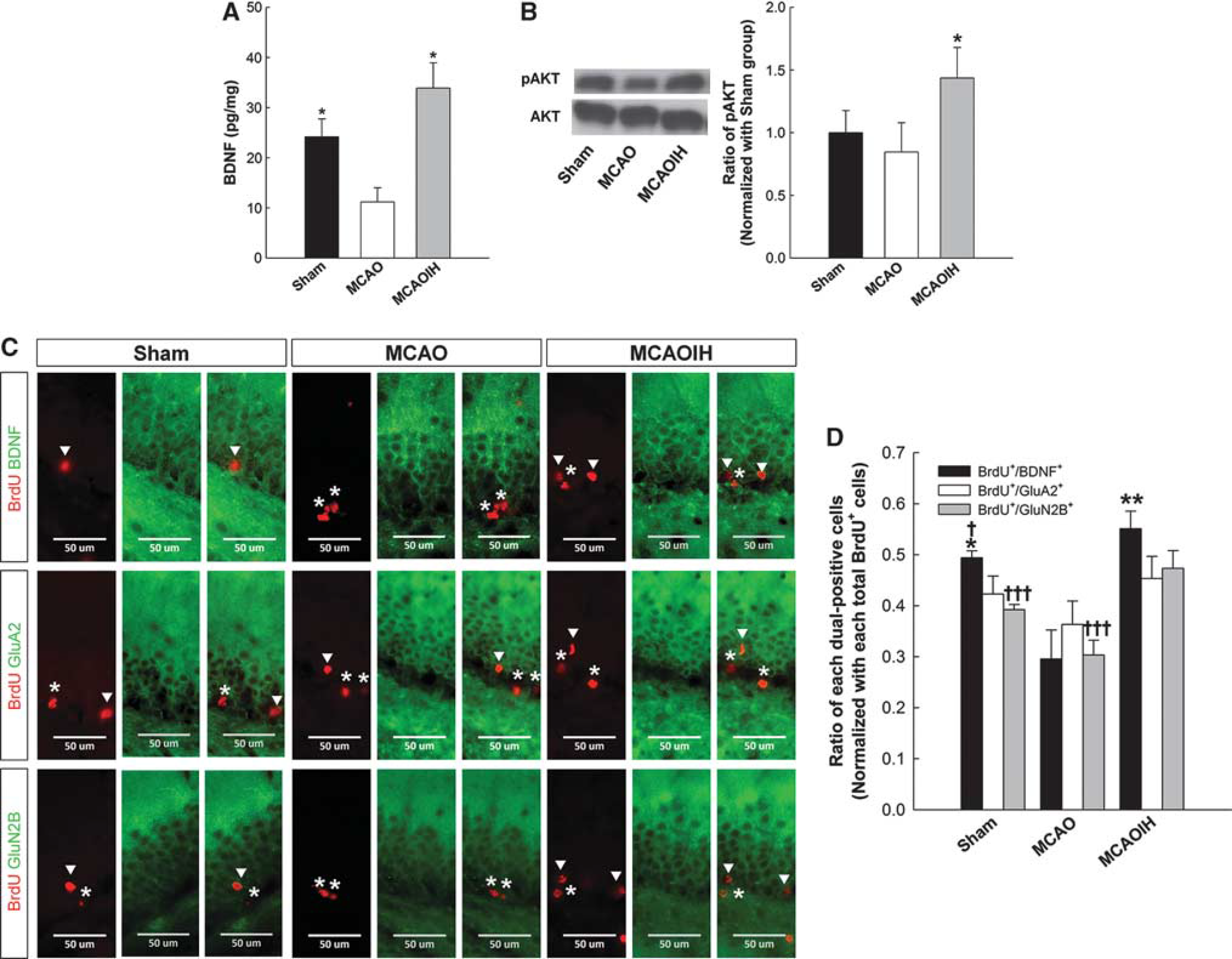
Intermittent hypoxia intervention induced synaptogenesis primarily through the BDNF/PI3K/AKT pathway in vitro. We hypothesized that BDNF and AKT expression increased following postischemia IH intervention. Enzyme-linked immunosorbent assay and immunoblotting analyses were used to probe the levels of hippocampal BDNF and AKT expression. We observed a significant decrease in BDNF expression in the MCAO group compared with the Sham and MCAOIH groups (P < 0.05 for both groups; Figure 6A). No difference in BDNF expression was observed in the Sham and MCAOIH groups (P = 0.126; Figure 6A), which suggests that postischemia IH induced hippocampal BDNF expression. We also observed a significant increase in pAKT expression in the MCAOIH group compared with the MCAO (P < 0.05) and no difference compared with the Sham groups (P = 0.078; Figure 6B). However, no difference in pAKT expression was observed between the Sham and MCAO groups (P = 0.082; Figure 6B). This result suggests that postischemia IH induced hippocampal AKT expression. Therefore, postischemia IH intervention induced both synaptogenesis and BDNF/PI3K/AKT expression in vivo.
Postischemia intermittent hypoxia (IH) intervention induces hippocampal neo-neurons expressing brain-derived neurotrophic factor (BDNF) and GluN2B. (
Brain-derived neurotrophic factor expression in newborn neurons was also investigated. We used dual-label immunohistochemical staining for BrdU and BDNF, and the number of BrdU+/BDNF+ cells was quantified (Figures 6C and 6D). A significant decrease in the number of BrdU+/BDNF+ cells was observed in the MCAO group compared with the MCAOIH (P < 0.01) and Sham groups (P < 0.05; Figures 6C and 6D). A significant increase in the number of BrdU+/BDNF+ cells was also observed in the MCAOIH group compared with the Sham group (P < 0.05; Figures 6C and 6D), indicating that postischemia IH intervention induced newborn neurons to express BDNF.
These findings indicate that postischemia IH intervention induced the expression of BDNF in newborn neurons and promoted synaptogenesis. These results suggest that postischemia IH intervention promoted newborn neuron proliferation and synaptogenesis.
Postischemia Intermittent Hypoxia Intervention Induces Newborn Neurons that Express Activity-Dependent Receptors
Postischemia IH intervention enhanced presynaptic function, as indicated by the phospho-synapsin expression and synaptophysin intensity. These findings suggest the occurrence of other alterations of the postsynapse density in addition to the postsynaptic function. Synaptic glutamate receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or N-methyl-D-aspartate (NMDA) receptors, are localized to the postsynaptic density complex where they are organized by scaffolding and adaptor proteins to bind neurotransmitters that are released from presynaptic terminals. 19 Receptor subtypes that contain GluN2B or GluA2 subunits are essential for LTP induction and memory formation.20,21 Chronic IH induces glutamate receptor-dependent plasticity in the hypothalamic paraventricular nucleus. 22 The activity-dependent postsynaptic changes were examining using immunoblot assays to quantify the expression of GluA2 (an AMPA receptor subunit) and GluN2B (an NMDA receptor subunit; Figure 5B). A marked reduction in GluA2 and GluN2B expression was observed in the MCAO group compared with the Sham group (P < 0.05 for all groups; Figure 5B). However, marked increases in GluA2 and GluN2B expression were observed in the MCAOIH group compared with the MCAO (P < 0.01 for all groups) and Sham groups (P < 0.05 for all groups; Figure 5B). These results suggest that postischemia IH promoted postsynaptic activity-dependent alterations.
We used dual-label immunostaining for BrdU, GluA2, and GluN2B and quantified the number of dual-immunoreactive cells to determine whether newborn neurons exhibited similar effects following IH intervention (Figure 6C). No differences in the numbers of BrdU+/GluA2+ cells were observed between groups (P = 0.176; Figures 6C and 6D). An increased number of BrdU+/GluN2B+ cells was observed in the MCAOIH group compared with the Sham (P < 0.001) and MCAO groups (P < 0.001; Figures 6C and 6D), which suggests that postischemia IH intervention induced the expression of GluN2B, but not GluA2, in the majority of newborn cells. These findings indicate that postischemia IH intervention induced activity-dependent alterations in newborn neurons.
DISCUSSION
This study demonstrated that postischemia IH intervention enhances newborn neuron synaptogenesis and function, which reverses spatial LTM impairment due to brain ischemia. Intermittent hypoxia was shown to regulate synaptogenesis, but not dendritic complexity, primarily via the BDNF/PI3K/AKT pathway. Postischemic IH intervention induces BDNF expression in hippocampal neo-neurons and enhances presynaptic function and postsynaptic activity-dependent changes.
The results from our laboratory and others provide evidence for the importance of neurogenesis in memory performance.1–4 As newborn neurons are produced, they differentiate, mature, and establish functional synapses on target cells. The maturation and establishment of synapses between new neurons and existing hippocampal circuits is important for memory performance.1,4 Brain-derived neurotrophic factor is an important growth factor for the enhancement of hippocampal progenitor cell proliferation, synapse formation, and stabilization as well as learning and memory formation, suggesting that BDNF may regulate newborn hippocampal neuron proliferation and synaptogenesis, both of which are essential for learning and memory.5,6,8,9 We observed a decrease in the neo-neuron maturation (the number of BrdU+/DCX+ cells) following ischemia that was rescued by postischemia IH intervention. These results are consistent with a report of hypoxic disruption of cell-cycle regulation and the induction of premature cell maturation. 13 We also demonstrated that synaptic densities were markedly reduced after ischemia and rescued by postischemia IH intervention from immunostaining results. These rescues were associated with the recovery of spatial LTM performance. Furthermore, postischemia IH induced hippocampal BDNF expression, which suggests that postischemia IH intervention induces synaptogenesis and increases BDNF expression concomitant with LTM improvement.
Integrated and functional synapses facilitate signaling from the presynaptic terminal to postsynaptic membranes. Brain-derived neurotrophic factor release enhances presynaptic activity and induces activity-dependent regulation of synaptic structure, particularly in glutamatergic synapses.8,23 Postsynaptic glutamate receptors ensure rapid and precise signal transduction and activate various second messenger pathways in learning and memory. These findings indicate that BDNF is a bidirectional modulator of presynaptic transmission and postsynaptic plasticity.
In our study, no difference on the synapsin and synaptophysin expression was noted from whole hippocampal lysate immunoblotting results among each group. However, a significant reduction on the presynaptic density and function was noted in MCAO rats but rescued after ischemic rats with IH intervention in DG from immunostaining data. These results suggest that postischemia induced presynaptic density and related to enhancement of presynaptic function in DG which was also confirmed by our pSynapsin expression from immunoblotting result consistent with previous study. 24 To keep the similar total amount of presynaptic protein, the larger size of the synaptic terminal in the MCAO group than other groups was suggested due to the different results exhibited in our immunoblotting and immunostaining for expression of presynaptic markers. Postsynaptic PSD95 regulates the maturation of excitatory synapses via glutamatergic receptor recruitment, such as GluN2B-containing NMDA receptors and GluA2-containing AMPA receptors, which are essential for LTP induction and memory formation and BDNF expression.20,21,25,26 Postischemia IH intervention was associated with the increased expression of GluN2B-containing NMDA receptors and GluA2-containing AMPA receptors in the hippocampus and memory recovery. Furthermore, postischemia IH induced the expression of BDNF- and GluN2B-containing NMDA receptors in newborn hippocampal neurons and was associated with LTM recovery, as previously reported.5,6,9 Postischemia IH-induced newborn hippocampal neurons were affected by presynaptic activity, which reflects the dynamic plasticity of glutamatergic receptors.
Brain-derived neurotrophic factor regulates synaptogenesis in hippocampal neurons via the BDNF/PI3K/AKT pathway.15,17 We demonstrated that the synaptic density (synapsin+/bassoon+ puntca) in primary neuron cultures decreased following IH intervention coupled with various inhibitors, and this decrease was not dependent on dendritic branch alterations. The reduction in the synaptic density ratios was greater in the HK (hypoxia with K252a treatment to abolish BDNF effect) and HL groups (hypoxia with LY294002 treatment to inhibit PI3K activation) than in the HP group (hypoxia with PD98059 treatment to repress MEK activation). These findings suggest that the BDNF/PI3K/AKT signaling pathway regulated synapse formation. We obtained similar results with postischemia IH intervention in vivo. However, K252a is a selective inhibitor of the Trk family, 27 which suggests that other neurotropic factors participate in synaptogenesis. Neurotrophins 3 and 4 play important roles in reinnervation or neurite outgrowth in the hippocampus. 28 The nerve growth factors promote the survival of hippocampal neo-neurons, which suggests that various neurotropic factors exert different effects on synapses. 29
In summary, postischemia IH intervention rescued ischemiainduced spatial learning and memory impairment by inducing hippocampal neurogenesis and functional synaptogenesis via the BDNF/PI3K/AKT pathway.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Yun-Chia Chou and Paulus S Wang for technical support.