
Abstract
Intracranial aneurysms (IAs) linger as a potentially devastating clinical problem. Despite intense investigation, our understanding of the mechanisms leading to aneurysm development, progression and rupture remain incompletely defined. An accumulating body of evidence implicates inflammation as a critical contributor to aneurysm pathogenesis. Intracranial aneurysm formation and progression appear to result from endothelial dysfunction, a mounting inflammatory response, and vascular smooth muscle cell phenotypic modulation producing a pro-inflammatory phenotype. A later final common pathway appears to involve apoptosis of cellular constituents of the vessel wall. These changes result in degradation of the integrity of the vascular wall leading to aneurysmal dilation, progression and eventual rupture in certain aneurysms. Various aspects of the inflammatory response have been investigated as contributors to IA pathogenesis including leukocytes, complement, immunoglobulins, cytokines, and other humoral mediators. Furthermore, gene expression profiling of IA compared with control arteries has prominently featured differential expression of genes involved with immune response/inflammation. Preliminary data suggest that therapies targeting the inflammatory response may have efficacy in the future treatment of IA. Further investigation, however, is necessary to elucidate the precise role of inflammation in IA pathogenesis, which can be exploited to improve the prognosis of patients harboring IA.
Introduction
Intracranial aneurysms (IAs) are common vascular abnormalities of the brain with a prevalence of 3.2% in the general population (Juvela, 2011) and an annual risk of rupture around 1% (ISUIA, 1998; Wermer et al, 2007). Despite recent diagnostic and therapeutic advances, an episode of subarachnoid hemorrhage remains lethal in up to 65% of cases (Wiebers et al, 2003) and significantly disabling in 50% of those who survive (Nieuwkamp et al, 2009). Preventive treatment of IA before rupture is therefore needed. Surgical clipping and endovascular therapy are invasive procedures with potentially serious complications. Moreover, the risk of these interventions may exceed the annual risk of rupture of IA (ISUIA, 1998; Wiebers et al, 2003). No safe and effective noninvasive therapies have, as of yet, been identified and implemented in clinical practice mainly because of a lack of knowledge of the mechanisms underlying formation, progression, and rupture of IA. Although several risk factors for IA have been identified, the inherent pathogenic pathways remain obscure. Recently, a growing body of evidence has supported the role of inflammation in the pathogenesis of IA. A series of experimental reports and animal studies have highlighted the critical role of various actors of inflammation in the formation and progression of IA. Moreover, experimental therapies targeting different molecular inflammatory effectors in animal models have proved efficacious and promising. In this article, we critically review the existing data from human and animal studies implicating inflammation in the pathogenesis of IA, outline potential therapeutic targets and highlight areas that require further investigation.
Pathology of Intracranial Aneurysms
Although there are different forms of IA, the present discussion will focus on saccular IA. Under physiological circumstances, there is a delicate balance between local hemodynamic stress and arterial wall integrity. Saccular IA is believed to develop as a result of disruption of this homeostatic balance. The preponderance of knowledge concerning the pathology of IA is derived from histological investigations from autopsy or surgical specimens and recently, to a lesser extent, from animal models. Collectively, it has been hypothesized that the pathogenesis of IA involves persistent pathological vascular remodeling (with proteolysis/extracellular matrix degradation via matrix metalloproteinases (MMPs) and apoptosis playing major roles) with concomitant vessel wall inflammation (Aoki et al, 2008a; Chyatte et al, 1999; Frosen et al, 2004; Hashimoto et al, 2006).
Atherosclerosis is a Common Pathological Feature in Saccular Intracranial Aneurysms
Loss or disruption of the internal elastic lamina (IEL) is a characteristic feature of all IA. Indeed, in animal models, disruption of the IEL defines early aneurysm formation or early aneurysmal change (Fukuda et al, 2000; Hazama and Hashimoto, 1987; Kim et al, 1993; Morimoto et al, 2002; Nagata et al, 1980; Sadamasa et al, 2003). Beyond disruption of the IEL, another common feature of saccular IA is atherosclerotic change within the aneurysm wall. Killer-Oberpfalzer et al (2011) and Kosierkiewicz et al (1994) found that atherosclerotic lesions were present in all saccular IA. In the smallest aneurysms, diffuse intimal thickening (proliferating smooth muscle cells (SMCs)) and macrophages/lymphocytes are seen. In the largest aneurysms, advanced atherosclerotic lesions are found with phenotypically modulated SMCs, lipid-laden macrophages, and lymphocytes (Kosierkiewicz et al, 1994). Additionally, progression of atherosclerosis correlated positively with aneurysmal growth (Kosierkiewicz et al, 1994). Atherosclerosis appears to contribute to abdominal aortic aneurysm development as well (Dobrin, 1989; Nordon et al, 2009; Reed et al, 1992; Xu et al, 2001), which further supports the association between atherosclerosis and aneurysm formation in general. The two pathologies also share similar risk factors including smoking and arterial hypertension. Atherosclerosis is a disorder in which chronic inflammation is a critical pathogenic feature (Ait-Oufella et al, 2011; Andersson et al, 2010; Lahoute et al, 2011; Libby et al, 2010; Packard and Libby, 2008; Packard et al, 2009; Tedgui and Mallat, 2006). The possible association of IA with atherosclerosis and the critical role of inflammation in the pathogenesis of atherosclerosis could provide a common link between IA and inflammation. We hypothesize that pathogenic mechanisms underlying atherosclerosis also fundamentally underlie IA formation, progression, and rupture and this will be developed in further detail below.
Changes in Constituents of the Cerebral Blood Vessel Wall in Association with Intacranial Aneurysms
Perturbations of homeostasis in the cerebral blood vessel wall ultimately contribute to IA development and progression. In general, the highly organized structure of the cerebral blood vessel wall is transformed into a disorganized array with fewer distinct layers within the aneurysm wall. As mentioned, there is a striking loss or fragmentation of the internal IEL. Other histopathological features include irregularity of the luminal surface within the aneurysm sac, intimal hyperplasia, loss of cellular components, and disorganization of muscle fiber structure (Abruzzo et al, 1998; Draghia et al, 2008; Kataoka et al, 1999; Santiago-Sim and Kim, 2011; Scanarini et al, 1978; Schlote and Gaus, 1994). As a consequence of vascular remodeling, other changes in composition of the vessel wall have been noted including collagen content and extracellular matrix components. In the aneurysm, type I collagen and fibronectin appear dispersed throughout the wall in stark contrast to normal vessel wall where expression of type I collagen is restricted to the adventitia and fibronectin to the media (Austin et al, 1993). Furthermore, expression of type III and IV collagen and laminin appears to be decreased in aneurysm wall compared with normal vessels (Kilic et al, 2005).
Intracranial Aneurysms and Pathological Alterations in the Endothelium
Pathological changes have been noted in the endothelium. A healthy endothelium has antiatherogenic properties and promotes vasodilation while inhibiting platelet aggregation and adhesion, smooth muscle proliferation, and leukocyte adherence/pro-inflammatory cascades (Dumont et al, 2001a, 2001b, 2003b). Endothelial dysfunction and pathophysiological changes in the endothelium may be the inciting event in IA formation (Tamura et al, 2009; Wei et al, 2011; Xu et al, 2011). The potential importance of the role of the endothelium in IA pathology is highlighted by the fact that a susceptibility locus for IA formation in certain populations is found on chromosome 8q in a region that is required for formation and maintenance of endothelial cells (Bilguvar et al, 2008).
Hemodynamic stress is known to be a key factor in IA pathology (Santiago-Sim and Kim, 2011). Endothelial cells appear to undergo significant changes in response to hemodynamic stress/changes in their local hemodynamic environment (Aoki et al, 2011c; Kadirvel et al, 2007; Sakamoto et al, 2010; Szymanski et al, 2008). Both functional and morphological changes are seen in response to hemodynamic stress. In response to shear stress, endothelial cells may elongate, change alignment with respect to the direction of flow while developing actin stress fibers and may change cell density/migrate (Sakamoto et al, 2010; Szymanski et al, 2008). Hemodynamic stress may also alter signaling within the endothelium thereby promoting aneurysm formation. For example, shear stress appears to activate the prostaglandin E(2)-E-prostanoid(2) PGE(2)–EP(2) pathway (a mediating pathway of inflammation) in endothelial cells and amplifies chronic inflammation via nuclear factor-κB (NF-κB) (Aoki et al, 2011c).
Damage to the endothelial layer is a universal pathological feature seen in both experimental and human IA (Draghia et al, 2008; Jamous et al, 2005b; Kataoka et al, 1999; Kimura et al, 2010; Nagata et al, 1981; Tada et al, 2010, 2011; Tamura et al, 2009; Zhang et al, 2003). Morphological and pathological events leading to aneurysm formation in experimental models feature endothelial abnormalities as critical changes (Draghia et al, 2008; Jamous et al, 2005a, 2005b, 2007; Kataoka et al, 1999; Kimura et al, 2010; Nagata et al, 1981; Tada et al, 2010, 2011; Tamura et al, 2009). Aneurysm formation and progression has been categorized into stages in experimental models with endothelial changes representing the earliest changes seen (including partial loss of endothelial cells and later intimal swelling).
Intracranial Aneurysms and Pathological Changes in Vascular Smooth Muscle Cells
The media layer provides structural integrity to the vessel wall. Dynamic changes in the media and eventual loss of this layer appear to contribute to aneurysm formation (Austin et al, 1993). Vascular SMCs (VSMCs) show plasticity unlike other SMC. Although principally concerned with contraction, in response to environmental cues/stimuli VSMC can undergo phenotypic modulation to a pro-inflammatory, pro-matrix remodeling/dedifferentiated phenotype (Owens, 1998, 2007; Owens et al, 2004; Yoshida and Owens, 2005). The VSMC phenotypic modulation appears to be critically important in atherosclerosis and its related sequelae (Owens, 1998, 2007; Owens et al, 2004; Yoshida and Owens, 2005). Accumulating data suggest that cerebral VSMC undergo phenotypic modulation and eventually degeneration which may be crucial to aneurysm formation and progression. Although VSMC undergo phenotypic modulation during both IA formation and atherosclerosis, it is unclear whether the mechanisms underlying these changes are similar. Specifically, the role of low-density lipoprotein, oxidized products, and free radicals among others in the initiation of VSMC phenotypic modulation during IA formation remains uncertain.
From a global perspective, thinning of the media layer is noted with aneurysm formation and progression (Meng et al, 2011). In addition, degenerative changes are seen within medial SMC (Hazama and Hashimoto, 1987). Upon closer inspection, an early event involves VSMC undergoing phenotypic modulation from differentiated SMC concerned with contraction (characterized by the significant expression of contractile genes/proteins such as smooth muscle-myosin heavy chain, and smooth muscle-α-actin) to cells with a pro-inflammatory, pro-matrix remodeling phenotype characterized by the increased expression of inflammatory factors and MMP (Figure 1; Supplementary Figure 1) (Aoki et al, 2007b, 2010; Ishibashi et al, 2010; Kilic et al, 2005; Kolega et al, 2011; Merei and Gallyas, 1980; Nakajima et al, 2000; Pera et al, 2010; Sakaki et al, 1997; Sibon et al, 2008; Tada et al, 2011). Phenotypic modulation of VSMC was an early morphological observation in some IA walls where spindlelike VSMC, arranged in a parallel manner, may dissociate from each other to be transformed into spiderlike cells (Merei and Gallyas, 1980). The VSMCs have been shown to migrate into the intima and proliferate producing myointimal hyperplasia (Kosierkiewicz et al, 1994). This may be an early event as this is seen in the smallest aneurysms (Kosierkiewicz et al, 1994). This appears to be analogous to the observation of phenotypically modulated SMC leading to myointimal hyperplasia in atherosclerosis (Owens et al, 2004). Nakajima et al (2000) used immunohistochemical methods to clearly show that phenotypic modulation of VSMC is present in IA walls. Specifically, they showed decreased expression of contractile proteins (indicative of the differentiated phenotype) in aneurysm walls. Moreover, phenotypic modulation appeared to be more pronounced in ruptured compared with unruptured aneurysms and control arteries. There appeared to be transition of VSMC from a contractile (differentiated) phenotype to a synthetic (modulated) phenotype and eventual loss of both phenotypes in ruptured aneurysms. The authors concluded that phenotypic modulation of VSMC in the aneurysm wall appears to be related to a remodeling of the aneurysm wall and to a rupture mechanism (Nakajima et al, 2000). Other authors have confirmed VSMC phenotypic modulation in IA (Kilic et al, 2005; Pera et al, 2010; Sibon et al, 2008) including a progressive reduction of expression of contractile proteins such as α-actin from control arteries to unruptured aneurysms to ruptured aneurysms (Kilic et al, 2005). Sibon et al (2008) showed that there was a dramatic reduction in semicarbazide-sensitive amine oxidase (a key regulator of VSMC differentiation) as well as smooth muscle-myosin heavy chain, and this correlated with elastic lamellae thinning in an experimental aneurysm model. Other changes indicative of phenotypic modulation in VSMC in IA include changes in expression of transcription factors (such as Ets-1; Aoki et al, 2010), p47phox (Aoki et al, 2009e), and MMP (Aoki et al, 2007b; Kolega et al, 2011), which promote inflammation, reactive oxygen species, and matrix remodeling. Furthermore, collagen biosynthesis and processing was seen to be significantly inhibited in VSMC in an experimental aneurysm model (Aoki et al, 2009c). It appears that subsequent changes in the media layer include apoptosis/loss of VSMC, changes in VSMC proliferation, and thinning of the aneurysm wall (Frosen et al, 2004; Guo et al, 2007; Jayaraman et al, 2008; Kondo et al, 1998; Meng et al, 2007; Sadamasa et al, 2007; Sakaki et al, 1997; Takagi et al, 2002).
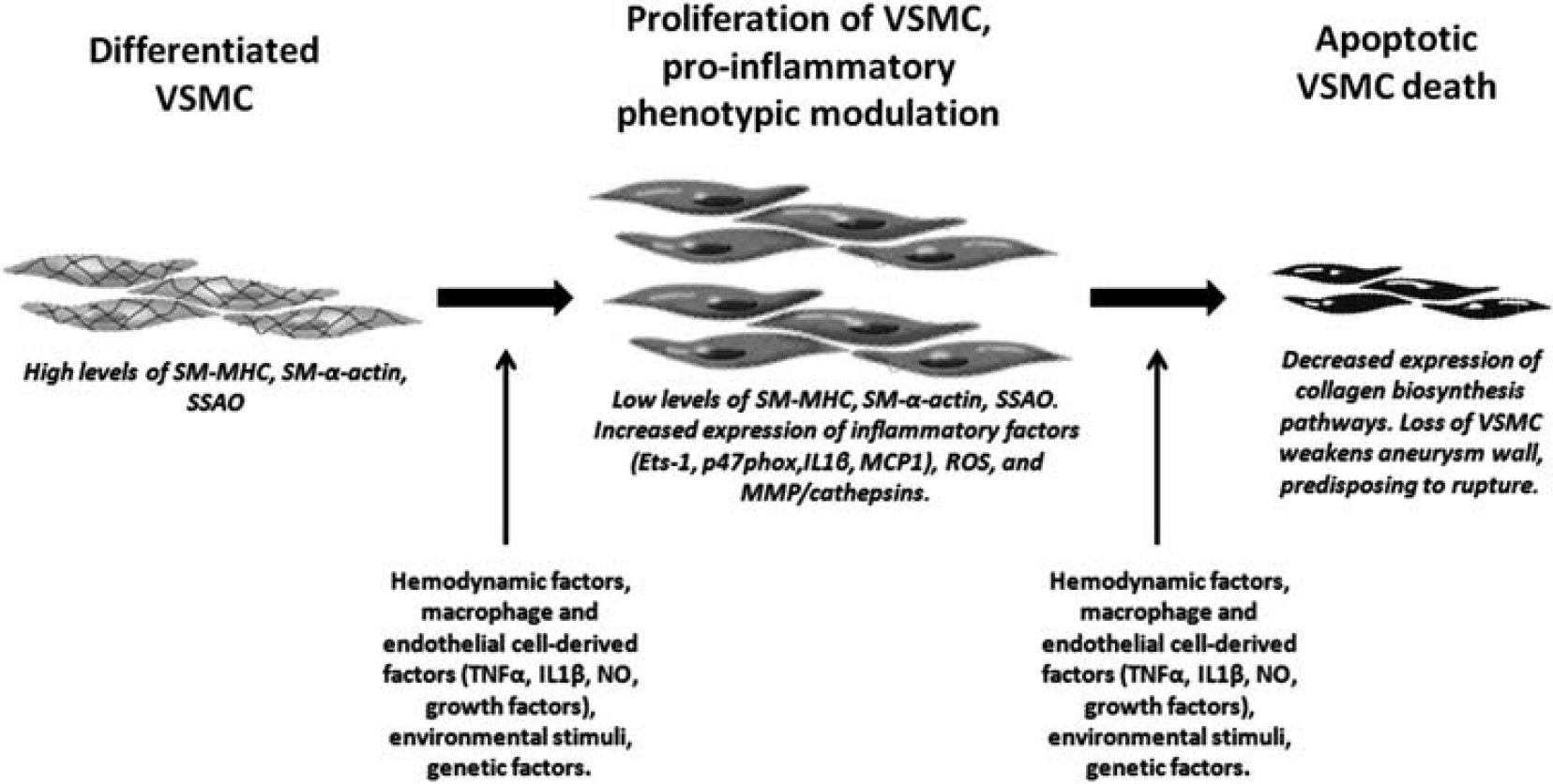
Vascular smooth muscle cells (VSMCs) in intracranial aneurysm (IA) wall. Phenotypic modulation of VSMC from a contractile to pro-inflammatory/pro-matrix remodeling phenotype within the aneurysm wall leads to myointimal hyperplasia, inflammation, and vessel wall degeneration. Subsequent apoptosis and VSMC death lead to a hypocellular thin wall with increased IA susceptibility to rupture. SM-MHC, smooth muscle-myosin heavy chain; SM-α-actin, smooth muscle-α-actin; SSAO, semicarbazide-sensitive amine oxidase; NO, nitric oxide; TNFα, tumor necrosis factor-α; MCP1, monocyte chemoattractant protein 1; IL1β, Interleukin 1β; ROS, reactive oxygen species; MMPs, matrix metalloproteinases.
The recent ability to culture cerebral VSMC from human IA (Bygglin et al, 2011) may provide additional insight into the role of the VSMC in IA pathology.
Intracranial Aneurysm and Inflammation
The central nervous system is an immunologically active environment where various constituents of the immune and inflammatory response, including leukocytes, cytokines, adhesion molecules, immunoglobulins (Igs), and complement (C) interact in a critically orchestrated manner (Dumont et al, 2003a). Although incompletely understood, the pathogenesis of IA seems to involve inflammation.
The Common Pathway
The common pathway for aneurysm formation involves endothelial dysfunction/injury, a mounting inflammatory response, VSMC phenotypic modulation, extracellular matrix remodeling, and subsequent cell death and vessel wall degeneration. Endothelial dysfunction initially and vascular remodeling subsequently are triggered by shear stress (Sho et al, 2002). This explains why IA is commonly found at arterial junctions, bifurcations or abrupt vascular angles where excessive hemodynamic stresses are exerted on arterial walls (Kondo et al, 1997). There is a close relation between wall sheer stress, endothelial dysfunction, and the downstream inflammatory reaction (Nixon et al, 2010). In fact, the activity of the inflammatory transcription factor NF-κB is intimately modulated by the magnitude of shear stress exerted on vessel walls (Mohan et al, 1997). Hemodynamic stress was also found to alter the expression of several other genes that contribute to IA formation including MMP and TIMP (tissue inhibitors of MMPs) in a spatially localized manner within the aneurysm wall (Kadirvel et al, 2007). Along these lines, Wang et al (2009) showed in canine models that in areas of high shear stress (arterial bifurcations), aneurysm wall remodeling is associated with interleukin 1β (IL1β) and MMP expression along with a loss of endothelial nitric oxide synthase expression. In line with these findings, endothelial cell injury was found by Jamous et al (2005a, 2007) to be the earliest change in aneurysm wall, followed by the formation of an inflammatory zone that leads to proteolytic destruction of the vascular extracellular matrix by MMP and ultimately to aneurysm formation. This disturbance in arterial homeostasis was elegantly illustrated by Kassam et al (2004) in a study that compared the expression of several molecules in samples from aneurysm domes and extracranial arteries. The ability to express proteins specific to flow modulation (prostacyclin-stimulating factor) and arterial repair (RAI) as well as the extracellular matrix (type III collagen) was found to be impaired within the ruptured domes when compared with control superficial temporal artery (STA) tissue. There was also a graded loss of expression of these molecules from different samples which was, according to the authors, suggestive of a continuum from aneurysm formation to rupture. As discussed above, phenotypic modulation of VSMC with subsequent cell death is also an integral part of the inflammatory reaction that leads to aneurysm formation and vessel wall degeneration.
Matrix metalloproteinases appear to be robustly produced by both leukocytes (such as macrophages; Aoki et al, 2007a) and VSMC (Aoki et al, 2007b; Ishibashi et al, 2010; Kolega et al, 2011) within the vessel/aneurysm wall. The MMP digests arterial wall extracellular matrix and causes further damage via upregulation of other proteinases and angiogenic factors (Tronc et al, 2000). Overexpression of MMP 9 in the wall of excised IA was first documented by Kim et al (1997) followed by Takemura et al (2010) who later showed via immunohistochemical staining the overexpression of MMP 1, 2, and 9 in aneurysm walls. Moreover, the levels of MMP 2 and 9 were found to be higher in ruptured compared with unruptured aneurysms in a series of 30 patients, suggesting that excessive breakdown of vessel extracellular matrix eventually leads to rupture (Jin et al, 2007). In their pioneering work on experimentally induced aneurysms in animal models, Aoki et al (2007a, 2008c) showed the overexpression of MMP 2, 9 and cysteine cathepsins (another type of proteinases) in aneurysm wall. Inhibitors of either MMP (Aoki et al, 2007a) or cathepsins (Aoki et al, 2008c) successfully blocked the progression of IA highlighting the crucial role of these collagenases in the pathogenesis of IA. In line with their previous findings, aneurysm progression was promoted in mice after knocking out TIMP (Aoki et al, 2007b).
Collectively, these data show that the common pathway for aneurysm formation begins with a hemodynamically induced endothelial dysfunction followed by the development of an inflammatory reaction and VSMC phenotypic modulation in the arterial wall that ultimately leads to the breakdown of extracellular matrix by various proteinases and eventual cell death (Table 1; Figure 2; Supplementary Figure 2).
Major inflammatory pathways and mediators involved in aneurysm formation

VSMC, vascular smooth muscle cell; NF-κB, nuclear factor-κB; MCP1, monocyte chemoattractant protein 1; IL, interleukin; NO, nitric oxide; MMP, matrix metalloproteinase; TNFα, tumor necrosis factor-α; Ig, immunoglobulin; TLR-4, Toll like receptor-4; PGE2, prostaglandin E2.
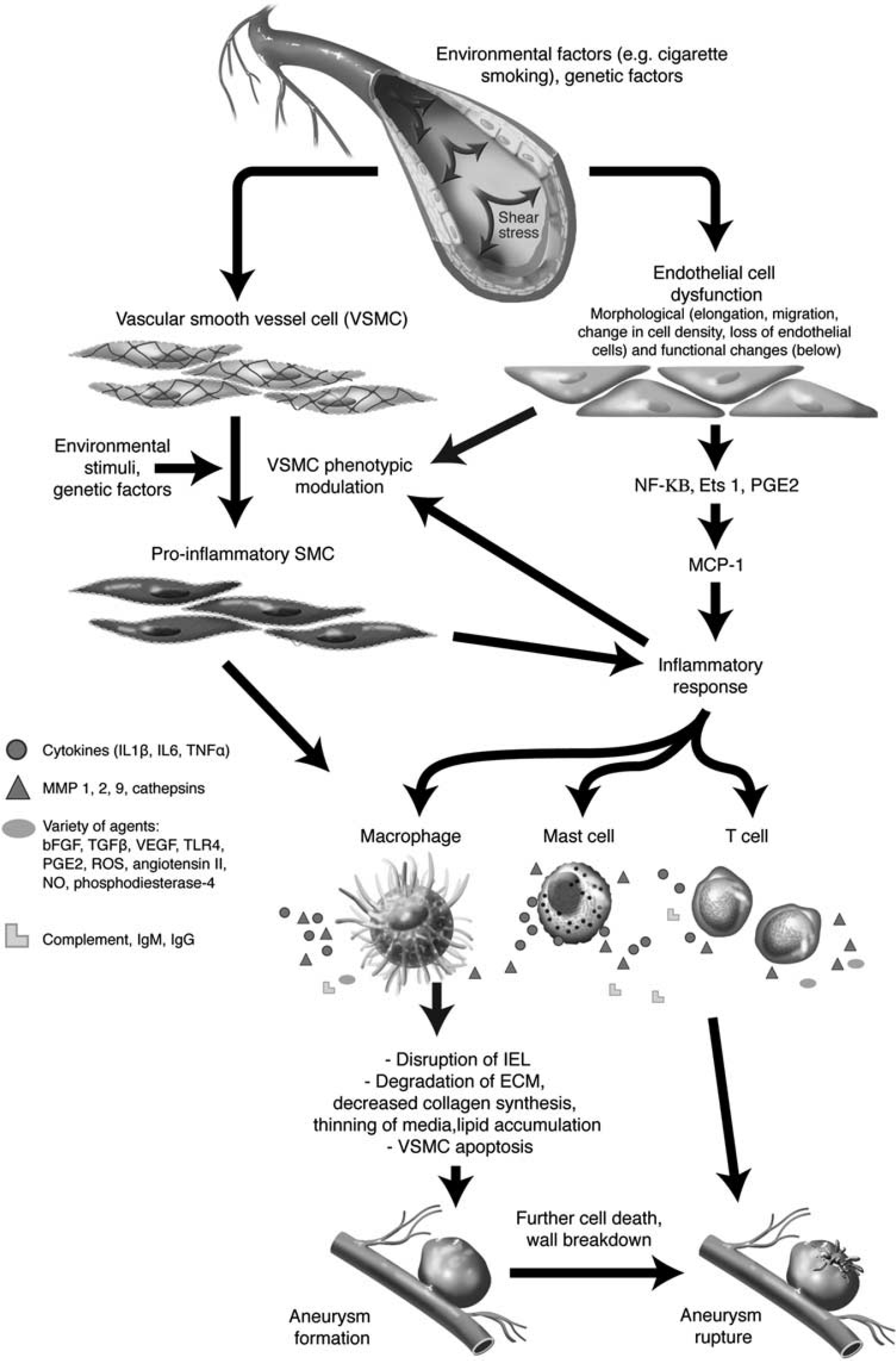
Inflammatory reaction associated with intracranial aneurysm (IA) formation. Aneurysm formation begins with a hemodynamically triggered endothelial dysfunction, followed by a mounting inflammatory response (implicating several inflammatory cells and mediators) and phenotypic modulation of VSMC from a contractile to pro-inflammatory/pro-matrix remodeling phenotype. The inflammatory response in vessel wall leads to disruption of IEL, extracellular matrix remodeling, and subsequent aneurysm formation. Further cell death and vessel wall degeneration ultimately culminate in IA rupture. VSMC, vascular smooth muscle cell; NF-κB, nuclear factor-κB; MCP1, monocyte chemoattractant protein 1; ECM, extracellular matrix; IEL, internal elastic lamina; IL, interleukin; TNFα, tumor necrosis factor-α; MMPs, matrix metalloproteinases; PGE2, prostaglandin E2; bFGF, basic fibroblast growth factor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; TLR4, Toll like receptor-4; ROS, reactive oxygen species; Ig, immunoglobulin; SMC, smooth muscle cell; NO, nitric oxide.
Leukocytes
The infiltration of inflammatory cells in general, and macrophages in particular, is a hallmark of IA (Chyatte et al, 1999; Frosen et al, 2004; Kataoka et al, 1999). Macrophages are thought to be the key factor in flow-induced vascular remodeling through the release of MMP (Nuki et al, 2009; Ota et al, 2009). In a recent well designed study by Kanematsu et al (2011), macrophage-depleted mice were found to have a substantially lower risk of developing IA compared with control mice (10% versus 60%, respectively). In addition, two studies have investigated the role of monocyte chemoattractant protein 1 (MCP1), a seemingly important chemoattractant of macrophages in atherosclerosis and abdominal aortic aneurysms (Egashira, 2003; Ishibashi et al, 2004), reporting a lower incidence of IA and inflammatory findings in MCP1-knockout mice (Aoki et al, 2009b; Kanematsu et al, 2011). MCP1 expression in VSMC of IA was found to be mediated by Ets-1, a transcription factor involved in several vascular inflammatory pathways (Aoki et al, 2010). Another key regulator of macrophage activation and recruitment is NF-κB. This transcription factor also specifically modulates the expression of several pro-inflammatory genes directly involved in aneurysm formation such as MMP, MCP1, and cytokines (Aoki and Nishimura, 2011). In a study by Aoki et al (2007c), NF-κB was overexpressed in walls of IA in rats and the formation of aneurysms was dramatically blocked after inhibition of NF-κB. The authors even hypothesized that the first step in IA formation is the activation of NF-κB in endothelial cells pursuant to hemodynamic stress (Aoki et al, 2007c). Aoki et al (2011a) extended their previous findings by showing that treating rats with chimeric decoy oligodeoxynucleotides that simultaneously inhibited NF-κB and Ets-1 1 month after aneurysm induction surgery decreased aneurysm size and thickened aneurysm walls of preexisting aneurysms. Furthermore, this treatment reduced expression of MCP1, macrophage infiltration and restored collagen biosynthesis in aneurysm walls (Aoki et al, 2011a). The role of MCP1, Ets-1, and NF-κB seems therefore pivotal in recruiting macrophages into the wall of IA, initiating the inflammatory reaction and contributing to aneurysm formation and progression.
Macrophages are not the only cells involved in the inflammatory reaction in the wall of IA. As previously noted, the endothelial cell is thought to be the initiator of the events. T cells also actively participate alongside macrophages in the inflammatory reaction (Chyatte et al, 1999; Frosen et al, 2004). In fact, infiltration of the vessel wall by macrophages and T cells associates with aneurysm rupture as reported by Frosen et al (2004) in a study that histologically compared 42 ruptured with 24 unruptured IAs. Furthermore, macrophage infiltration was associated with VSMC proliferation in the aneurysm wall. Based on these data, the authors hypothesized that macrophages may induce phenotypic modulation (and proliferation) in VSMC. Leukocyte infiltration and VSMC proliferation were seen in aneurysm samples resected <12 hours after rupture. It is unclear whether these changes were present before rupture or were simply a response to aneurysm rupture.
Mast cells are leukocytes involved in various inflammatory conditions such as allergy that act mainly through the release of mediators and cytokines (Krishnaswamy et al, 2006). Their role in the pathogenesis of atherosclerosis and abdominal aortic aneurysms has been well defined (Mayranpaa et al, 2009; Tang et al, 2009). Recently, Ishibashi et al (2010) assessed the role of these cells in IA formation using an experimental rat model. They found an increased number of mast cells during the formation of IA and an induction of MMP in VSMC after mast cell degranulation. Progression of IA was also halted after administration of a mast cell degranulation inhibitor through the blockade of different components of the inflammatory response. Based on the results of this study, it seems that mast cells constitute an integral part of the inflammatory response that leads to IA formation.
Complement and Immunoglobulins
Complement and Ig have also been studied as possible contributors to the pathogenesis of IA. Their presence in the walls of IA has been reported in human and animal studies (Aoki and Nishimura, 2011; Chyatte et al, 1999). The distinguished work of the Helsinki group from Finland has pointed to the importance of C in the pathogenesis of rupture of IA. In one study that compared ruptured with unruptured IA, the expression of membrane attack complex (C activation end product) was greater in ruptured samples and associated significantly with aneurysm wall degeneration and inflammatory cell infiltration (Tulamo et al, 2006). Later, the same group showed that activation of C occurs via the classical pathway as evidenced by the presence of classical pathway activators (IgG, IgM, C-reactive protein, oxidized low-density lipoprotein) in the IA wall (Tulamo et al, 2010a). In a recently published paper, they showed that in contrast to the luminal part, the outer wall of IA lacked C inhibitors and was subject to full-blown C activation (Tulamo et al, 2010b). However, the authors failed to provide a reasonable explanation as to how C activation can lead to cell wall degeneration and ultimately rupture of IA. These data favor a substantial role for C in the pathogenesis of IA progression and rupture. However, the need for further investigation of this facet of the inflammatory response is apparent.
Cytokines, Growth Factors, and other Mediators
Cytokines are a heterogeneous group of soluble short acting proteins and peptides that are powerful mediators of the immune and inflammatory response (Ait-Oufella et al, 2011; Sprague and Khalil, 2009; Tedgui and Mallat, 2006). Several cytokines have been found to be involved in the pathogenesis of IA especially IL1β, IL6, and tumor necrosis factor-α (TNFα). The IL1β may be produced by monocytes, macrophages, T and B cells, dendritic cells, endothelial cells, and VSMC (Sprague and Khalil, 2009). The IL1β is induced in the early stages of IA formation in mice and prominently localizes to VSMC (Moriwaki et al, 2006). Its presence promotes VSMC apoptosis (Moriwaki et al, 2006). The IL1β−/− mice were compared with age-matched wild-type mice. Although no difference was detected in the rate of aneurysm development between IL1β−/− and wild-type mice, there were significantly fewer advanced aneurysms seen in IL1β−/− mice. Moreover, apoptosis was significantly reduced in aneurysms in IL1β−/− compared with wild-type animals (Moriwaki et al, 2006). Collectively, these data suggest that IL1β is important in aneurysm progression (Moriwaki et al, 2006).
Case–control studies using allelic-association models have provided indirect evidence for a potential role of IL6 in IA pathogenesis. Specifically, IL6 gene polymorphisms were shown to be associated with IA. The 572C/174G haplotype and the GG genotype predisposed to IA in a British (Morgan et al, 2006) and Chinese (Sun et al, 2008) population, respectively. More recently, Zhang et al (2011) showed an association of IL6-572G/C gene polymorphisms with IA in a Cantonese population, suggesting that the IL6-572G/C gene is an important candidate gene for IA. To overcome issues related to sample size and variability of results, McColgan et al (2010) conducted a meta-analysis based on all available published data. They found that the IL6 gene G572C polymorphism associated with IA (odds ratio 7.08, 95% confidence interval 2.85 to 17.57). Interestingly, the IL6/G174C polymorphism was associated with a significant protective effect against IA (odds ratio 0.49, 95% confidence interval 0.25 to 0.95). Taken together, it appears that IL6 is a candidate gene that contributes to IA pathogenesis. Further investigation into this association, however, is required to define the role of IL6 in IA biology.
Tumor necrosis factor-α is a potent pro-inflammatory cytokine that is also an inductive signal in initiation of apoptosis (Ait-Oufella et al, 2011; Jayaraman et al, 2005, 2008; Sprague and Khalil, 2009; Tedgui and Mallat, 2006). In addition, TNFα is known to activate MMP (Polavarapu et al, 2005; Thomson et al, 2012), which is critical in aneurysm pathology. Risk factors associated with aneurysm development have been associated with TNFα induction (Jayaraman et al, 2008), including hypertension (Munoz et al, 1999), hemodynamic flow (Gonzalez et al, 1992; Hoi et al, 2004; Jou et al, 2005; Kapadia et al, 2000), aging (Hartel et al, 2005; Kirwan et al, 2001; Paolisso et al, 1998), gender (Ferreri, 2007; Huang et al, 2005), alcohol (Lanzke et al, 2007; Luedemann et al, 2005), and smoking (Anto et al, 2002; Sun et al, 2007; Wright et al, 2007). In a study by Jayaraman et al (2005), the expression of TNFα and its proapoptotic downstream target FAS were increased in IA, indicating that TNFα has a proapoptotic and pro-inflammatory action in the wall of aneurysms. Building on this prior work, Jayaraman et al (2008) report that elevated TNFα in human IA correlates with increased expression of intracellular calcium release channels that regulate intracellular calcium, Toll like receptors that mediate innate immunity and a reduction in expression of TIMP-1. Further indirect support of a role for TNFα in aneurysm pathogenesis includes association of TNFα gene polymorphisms with IA (Fontanella et al, 2007; Low et al, 2011). In summary, data suggest that TNFα may be an important factor in IA pathogenesis but additional investigation is necessary to more clearly elucidate its role.
A variety of growth factors may have a basic role in aneurysm formation and rupture through wall remodeling. Receptors for vascular endothelial growth factor, basic fibroblast growth factor, and transforming growth factor β were found to be associated with the inflammatory reaction and the remodeling process in the walls of IA (Frosen et al, 2006). A decrease in the levels of transforming growth factor α may also contribute to aneurysm formation although the mechanisms involved remain speculative (Kilic et al, 2005).
Angiotensin II is a potent vasoconstrictor, a mediator of atherosclerosis, and a potent pro-inflammatory stimulant (MacKenzie, 2011; Montecucco et al, 2009). A potential role of angiotensin II in IA, however, has not been clearly elucidated. Angiotensin II has an important role in aortic aneurysms. Based on this premise, Aoki et al (2009d) examined a role for angiotensin II in a rodent model of IA. They found that angiotensin type I receptor was not upregulated and subcutaneous administration of an angiotensin type I receptor antagonist did not inhibit inflammation nor cerebral aneurysm formation (Aoki et al, 2009d). A recent review suggested that there is strong, albeit circumstantial, evidence for a relationship between the brain renin–angiotensin system and the pathogenesis of IA (Shoja et al, 2011). They emphasized the need for research focused on the association between polymorphisms in renin–angiotensin system-related genes and the incidence of IA (Shoja et al, 2011).
Recently, endothelial and neuronal nitric oxide synthase, two enzymes that generate nitric oxide, were shown to have a protective role against IA formation (Aoki et al, 2011b). This could be explained by the vasodilator action of nitric oxide that relieves hemodynamic stress and prevent the subsequent inflammatory reaction in vessel walls (thereby maintaining normal critical endothelial function).
Gene Expression Profile
Numerous studies have recently profiled gene expression in IA, using microarray or PCR techniques, in an attempt to characterize the pathogenesis of formation and rupture of these lesions. These studies are potentially powerful and informative but have limitations including the status of the sample aneurysm (ruptured versus symptomatic unruptured versus asymptomatic unruptured, timing of harvest versus onset of rupture, location, etc.), the tissue used for control (extracranial versus intracranial vessel, small versus large vessel, etc.), the size of the cohort examined, and the techniques used for analysis. Not surprisingly, the results of these studies are heterogeneous and leave considerable room for future investigation (Table 2).
Gene expression profile of IA
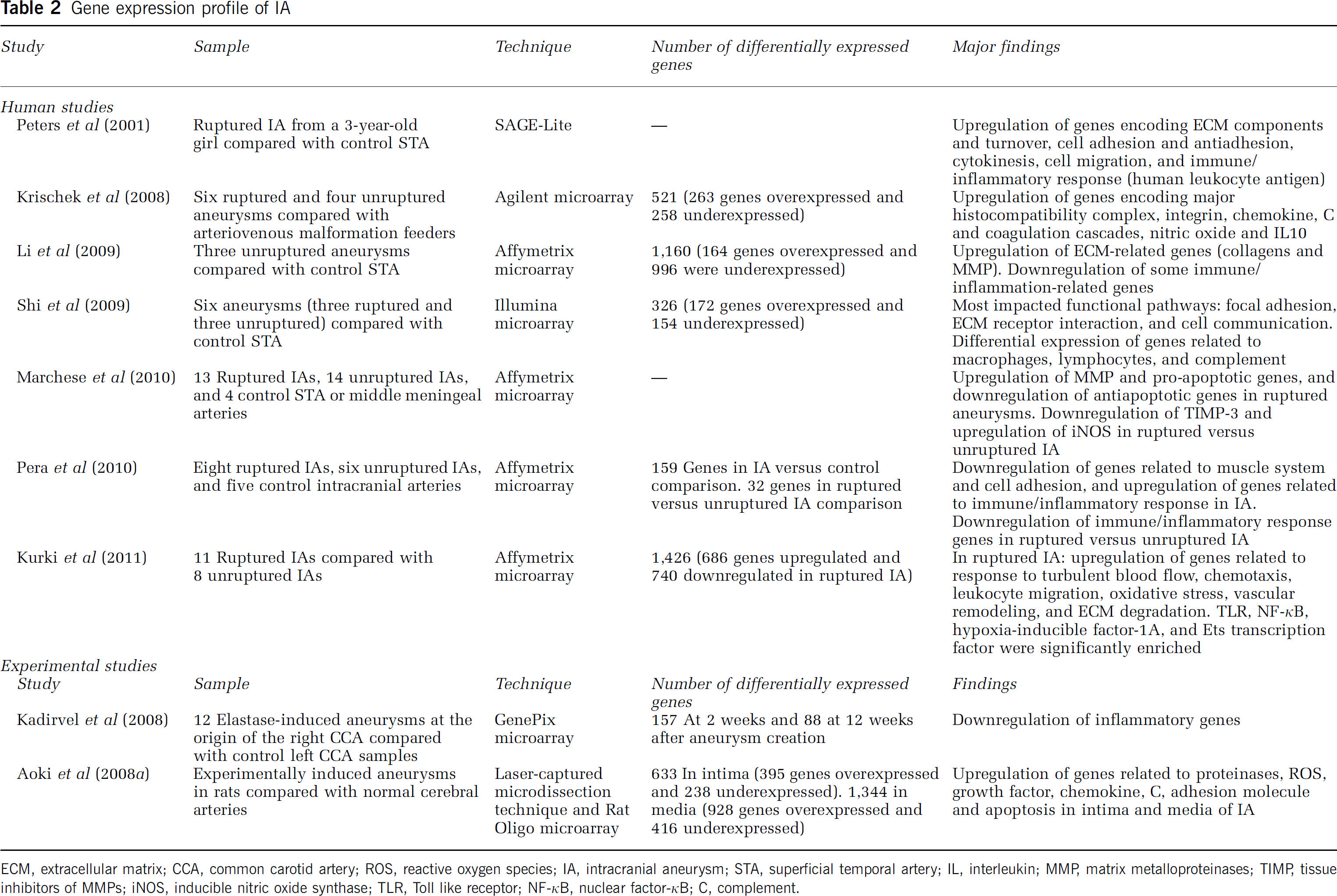
ECM, extracellular matrix; CCA, common carotid artery; ROS, reactive oxygen species; IA, intracranial aneurysm; STA, superficial temporal artery; IL, interleukin; MMP, matrix metalloproteinases; TIMP, tissue inhibitors of MMPs; iNOS, inducible nitric oxide synthase; TLR, Toll like receptor; NF-κB, nuclear factor-κB; C, complement.
In an early preliminary study by Peters et al (2001), the authors compared a single aneurysm from a 3-year-old girl with control STA using a global gene expression analysis approach (SAGE-Lite). They found overexpression of genes encoding extracellular matrix components, genes involved in extracellular matrix turnover, cell adhesion and antiadhesion, cytokinesis, and cell migration. Upon careful inspection, the authors found changes in expression of messenger ribonucleic acid involved with inflammation and immune response including major histocompatibility complex class I and II markers and Ig. It is unclear whether a single aneurysm in a 3-year-old patient can be generalized to IA in the general population. As an extension of this prior work, this same group examined changes in expression of genes between aneurysm domes and control STA or occipital artery vessels in a much larger cohort (Kassam et al, 2004). However, this study only focused on select genes concerned with flow modulation and arterial repair identified in the aforementioned preliminary study and did not examine inflammatory genes.
Next, Krischek et al (2008) conducted a networked-based gene expression analysis of IA tissue using oligonucleotide microarrays. They examined six ruptured and four unruptured aneurysms and compared these with control intracranial arteries (obtained from feeding arteries to arteriovenous malformations at the time of craniotomy). This use of an intracranial vessel as a control (even though it was on a feeding vessel to an arteriovenous malformation) is a strength as extracranial vessels (which are easier to obtain) were used in previous work and may not be representative control vessels. They found 521 differentially expressed genes. The two most significantly associated gene ontology terms were antigen processing and immune response. Upon further analysis, the most significant canonical pathways involved in aneurysm tissue included antigen presentation, integrin signaling, chemokine signaling, C and coagulation cascades, nitric oxide signaling in the cardiovascular system, and IL10 signaling. Real-time RT-PCR provided confirmation of these results. The authors provided strong evidence for major histocompatibility complex class II gene overexpression in human IA tissue and concluded that antigen-presenting cells (macrophages and monocytes) have a key role in IA formation.
Kadirvel et al (2008) conducted a gene expression analysis of experimental saccular aneurysms using deoxyribonucleic acid microarrays in their established rabbit elastase model. They found that multiple genes in diverse pathways were differentially expressed in the rabbit aneurysm model. Inflammation-related genes were not found to be upregulated. It is important to note that inflammation is rarely observed in this model. In addition, only 209 genes were examined using this custom-made rabbit microarray (which is a limited repertoire of genes examined compared with current microarrays in other species).
Aoki et al (2008a) conducted an interesting experiment where they examined a gene expression profile of the intima and media in an experimental rodent model of IA using laser-captured microdissection technique and microarray analysis. The microarray used examined 41,012 genes. They showed that 633 genes were differentially expressed between normal cerebral artery and cerebral aneurysm in the intima (395 showing increased and 238 showing decreased expression). In the media, 1,344 genes were differentially expressed (with 928 showing increased and 416 showing decreased expression). Further analysis of genes that showed increased expression indicated that these genes were involved in pathways concerned with proteinases, reactive oxygen species, growth factor, chemokine, C, adhesion molecules, and apoptosis in both the intima and media of aneurysmal walls. Additionally, some genes showed an opposite expression pattern in the intima versus media suggesting a different role between endothelial cells and VSMC in the process. The authors concluded that IA formation and progression appears to be closely related to vascular inflammation, degeneration of extracellular matrix, and apoptosis.
Li et al (2009) conducted a transcriptome-wide characterization of gene expression associated with unruptured IA. They examined three unruptured aneurysms and compared these with STA control tissue. They found that 1,160 genes were differentially expressed (164 were upregulated and 996 were downregulated). Extracellular matrix-related genes and MMP were significantly upregulated. However, in contrast to prior reports, a number of immune/inflammation-related genes were found to be downregulated in IA. The authors suggested that the observed downregulation of many immune/inflammation-related genes may be due to differences in aneurysm samples compared with prior studies (unruptured aneurysms in this case compared with ruptured aneurysms in others). Furthermore, the authors suggest that inflammation may be a response to hemorrhage rather than a contributor. An alternative explanation may be that inflammation leads to rupture (previous authors show that the inflammatory response is present shortly after rupture (Frosen et al, 2004) and not simply a response to rupture). Again, this study examined only three aneurysm samples and the control was the extracranial STA.
Shi et al (2009) conducted Illumina microarray analysis on human IA via comparison of aneurysm wall and STA tissue from six consecutive patients (three ruptured and three unruptured lesions). They found that 326 genes were differentially expressed between IA and STA (>2-fold change with a false discovery rate <0.01). The authors found differentially expressed genes involved focal adhesion, extracellular matrix receptor interaction, cell communication, inflammatory response, and apoptosis. This study was limited by its small sample size and use of STA as control tissue. However, they conducted a careful statistical analysis using stringent criteria to denote significance.
Marchese et al (2010) conducted another deoxyribonucleic acid microarray analysis of gene expression in aneurysm walls versus control (STA or middle meningeal) arteries. They included both ruptured and unruptured aneurysms. In ruptured aneurysms, genes showing differential expression included structural proteins of the extracellular matrix, members of the MMP family and genes involved with apoptosis. When comparing ruptured with unruptured aneurysms, TIMP-3 was seen to be markedly downregulated and inducible nitric oxide synthase was significantly overexpressed. The authors conclude that the inhibitor of MMP, the pathway of nitric oxide, and the apoptotic process have an important role in weakening of the vessel wall that can result in formation and rupture of IA. Changes in specific inflammation-related genes were not emphasized.
In a well-designed microarray study, Pera et al (2010) examined transcription profiles in ruptured (n=6) and unruptured (n=4) IA compared with control middle meningeal arteries. There were changes in expression of 159 genes compared with control with 131 genes showing common directions of change in both ruptured and unruptured IA. They found consistent downregulation of genes involved with the muscle system and cell adhesion with upregulation of genes involved with the immune system and inflammatory response. Interestingly, the expression of genes related to the immune system and inflammation was found to be reduced in ruptured compared with unruptured aneurysms, suggesting a protective role of inflammation against rupture. This latter observation leaves the role of inflammation unclear; the authors suggest that inflammation may participate in the healing process within IA protecting against IA rupture. Limitations such as the limited number of samples, the use of the noncerebral middle meningeal vessel, and a potential discrepancy in baseline risk factors (including high incidence of hypertension in patients with unruptured aneurysms) should be noted.
In a recent study, Kurki et al (2011) compared gene expression patterns in 11 ruptured and 8 unruptured IA using oligonucleotide microarrays. A total of 686 genes were found to be significantly upregulated and 740 were downregulated in ruptured IA. Upregulated pathways included response to turbulent blood flow, chemotaxis, leukocyte migration, oxidative stress, vascular remodeling, and extracellular matrix degradation. Significantly enriched genes were Toll like receptor, NF-κB, hypoxia-inducible factor-1A, and Ets transcription factor binding sites. These data add to the growing body of evidence, suggesting that the biology of ruptured IA is distinct from that of unruptured IA, with inflammation being a key factor in the pathway leading to IA rupture. Among the limitations of this study are the specific ethnicity of the studied population (genetically homogeneous Finnish population) and the small sample size (though larger than previous studies). Additionally, it is hard to conclude confidently based on the results of this study that inflammation was the cause rather than the consequence of aneurysm rupture. The findings of this study are in line with those of previous studies comparing ruptured and unruptured aneurysms. Kataoka et al (1999) compared histologically 44 ruptured with 27 unruptured aneurysms and found that ruptured aneurysms manifested significantly more endothelial damage, structural changes, and inflammatory cell invasion compared with unruptured aneurysms. There was also a significant correlation between the degree of inflammatory cell invasion and the level of fragility of the wall. Importantly, the time from rupture to resection did not show any correlation with the degree of inflammation, which indicates that the inflammatory response was probably present before IA rupture (since an acute inflammatory response would be expected to change over time). Along similar lines, Frosen et al (2004) compared histologically 42 ruptured with 24 unruptured IA and noted that apoptosis, deendothelialization, luminal thrombosis, SMC proliferation, and T-cell/macrophage infiltration associated with IA rupture. Collectively, these studies indicate that the pathobiology of ruptured aneurysms could be substantially different from that of unruptured aneurysms which potentially explains why only a minority of IA progresses to rupture.
Overall, it appears that alterations in expression of immune system and inflammation-related genes are frequently observed in IA from human and experimental models and highlight their importance in the pathogenesis of IA formation, progression, and rupture. Differences in technique of ribonucleic acid harvest/extraction, microarray used, statistical analysis, type of aneurysm studied, and type of control vessel used may explain the heterogeneous results across studies.
Therapies Targeting the Inflammatory Cascade
A multitude of therapeutic strategies have been investigated in animal models with promising results (Table 3). Through their inhibitory action on NF-κB, three different statins were found to block various stages of the inflammatory reaction, decrease degenerative changes in the vessel wall and subsequently halt IA progression (Aoki et al, 2008b, 2009a; Kimura et al, 2010). However, despite these early promising results recent data question the efficacy of statins in reducing aneurysm development/progression. Tada et al (2011) have shown that a lower dose of pravastatin (5 mg/kg per day) reduced endothelial damage and inhibited aneurysm formation in an experimental rodent model. However, doses of 25 and 50 mg/kg per day and simvastatin (at 5 mg/kg per day) promoted aneurysm growth and high-dose pravastatin induced aneurysm rupture. These deleterious effects of statins were associated with an increase in apoptotic caspase-3 levels and TUNEL-positive cells, suggesting that statins exert dose-dependent disparate effects. Marbacher et al (2011) conducted a case–control study to examine whether statins reduced the risk of aneurysm development. Although hypertension and smoking significantly increased the risk of IA development, no overall association was found between statin use and incidence of IA formation. Overall, statins appear to show promise in altering IA biology, however, further investigation is necessary to clarify some recently emerging conflicting data.
Targeted therapies for intracranial aneurysms (IAs)

ODN, oligodeoxynucleotide; ARB, angiotensin-II receptor blocker; IEL, internal elastic lamina; MCP1, monocyte chemoattractant protein 1; SAH, subarachnoid hemorrhage.
Promising results were reported with an inhibitor of phosphodiesterase-4 (Ibudilast), a cyclic adenosine monophosphate-specific enzyme involved in various inflammatory diseases (Yagi et al, 2010). Several angiotensin receptor blockers were also tested with varying results (Aoki et al, 2009d; Kimura et al, 2010; Tamura et al, 2009). Perhaps, the most impressive strategy, thus far, involves NF-κB inhibition that caused a dramatic decrease in the inflammatory response and IA incidence in rats (a 60% decrease; Aoki et al, 2007c). Whether these results from animal studies can translate into safe and effective therapies in clinical practice is unknown. Additionally, as data accrue concerning which constituents of the inflammatory response appear critical in aneurysm formation, progression, and rupture, more specific and efficacious therapeutic strategies can be devised and tested. It is worth noting that none of these animal studies has assessed the risk of rupture of IA, which keeps questions related to their capacity to prevent subarachnoid hemorrhage unanswered, even in animals.
In a recent case–control study from subjects enrolled in the International Study of Unruptured Intracranial Aneurysms, Hasan et al (2011) found that patients with unruptured aneurysms who used aspirin three times weekly or more had a lower risk of hemorrhage compared with those who never used aspirin. The authors postulated that this protective effect of aspirin stems from its inhibitory effects on several inflammatory mediators implicated in the pathogenesis of IA. This interesting finding could open new therapeutic perspectives in the prevention of aneurysmal subarachnoid hemorrhage.
Conclusion
Despite obvious limitations of animal models and difficulties in performing human studies, there is a growing body of evidence suggesting that inflammation underlies the development of IA. The mechanisms seem at least complex and further investigation is needed to answer basic questions regarding the role of inflammation in IA pathogenesis in general and rupture in particular. We believe that future research and efforts should be primarily directed toward revealing the genetic and molecular platform associated with rupture of IA. Such knowledge may pave the way for the development of novel targeted therapies or even the conception of molecular-based imaging studies identifying rupture-prone IA.
Footnotes
Acknowledgements
The authors would like to thank Mr Paul Schiffmacher for his elegant work on the illustrations.
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.