
Abstract

P068. Identification of a pre-clinical in vivo receptor occupancy tracer molecule for the MCH1 receptor
Eli Lilly & Company, Indianapolis, Indiana, USA
Background
Due to the MCH1 receptor being a target of long standing interest in the pharmaceutical industry, there is interest in generating a MCHR1 receptor occupancy (RO) ligand suitable for in vivo imaging experiments to facilitate drug discovery efforts and ultimately for translation to a human PET tracer. However, the identification of robust clinical and pre-clinical target engagement biomarkers for this target has remained a challenge. Recently, the BANYU group reported high affinity radiotracer with low non-specific binding, however it suffered from low brain penetration making it unsuitable as an in vivo RO ligand. A subsequent publication from the BANYU group reported molecules with improved brain penetration.
Methods
Herein is an analysis of the SAR disclosed by BANYU combined with the use of computational models to predict parameters related to brain penetration and non-specific binding.
Results
This analysis led us to propose the generation of a matrix of molecules.
Conclusions
An MCHR1 tracer molecule was identified.
P069. GMOM analogs for PET imaging of the NMDA receptor: synthesis and evaluation
VU University Medical Center, Department of Nuclear Medicine & PET Research, Amsterdam, The Netherlands
Background
N-methyl-D-Aspartate receptors (NMDAr) are a subtype of glutamaterigic ligand-gated ion channels. The NMDAr is involved in many physiological processes and neurological disorders, including Alzheimer's disease, Parkinson's disease, stroke, epilepsy and schizophrenia. Imaging of the NMDAr in the living human brain is warranted to further elucidate the role of NMDA receptors in health and disease [1,2]. To date, the SPECT ligand [123I]CNS 1261 is the only tracer that has been used in in patients affected by schizophrenia. Unfortunately, using this tracer, small changes in NMDAr distribution could not be quantified [3]. [11C]CNS 5161, [18F]fluoromemantine, [123I]iodo-MK-801 and [11C]ketamine showed promising characteristics in vitro, but in human results were not encouraging [4].
The ultimate purpose of the present study is to develop new PET ligands for the NMDAr ion channel. To this end, the 1-(2-chloro-5-(methylthio)phenyl)-3-phenylguanidine structure of GMOM [2] was used as a template for a new series of substituted N,N-diarylguanidines. Compounds showing high affinity for the NMDAr ion channel will be selected for labelled with either carbon-11 or fluorine-18.
Methods
Analogs of GMOM were synthesized using existing methods [5]. Using rat forebrain membranes, competition binding assays on NMDAr were performed against [3H]MK-801.
Results
A series of 18 substituted N,N-diarylguanidines was synthesized in yields comparable with other analogs synthesized previously [6]. Ki values are shown in the Table (nM).
Conclusions
Introducing either one or more methylene groups between R1 and the guanidine moiety, or pyridinyl in R1 was not tolerated, as binding affinity decreased dramatically. In contrast, a methyl group at R2 increased binding affinity. Replacing methyl at the methoxy moiety with 1-fluoropropane decreased binding affinity 10 fold. Substitution of hydrogen by fluorine in the methoxy moiety increased binding affinity up to two-fold compared with GMOM, therefore these two analogs are selected for labeling with carbon-11.
Acknowledgements
This work was supported by CTMM LeARN, work package 02N-101-01.
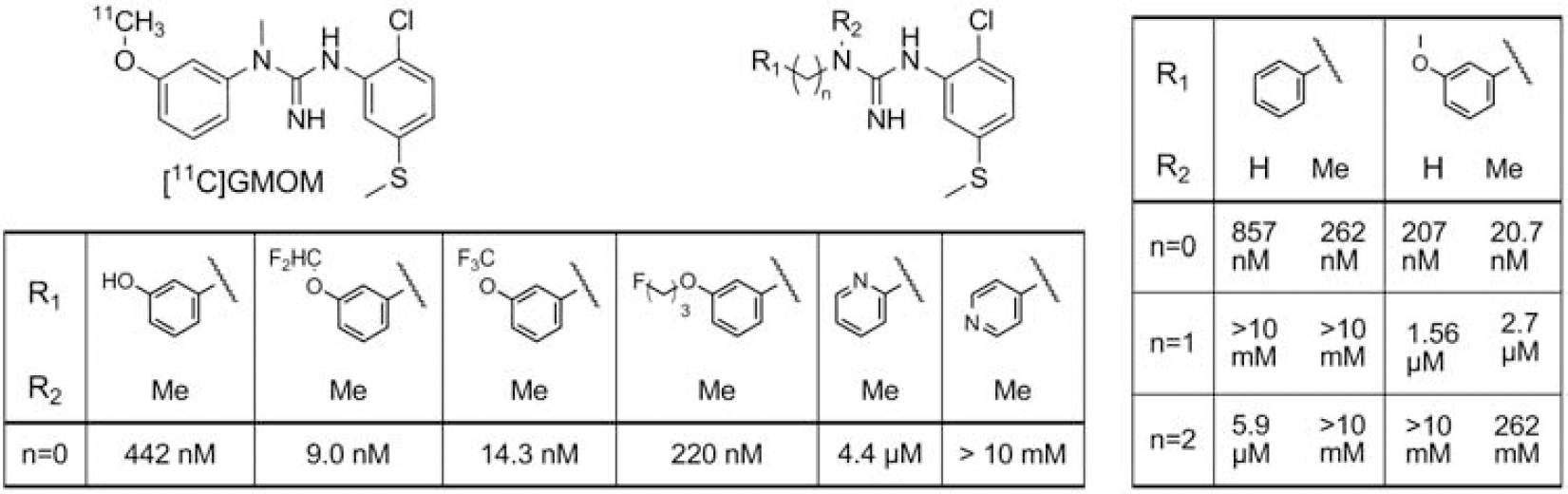
Chemical structures of [11C]GMOM and analogs and Ki values of substituted N,N-diarylguanidines for the NMDAr ion channel site (nM).
P070. Synthesis and PET evaluation of a novel F-18 labeled deuterated fluorodeprenyl ([18F]fluorodeprenyl-D2) radioligand to study MAO-B activity
1Karolinska Institutet, Department of Clinical Neuroscience, Stockholm, Sweden; 2Bayer Healthcare AG, Global Drug Discovery, Berlin, Germany
Background
Monoamine oxidases (MAO) are important enzymes regulating the levels of monoaminergic neurotransmitters and bioactive monoamines by catalyzing their deamination. MAO-B inhibitors are widely used in the treatment of e.g. Parkinson's disease (PD) and depression. L-Deprenyl is a selective MAO-B inhibitor, has been labeled with 11C and used in PET studies to image the distribution of available MAO-B in the human brain. 1 In the brain regions being rich in MAO B, the tracer is trapped at a rate which exceeds the rate at which it is delivered by the plasma. Bis-deuterium-substituted L-deprenyl ([11C]L-deprenyl-D2) has been introduced to reduce the rate of radiotracer trapping in the human brain. 2 Carbon-11 labeled compounds are less suitable for distribution to external clinics because of its relatively short half-life. Recently, we reported a novel fluorine-18 labeled fluoro analogue of L-deprenyl to image MAO-B in monkey brain.3,4 In this project our aim was to develop a fast and efficient synthetic method for labeling novel bis-deuterium substituted L-deprenyl analogues with fluorine-18 towards N-[(2S)-1-[18F]fluoro-3-phenylpropan-2-yl]-N-methyl(1,1-2H2)prop-2-yn-1-amine ([18F]fluorodeprenyl-D2).
Methods
In vitro MAO inhibition was determined for 19F-fluorodeprenyl-D2 in an enzymatic assay with kynuramine as substrate using pargyline and L-deprenyl as control inhibitors for MAO-B and clorgyline as control inhibitor for MAO-A. Precursor (
Results
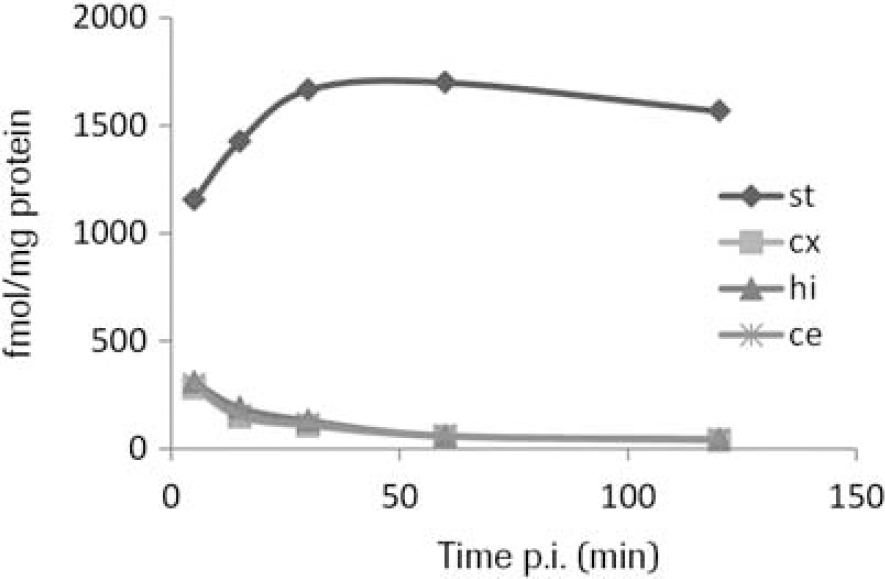
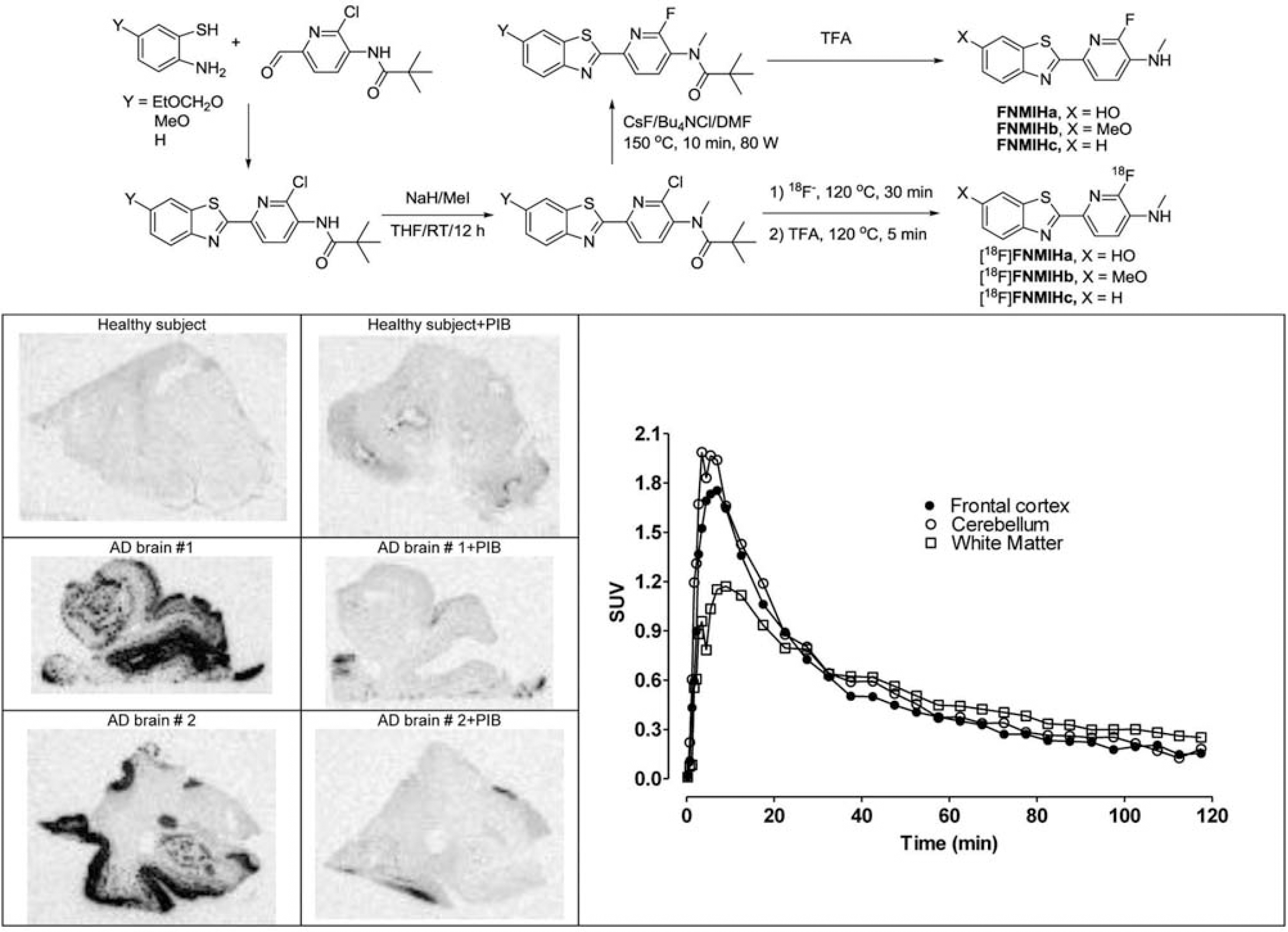
Fluorodeprenyl-D2 inhibited the MAO-B activity with an IC50 of 227±36.8 nM (L-deprenyl=13±0.4 nM) and the MAO-A activity with an IC50 of >50 μM. The radiolabeling was achieved by one step nucleophilic substitution reaction of the chloride precursor (5) by [18F]fluoride in anhydrous DMSO. The overall radiosynthesis including fluorination, HPLC purification and radiotracer formulation was completed in 80 minutes. The incorporation yield of the fluorination reactions was >70%, the radiochemical purity was >99% and the specific radioactivity >200 GBq/μmol at the time of administration. The radioligand [18F]fluorodeprenyl-D2 was found to be stable, with a radiochemical purity of more than 99% at 3 h after formulation in a sterile phosphate buffered solution (pH=7,4). In vivo there was a high uptake in the monkey brain (240 SUV (%) at 4 min) with higher amounts in the striatum and thalamus compared to the cortex and cerebellum (Figure 2). Metabolite studies demonstrated 40% unchanged radioligand at 120 min post injection.

Synthesis scheme.
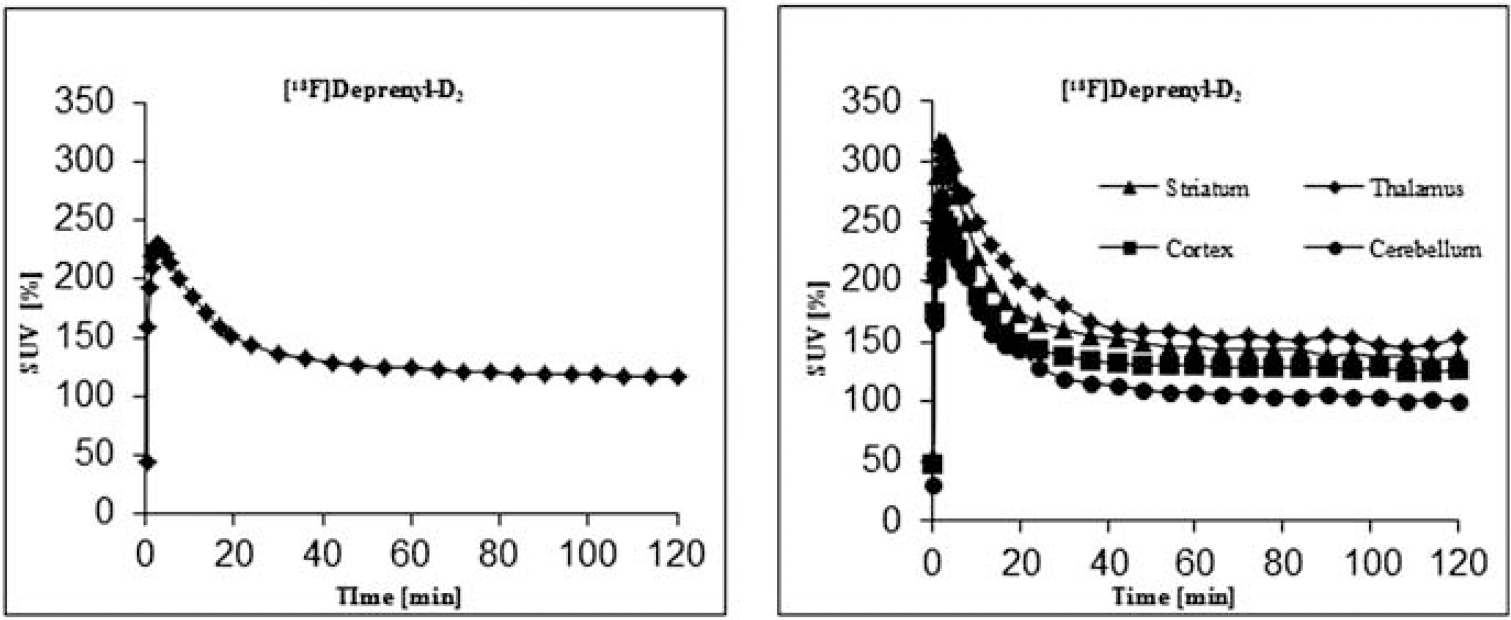
Brain uptake expressed as percent standardised uptake value (%SUV). Time activity curves in whole brain and in different brain regions.
Conclusions
Radiolabeling of a novel fluorine-18 analogue of bis-deuterium substituted L-deprenyl (
P071. Specificity assessment of the GABAA receptor α5 subtype preferring radiotracer [3H]RO15-4513
F. Hoffmann-La Roche Ltd, Basal, Switzerland
Background
RO15-4513 is a well-established partial inverse agonist at the GABAA receptor showing preferential binding to the α5-subunit containing receptor subtype (GABRA5). Tritiated RO15-4513 as well as [11C]-labeled RO15-4513 are widely used tools to analyze the GABRA5 subtype in vitro and in vivo. The aim of this study was to investigate in detail the in vitro binding specificity of RO15-4513 for the GABRA5 subtype.
Methods
A CEREP® screen of RO15-4513 was performed to test ligand binding to a battery of more than 80 CNS targets. Sagittal brain sections of GABRA5 knockout mice and wildtype controls were used for specificity assessment of the tritiated radioligand by in vitro autoradiographical analysis. [3H]RO15-4513 binding (at 0.1 nM) was quantified in absence and presence of various blockers with different subunit specificity and compared with the non-selective GABAA receptor radiotracer [3H]RO15-1788 (Flumazenil).
Results
The pharmacological specificity of RO15-4513 for the GABAA receptor was confirmed by a broad CEREP® screen where 10 μM RO0154513 showed binding potency only at the GABAAreceptor. In wildtype mouse brains highest binding of [3H]RO15-4513 was identified in the hippocampus followed by cortex and pons (Figure 1A). In brain sections of GABRA5 knockout animals radioligand binding in the hippocampus was reduced by 84% whereas considerable residual binding of [3H]RO15-4513 (>50% of total binding) was found in other brain regions (Figure 1B). Co-incubation with Zolpidem, a ligand with high affinity for α1, α2 and α3-subunit containing receptor subtypes and no affinity for GABRA5, revealed a fairly pure GABRA5 binding signal in wildtype mouse brains (Figure 1C). This finding was corroborated by an almost complete blockade of [3H]RO15-4513 binding by Zolpidem in GABRA5 knockout brains only leaving some minimal binding to α4 and α6-subunit containing receptor subtypes expressed in the cerebellum (Figure 1D). Flumazenil binding in GABRA5 knockout brains was marginally reduced compared to wildtype controls confirming that GABRA5 represents a minor subtype compared to the total GABAA receptor population.
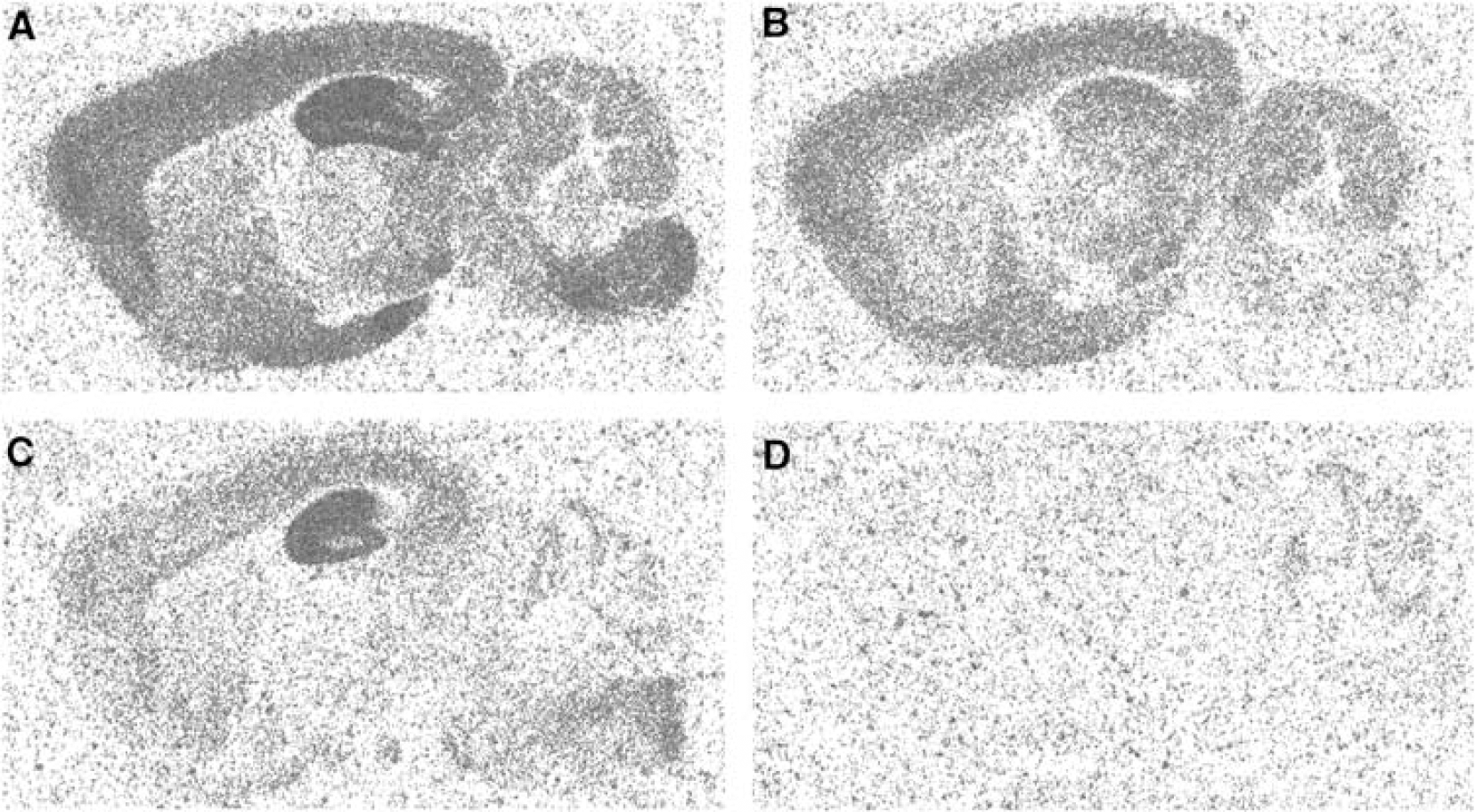
Conclusions
This study confirms the preferential binding of [3H]RO15-4513 to GABRA5 in vitro. However, in brain regions with relatively low abundance of GABRA5 (i.e. all brain regions except the hippocampus) the in vitro specificity of [3H]RO15-4513 for GABRA5 is limited even at concentrations well below the Ki value of the radioligand for GABRA5. Co-administration of Zolpidem may provide an opportunity to target and quantify GABRA5 more specifically. Novel tracers with selectivity ratios of more than 20 fold for GABRA5 over other GABAA receptor subtypes will be desirable for a more reliable quantification of GABRA5 in vitro and in vivo.
P072. Radioiodinated reboxetine analogues for imaging of the norepinephrine transporters in brain using single photon emission computed tomography
1Institute of Neuroscience and Psychology, College of Medical, Veterinary and Life Sciences, University of Glasgow, UK; 2WestCHEM, School of Chemistry, The Joseph Black Building, University of Glasgow, UK; 3West of Scotland Radionuclide Dispensary, University of Glasgow and North Glasgow University Hospital NHS Trust, Glasgow, UK; 4Molecular NeuroImaging, LLC, New Haven, Connecticute, USA; 5School of Medicine, Yale University, New Haven, Connecticut, USA; 6Vet Affairs, Connecticut Healthcare System, West Haven, Connecticut, USA
Background
123I-INER, a radioiodinated (S,S) reboxetine analogue, and 123I-NKJ64, a radioiodinated (R,S) stereoisomer of 123I-INER, are the most promising radiotracers developed to date for in vivo imaging of the norepinephrine transporter (NET) using single photon emission computed tomography (SPECT) [1–4]. The present study aimed to compare the brain distribution and pharmacokinetics of 123I-NKJ64 with that of 123I-INER in baboons and to investigate the occupancy of NET in baboons using SPECT in order to determine the most promising radioiodinated reboxetine analogue.
Methods
Adult female baboons (Papio anubis) were anesthetized and imaged on a Neurofocus SPECT camera (NeuroPhysics, Sharon, MA, USA). For the first experiments, baboons received a bolus of either 123I-INER or 123I-NKJ64 intravenously and arterial blood was sampled during the SPECT acquisition. Image analysis; invasive and noninvasive kinetic modelling were performed using PMOD 3.203 software (PMOD technologies, Zurich, Switzerland). 123I-INER was also used to estimate receptor occupancy by bolus plus constant infusion studies with displacement at equilibrium using six different doses of atomoxetine (0.03 to 0.85 mg/kg) and four different doses of reboxetine (0.5 to 3.0 mg/kg).
Results
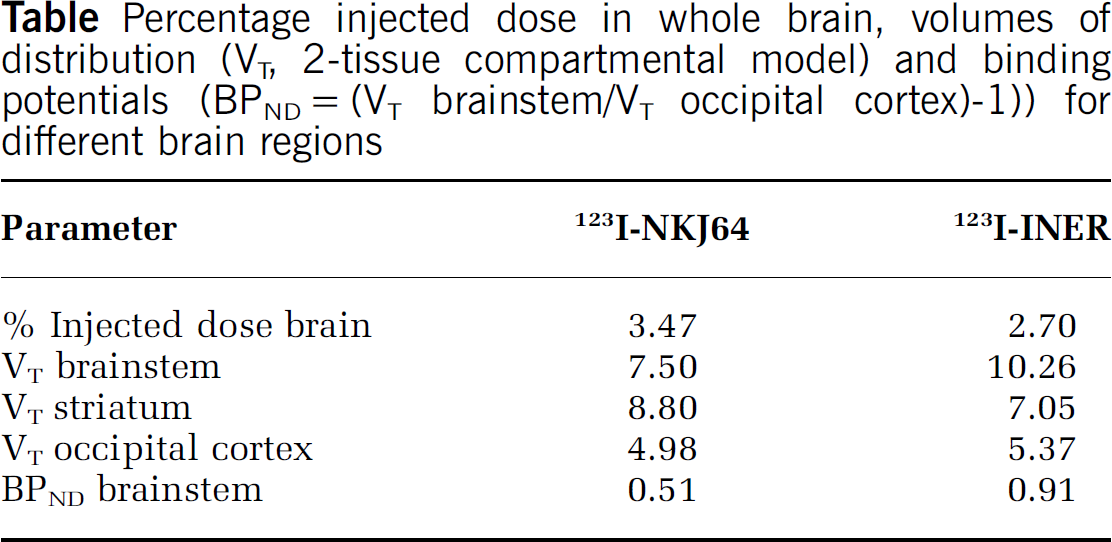
Following bolus injection, both radiotracers rapidly and avidly entered the baboon brain. The regional brain accumulation of 123I-NKJ64 did not match the known distribution of NET in baboon brain (highest in brainstem), contrasting with previous results obtained in rats [4]. Conversely, the regional distribution of 123I-INER was consistent with the known distribution of NET in baboon brain (Table). Since 123I-INER was found to be the preferred radioiodinated reboxetine analogue, occupancy measures of NET in baboon brain were determined using SPECT. After administration of atomoxetine or reboxetine, a dose-dependent occupancy was observed in brain regions known to contain high densities of NET (atomoxetine ED50=0.10 mg/kg and reboxetine ED50=1.07 mg/kg).
Conclusions
These data suggest that 123I-NKJ64 may lack affinity and selectivity for NET in baboon brain. 123I-INER is the most promising SPECT radioiodinated reboxetine analogue developed to date for in vivo imaging of NET in brain. Dose-dependent NET occupancy for two selective norepinephrine reuptake inhibitors was successfully measured in vivo in baboon brain using SPECT and 123I-INER. These results highlight the importance of species differences during radiotracer development and the enantioselectivity of radioiodinated analogues of reboxetine in vivo.
Percentage injected dose in whole brain, volumes of distribution (VT, 2-tissue compartmental model) and binding potentials (BPND=(VT brainstem/VT occipital cortex)-1)) for different brain regions
Acknowledgements
Adriana Tavares was funded by a scholarship from the Scottish Imaging Network: A Platform for Scientific Excellence (SINAPSE) Collaboration.
P073. Characterization of the radiotracers [3H]- and [11C]-RO6802488 for imaging PDE10A
1F. Hoffmann-La Roche Ltd, Basel, Switzerland; 2Division of Nuclear Medicine, Department of Radiology, Johns Hopkins School of Medicine, Baltimore, Maryland, USA
Background
Phosphodiesterase 10A (PDE10A) is a dual phosphodiesterase that hydrolyzes both cyclic nucleotides cAMP and cGMP. PDE10A is expressed in the testes to a low level in contrast to the brain where PDE10A is abundantly expressed and restricted in the striatum within the GABAergic medium spiny neurons. This highly localized expression pattern stimulates immediate interest in a potential role of the inhibition of PDE10A as a new therapeutic approach in treating neurological and psychiatric disorders. RO6802488 is a potent PDE10A inhibitor (pIC50=8.3) with favorable lipophilicity (logD=2.9) for a CNS radiotracer. The aim of this study was to evaluate RO6802488 in vitro and in vivo for its use as radiotracer to specifically visualize and quantify PDE10A in the brain.
Methods
[3H]-RO6802488 and [11C]-RO6802488 were prepared from a common precursor by a methylation reaction with [3H]MeNs or [11C]MeI. Sagittal rat brain sections were used for specificity assessment of the tritiated radioligand by in vitro autoradiographical analysis. In vitro autoradiography studies with [3H]-RO6802488 were compared on rat, Cynomolgus monkey, baboon and human brain slices. Ex vivo autoradiography of [3H]-RO6802488 and tissue sampling were performed to assess in vivo tracer distribution and kinetics in the rat brain. Blockade experiments were performed both in vitro and in vivo with a structurally different PDE10A inhibitor. These data were compared with [11C]-RO6802488 PET data obtained in rat and in baboon.
Results
[3H]-RO6802488 showed predominant in vitro binding in the striatum, the substantia nigra and the tubercule olfactorium. A comparable distribution was observed in Cynomolgus monkey, baboon and human brain samples. Specificity of the radioligand for PDE10A was demonstrated by a full blockade of binding using co-incubation with a structurally dissimilar PDE10A inhibitor. In vivo, [3H]- and [11C]-RO6802488 showed good uptake into the rat brain with a regional distribution in agreement with the in vitro experiments. Tracer peak concentrations were reached at 30–40 min post administration followed by a washout phase. Significant blockade of tracer uptake was achieved by pretreatment of the animals with a structurally different PDE10A inhibitor. In baboons, [11C]-RO6802488 demonstrated moderate brain uptake and slow washout kinetics. HPLC analysis of plasma samples indicated rapid metabolism of [11C]-RO6802488, with approximately 50% of parent radiotracer left after the first 10 min and one single more polar radiometabolite being formed in minor amounts. Despite the favorable metabolic pattern in plasma, a constant increase in radioactivity was observed in regions with low PDE10A density which might be attributed to a brain penetrant radiometabolite.
Quantification of the ex vivo autoradiography of [3H]RO6802488 in the rat. i.v. dosing, 18.5 MBq/kg; 3.6 μg/kg.
Conclusions
RO6802488, labeled with [3H] or [11C], is a specific radioligand/tracer for PDE10A with favorable characteristics for in vitro and in vivo evaluation of PDE10A in the rodent brain. The presence of a putative brain penetrant radiometabolite in the baboon suggests that this tracer is not suitable for in vivo evaluation of PDE10A in non-human primate brain.
P074. Modelling of the novel P-gp expression tracer [11C]laniquidar: a test-retest study in healthy volunteers
1Department of Nuclear Medicine & PET Research, VU University Medical Center, Amsterdam, The Netherlands; 2SEIN – Stichting Epilepsie Instellingen Nederland, Heemstede, The Netherlands; 3Department of Neurology, VU University Medical Center, Amsterdam, The Netherlands; 4Department of Clinical Pharmacology & Pharmacy, VU University Medical Center, Amsterdam, The Netherlands
Background
Overexpression of P-glycoprotein (P-gp) is thought to play a role in drug resistance, affecting 30% of patients with epilepsy. Laniquidar is supposed to be an inhibitor of P-gp and, therefore, overexpression of P-gp should lead to an increased signal. The aims of this first-in-man study were to characterize [11C]laniquidar kinetics in human subjects and to develop a tracer kinetic model for analyzing [11C]laniquidar data.
Methods
Dynamic 60 minutes [11C]laniquidar PET scans (24 frames) were performed in 11 healthy volunteers using an HR+ scanner (CTI/Siemens). Plasma input functions were obtained using an on-line sampling device, with metabolite correction based on up to 7 manual HPLC processed samples. T1-weighted MRI scans were acquired on a SONATA 1.5T scanner (Siemens) and used for co-registration and volume of interest delineation. Regional time activity curves were fitted to various one and two tissue compartment models in order to identify the best model describing [11C]laniquidar kinetics.
Results
Modelling was compromised by the low first-pass extraction fraction of [11C]laniquidar, which was estimated to be 2–3%. Metabolism of [11C]laniquidar in plasma was fast (only 50% of parent tracer remaining after 10 minutes, 20% after 60 minutes). Both standard single and two tissue compartment models did not fit the data well, with k2 values that were too small to be estimated reliably. In all subjects, cerebral [11C]laniquidar concentrations started to increase after 5–10 minutes. The quality of the fits improved significantly by adding a second parallel (single tissue) compartment with one of the labelled plasma metabolites as input function, suggesting that metabolites enter the brain. The most robust model was found to be an irreversible single tissue compartment model providing estimates for K1 and Vb and using only the first 3 minutes of PET data in order to prevent the contribution of labelled metabolites. For this model, the mean (±SD) of K1 was 0.019±0.004. Initial test-retest studies showed a significant increase in K1 in the afternoon scan. Interestingly, however, after standardizing breakfast and lunch, reproducibility of K1 improved.
Conclusions
[11C]laniquidar kinetics suggest that labelled metabolites enter the brain. Indeed, a dual input model with both parent tracer and metabolite as input functions provided the best fits to full 60 minutes data. If parent plasma is used as input function, robust estimates of K1 can still be obtained by fitting only the first 3 minutes of PET data. The relationship between diet and test-retest variability of K1 requires further investigation. P-gp blocking studies are needed to assess whether [11C]laniquidar is a pure inhibitor of P-gp.
Acknowledgements
European Project (FP7) EURIPIDES, J&J.
P075. [11C]N-Desmethyl-Loperamide as a marker of Pgp function in patients with gliomas
1Neuro-Oncology Branch, National Cancer Institute, Bethesda, Maryland, USA; 2Molecular Imaging Branch, National Institute of Mental Health, Bethesda, Maryland, USA; 3PET Department, National Institutes of Health Clinical Center, Bethesda, Maryland, USA
Background
[11C]N-desmethyl-loperamide ([11C]dLop) is a radiotracer substrate for Pgp and may provide a non-invasive, in vivo method of examining Pgp function in brain tumor patients.
Methods
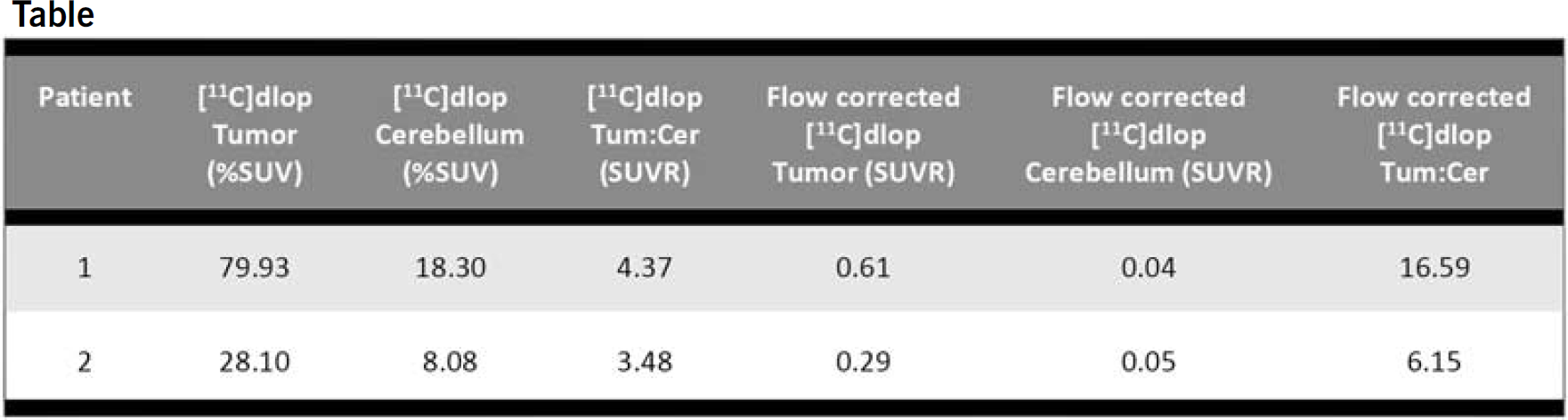
This is a pilot study to characterize [11C]dLop uptake in patients with histologically confirmed gliomas. Concurrent use of a Pgp inducer within 2 weeks and loperamide within 3 days of the study scan were not allowed. A perfusion MRI was obtained within 14 days, and FDG PET scan within 4 weeks of [11C]dLop PET imaging. Patients received 16 mCi of [15O]H2O for a 60 sec acquisition to correct for blood flow, and 20 mCi of [11C]dLop with a 60 min acquisition. A volume of interest (VOI) was determined base on FLAIR abnormality for the entire brain volume. This volume was refined by creating automated isocontours determined by an intensity threshold for the coregistered [11C]dLop study. The ipsilateral cerebellar hemisphere was used as a reference VOI. Average values within the target VOI were compared to the reference for the [11C]dLop, flow-corrected [11C]dLop, and FDG PET scans using unpaired t-test.
Results
Two patients have participated in the study. There was a 3–4 fold increase in [11C]dLop uptake (7–16 fold increase for the flow-corrected study) in areas of tumor vs. cerebellum. [11C]dLop uptake was not predicted by the areas of enhancing and non-enhancing disease on the coregistered MRI.
Conclusions
[11C]dLop is a useful PET tracer for Pgp function and blood brain barrier integrity in patients with gliomas. This tracer has the potential to be a more sensitive indicator of BBB disruption than gadolinium MRI.
P076. Prediction and identification of PET radiometabolites by cytochrome P450, UPLC/Q-ToF-MS and fast radio-LC: application to [18F]FE-PE2I
1Karolinska Institutet, Department of Clinical Neuroscience, Stockholm, Sweden; 2AstraZeneca R&D, Innovative Medicines, CNSP iMed Södertälje, Sweden
Background
Positron emission tomography (PET) is a sensitive in vivo imaging technique in which compounds with short lived radionuclides such as 18F and 11C are administered via intravenous injection and used for quantitative measurements of biochemical processes. However, a drawback with PET is that it only reflects the total amount of radioactivity in tissue and does not provide any information on its chemical form. Therefore it is important to develop methods by which radiometabolites of radiotracers, especially ones with the potential of crossing the blood brain barrier, can be identified and measured over time. [18F]FE-PE2I is a newly developed dopamine transporter (DAT) PET radioligand superior to [11C]PE2I 1 .
Methods
Carrier-added [18F]FE-PE2I was incubated with rat, monkey and human liver microsomes (BD). The metabolism was stopped by addition of cold acetonitrile at several time points and analyzed by fast radio-LC (XBridge C18 column, Waters, 10 × 50 mm i.d., 2.5 μm) at a flow rate of 6 mL/min using an Agilent binary pump coupled to a fraction collector (Agilent 1200 series) and radiation detector (Oyokoken, S-2493Z). The mobile phase was (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile with an elution profile starting from 15% B and continuing isocratic for 4 min and then reaching 40% B after 4 min. Peaks corresponding to major radiometabolites were collected and identified by UPLC/Q-ToF-MS (Waters 2.1 × 50 mm i.d., 1.7 μm BEH C18 column). Positive electrospray ionization (ESI) in V-mode with extended dynamic range was used. Two scan functions, MS and MSE, were performed simultaneously. Rat, monkey, human plasma and rat brain extract samples were analyzed in the same fashion after intravenous injection of [18F]FE-PE2I.
Results
Major radiometabolites of [18F]FE-PE2I produced by CYP450 were identified using accurate mass/charge measurements of both the parent and fragment ions and were: hydroxyl [18F]FE-PE2I, carboxyl [18F]FE-PE2I, desiodopropenyl [18F]FE-PE2I, hydroxyl desiodopropenyl [18F]FE-PE2I and carboxyl desiodopropenyl [18F]FE-PE2I. Based on these findings and the use of fast radio-LC 2 , which allowed the achievement of higher peak resolution and sensitivity in a shorter time frame compared to conventional methods, it was possible to further identify and determine the relative abundance of [18F]FE-PE2I and its radiometabolites over time in plasma of different species as well in rat brain. In plasma samples of all the studied cases carboxyl [18F]FE-PE2I was the most abundant radiometabolite found. However, in rat striatum hydroxyl [18F]FE-PE2I had the highest presence.
Conclusions
The work flow presented here can be used to successfully identify and measure radiometabolites of PET radioligands in general. This enables us to predict the presence and degree of significance of the more lipophilic radiometabolites, which have the potential of crossing the blood brain barrier.
P077. Imaging of the brain serotonin transporters (SERT) with a new fluorine-18 labelled fluoromethyl-analogue of McN5652 and PET in humans
Swen Hesse1, Peter Brust2, Peter Mäding2, Georg-Alexander Becker1, Marianne Patt1, Anita Seese1, Dietlind Sorger1, Julia Luthardt1, Jörg Steinbach2 and
1Department of Nuclear Medicine, University of Leipzig, Leipzig, Germany; 2Helmholtz-Zentrum Dresden-Rossendorf, Institute of Radiopharmacy, Dresden and Leipzig, Germany
Background
Due to limitations by the short half-life of the gold-standard SERT-radiotracer [11C]DASB, its moderate cortical test retest reliability, and the lack of options for quantifying endogenous serotonin, fluorine-18-labelled compounds for imaging the brain SERT are of unmet interest. Here, we report on our first application in man of a new fluorine-18 labelled fluoromethyl analogue of (+)-McN5652 ((+)-[18F]FMe-McN5652) and its potential for SERT quantification.
Methods
Five healthy volunteers (3 male, 2 female, age 39±10 years) were included in the study. The PET data were analysed by individual MRI-based volume-of-interest analysis (PMOD). Rate constants and total distribution volumes (VT) were calculated using a 2-tissue compartment model and arterial input function measurements corrected for metabolites. Standardized uptake region-to-cerebellum ratios (SUVR) as measure of specific radiotracer accumulation were compared with those of a new 3D [11C]DASB-PET data set of 5 healthy age- and gender matched subjects.
Results
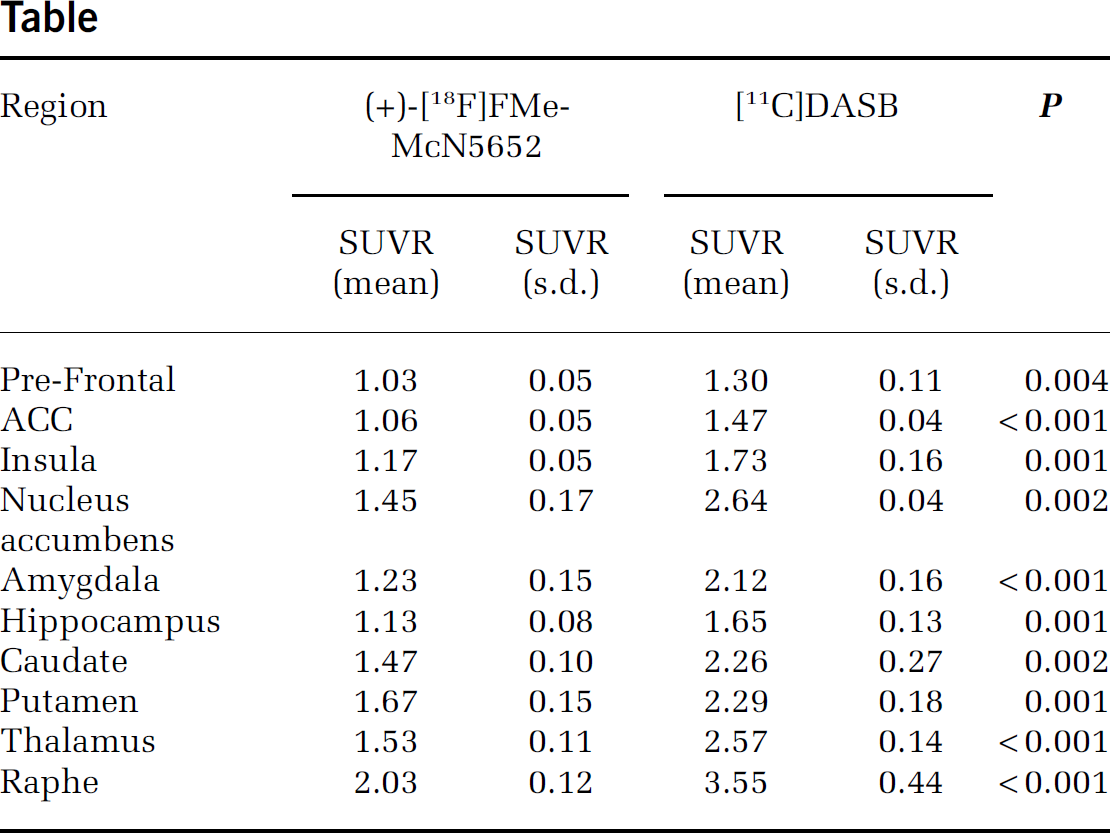
Cerebral (+)-[18F]FMe-McN5652 uptake corresponds well with the known SERT distribution in humans (Figure). The 2-tissue compartment model provided adequate fits to the data. Estimates of total distribution volume (VT) demonstrated good identifiability based on coefficient of variation (COV) for the volumes of interest (VOIs) in SERT-rich and also for cortical areas (COV VT<10%). Regional SUVR of (+)-[18F]FMe-McN5652 and [11C]DASB were highly correlated (R=0.94, P<0.001) and significantly different with lower values of the 18F-labelled derivative and less standard deviation in most of the regions, in particular in the raphe region, but not in the nucleus accumbens (Table).
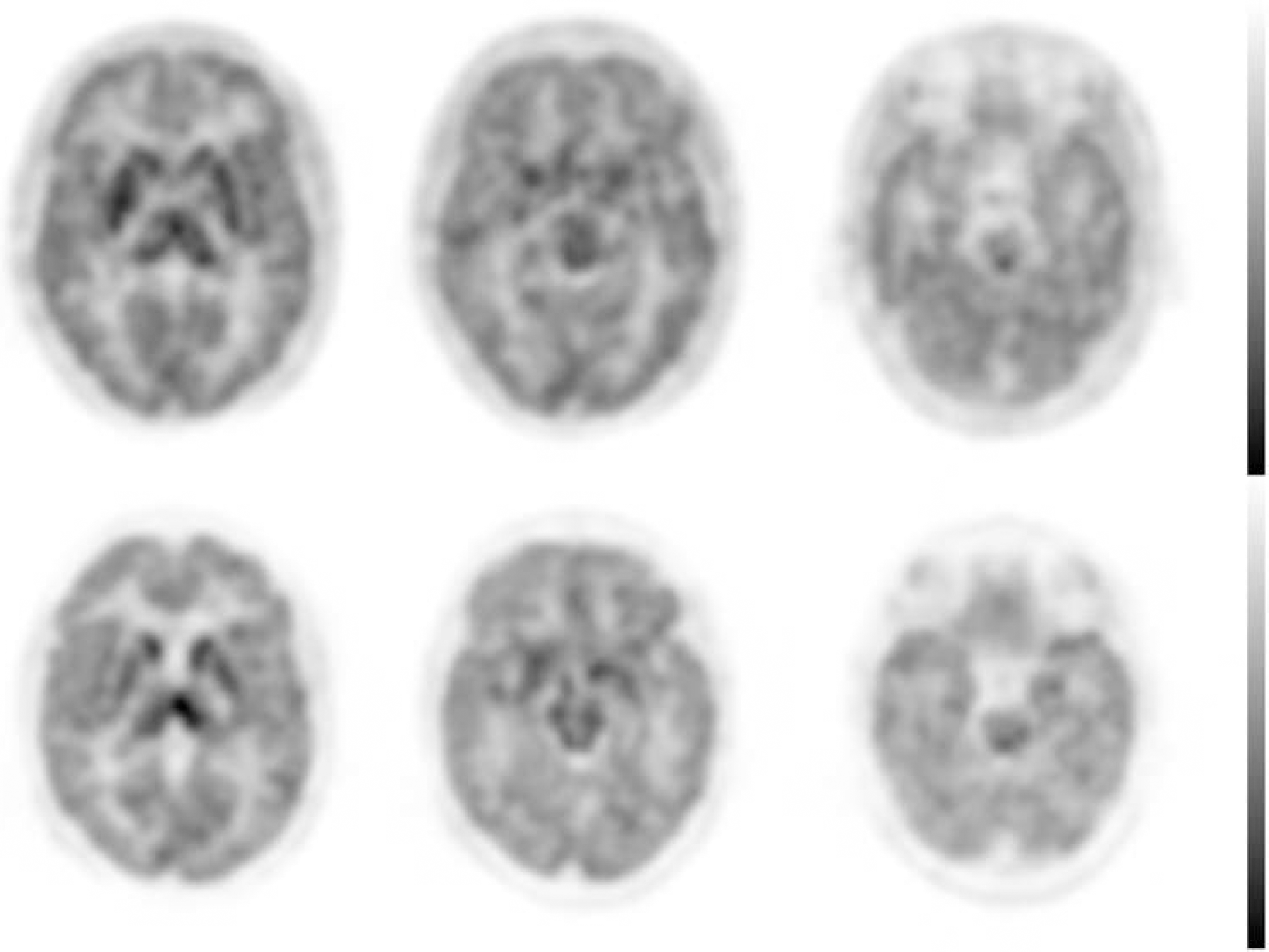
Summed iterative reconstructed 3D PET images of (+)-[18F]FMe-McN5652 (top row) and [11C]DASB (bottom row) at different levels indicating SERT-distribution.
Conclusions
The results showed that (+)-[18F]FMe-McN5652 is suitable for in vivo visualisation and quantification of SERT with PET bearing also potential for widespread use and further tracer kinetic studies, i.e. to reveal its sensitivity for endogenous serotonin.
P078. Measuring non-specific binding of novel PET radioligands to determine structure-activity relationships between NSB and their physiochemical properties
Chloe Child1,
1Kings College London, UK; 2Imperial College, London, UK
Background
The non-invasive imaging modality positron emission tomography (PET) is used extensively in clinical settings and is increasingly being used by the pharmaceutical industry in drug development. Molecules of biological interest are labelled with positron emitting isotopes e.g. 11C, allowing their biodistribution and kinetics to be followed in vivo. A major factor in the failure of radioligands is the magnitude of unwanted background signal, non-specific binding (NSB) obscuring binding to the desired target. Assumptions have previously been made as to the physiochemical and pharmacological properties of radioligands that can affect NSB. However, little work has been carried out to quantify NSB with regard to determining structure-activity relationships (SARs) in order to optimise efficient radiotracer discovery.
Methods
Non-specific binding is a poorly understood process but is believed to be related to the non-saturable binding of labelled molecules with tissue membranes. In this work the synthesis of novel radiolabelled molecular libraries has been conducted, their physicochemical properties determined and their non-specific binding measured in vitro using autoradiographical and cell based mass spectrometry assay methods. Structure-activity relationships have been formed between partition coefficient properties, acid dissociation constants, interaction energies and molecular weight in order to determine the effect each of these properties has on non-specific binding.
Results
Traditionally lipophilicity, log P, of a radioligand is the main predictor to its non-specific binding properties. However, from this work, it has been shown that a single physicochemical property cannot be relied on to predict the NSB of a radioligand.
Conclusions
Multiple properties must be considered.
P079. Development of an 18F-labelled 5-HT2A receptor agonist PET radioligand
1Neurobiology Research Unit and Center for Integrated Molecular Brain Imaging (Cimbi), Copenhagen University Hospital, Denmark; 2Department of Medicinal Chemistry, Faculty of Pharmaceutical Sciences, University of Copenhagen, Denmark; 3PET and Cyclotron Unit, Copenhagen University Hospital, Denmark
Background
We have recently validated a series of N-benzylphenethylamine-based agonists as promising positron emission tomography (PET) tracers for quantification of serotonin 2A (5-HT2A) receptor binding in the living brain (1,2). An agonist 5-HT2A receptor radioligand is hypothesized both to provide a more relevant measure of the 5-HT2A receptors and also to be more susceptible to changes in endogenous serotonin as compared with 5-HT2A receptor antagonist tracers. Thus, a 5-HT2A receptor agonist PET radioligand may reflect endogenous serotonin levels in the living brain. So far, all these radioligand have been labelled with C-11. However, labelling a PET radioligand with the longer-lived 18F-isotope offers distinct advantages including better count statistics in longer scans making e.g. bolus-infusion paradigms more easily applicable. Furthermore, 18F-labelled radioligands can be distributed to PET centers that do not have an on-site cyclotron. Here, we report the radiosynthesis and evaluation of three novel N-benzylphenethylamine 18F-labelled 5-HT2A receptor agonist PET tracers.
Methods
We synthesized three PET radioligands 2-(4-bromo-2-(2-[18F]-fluoroethoxy)-5-methoxyphenyl)-N-(2-methoxybenzyl)ethanamine ([18F]Cimbi-360), 2-(4-bromo-2,5-dimethoxyphenyl)-N-(2-(2-[18F]-fluoroethoxy)benzyl)ethanamine ([18F]Cimbi-362), and 2-(4-(2-[18F]-fluoroethyl)-2,5-dimethoxyphenyl)-N-(2-methoxybenzyl)ethanamine ([18F]Cimbi-320) by radio-fluorination of the N-Boc-protected precursors. We studied the in vivo brain distribution of the tracers in three female Danish Landrace pigs. After intravenous administration of each tracer, the pigs were scanned for 150 minutes in list-mode in a high resolution research tomography (HRRT) scanner. In all scans, the 5-HT2A receptor antagonist ketanserin was injected i.v. after 90 min as a within scan challenge. Arterial blood samples and radiometabolite analyses were done in all pigs to compute arterial input functions, and brain regional radioactive concentrations were obtained after co-registration to a MRI-based atlas of the pig brain. Furthermore, in vitro receptor autoradiography was perfomed with [18F]Cimbi-320 on pig and rat brain sections.
Results
The in vitro receptor autoradiography showed that [18F]Cimbi-320 binding on pig brain sections was in accordance with the expected 5-HT2A receptor distribution. Furthermore, the [18F]Cimbi-320 binding was completely displaced by ketanserin confirming the 5-HT2A receptor affinity of this compound in vitro. The in vivo PET scans showed that [18F]Cimbi-362, [18F]Cimbi-360, and [18F]Cimbi-320 entered the pig brain in insufficient amounts, and the peak brain uptake was 1 SUV, 0.7 SUV, and 1.3 SUV, respectively. Furthermore, the PET scan of [18F]Cimbi-320 showed extensive skull uptake indicating rapid defluorination in vivo. For all three compounds, the uptake was lower in brain tissue as compared to the surrounding tissue. This suggest that these PET radioligands undergo rapid defluorination in vivo (to yield free 18F-fluoride which is deposited in bone) or that they interact with an active efflux transporter such as P-glycoprotein. The insufficient brain uptake could also arise from to a combination of these factors.
Conclusions
Although [18F]Cimbi-320 binds the 5-HT2A receptors in vitro, neither [18F]Cimbi-362, [18F]Cimbi-360, nor [18F]Cimbi-320 were successful in imaging 5-HT2A receptor agonist binding in the pig brain due to low brain uptake with these compounds.
P080. Determination of in vivo kinetic properties of α4β2∗ nAChR specific [18F]nifene in the nonhuman primate
1University of Wisconsin, Madison, USA; 2University of California, Irvine, USA
Background
The PET radioligand [18F]nifene binds with high affinity to α4β2∗ nicotinic acetylcholine receptors (nAChR) with fast in vivo equilibration times (∼40 minutes). This work analyzes the in vivo kinetic properties of [18F]nifene with kinetic modeling and reference region methods.
Methods
Dynamic PET experiments were performed on four rhesus monkey subjects (4F, 9–13 yr) with a MicroPET P4 scanner. Experiments began with a high specific activity [18F]nifene injection (89–126 MBq) followed by a coinjection of [18F]nifene with varying masses of nifene (1–17 nmol/kg) at t=60 min. Assay of radioactivity in arterial blood, including corrections for radiolabeled [18F]nifene metabolites, was conducted to provide a parent radioligand input function. Regions examined include the antereoventral thalamus (AVT), lateral geniculate body (LG), frontal cortex (FC), subiculum (SB), and cerebellum (CB). Data from the high specific activity injection was modeled with one-tissue and two-tissue compartment models (1TCM and 2TCM) and Logan graphical methods (both with and without blood sampling). The apparent dissociation constant (KDapp) was estimated for A) all subjects by calculating the change in receptor occupancy between the two injections with the Logan graphical method for each subject to create a Scatchard plot and B) two individual subjects using compartment modeling of the two-injection procedure to uncouple KDapp and receptor density (Bmax) parameters.
Results
The time course of [18F]nifene in all brain regions was appropriately modeled by the 1TCM. No significant levels of specific binding were detected in the CB, validating its use as a reference region. Using the 1TCM, a total distribution volume (VT) of 6.91±0.61 ml/cm3 was observed in the CB, with a plasma-to-tissue transfer parameter (K1) of 1.55±0.39 ml/min/ml and a tissue- clearance constant (k2) of 0.23±0.07 min−1. Distribution volume ratios (DVR) using the CB as a reference region calculated with the 1TCM were 2.60±0.17, 2.35±0.16, 1.26±0.08, and 1.30±0.07 in the AVT, LG, FC, and SB, respectively. Agreement within 0.04 absolute DVR values was observed between the 1TCM and Logan methods. Linearization of Logan plots was sufficiently rapid to evaluate binding (BPND=DVR-1) for each injection, and the ratio of binding between the two injections was taken as receptor occupancy to create a Scatchard plot. With this method, KDapp of all subjects grouped together was estimated at 3±1 pmol/ml. Compartment modeling techniques yielded KDapp of 2.0 and 2.3 pmol/ml, while the average estimated Bmax was 2.8, 3.2, 0.8, and 0.9 pmol/ml in the AVT, LG, FC, and SB, respectively.
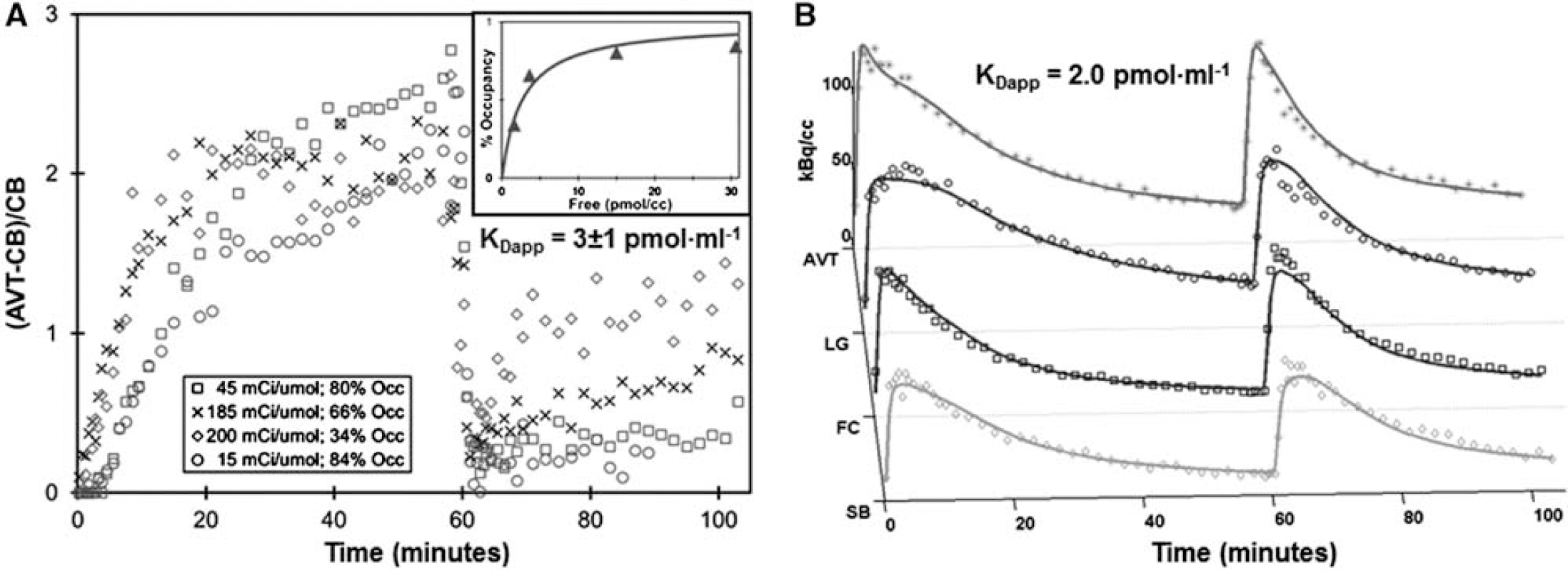
Determination of [18F]nifene KDapp.
Conclusions
The fast kinetic properties and specific regional binding of [18F]nifene demonstrate favorable properties for assay of α4β2∗ nAChR binding and promote its extension into human subjects.
P081. In vivo assessment of [123I]MNI420, an adenosine 2a SPECT radioligand, in nonhuman primates
1Molecular NeuroImaging, LLC, New Haven, Connecticut, USA; 2Institute for Neurodegenerative Disorders, New Haven, Connecticut, USA; 3Molecular NeuroImaging, LLC and School of Medicine Yale University, New Haven, Connecticut, USA
Background
As the adenosine 2a (A2a) receptor has been implicated in a variety of pathophysiologic processes, in vivo imaging of A2a would be a valuable tool in evaluating: neurodegenerative disease and efficacy of both existing and novel therapeutics. We recently developed 7-(2-(4-(2-fluoro-4-[123I]iodophenyl)piperazin-1-yl)ethyl)-2-(furan-2-yl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5c]pyrimidin-5-amine ([123I]MNI420), an A2a SPECT radioligand. To assess the in vivo characteristics of [123I]MNI420, we conducted a series of SPECT scans in cynomolgus macaques.
Methods
Two male cynomolgus macaques (6 years old) were each scanned 6 times following IV bolus administration of [123I]MNI420 (injected dose: 131.7±55.2 MBq). Both animals were scanned once under baseline conditions. Preladenant, a selective A2a antagonist, was given as an IV bolus 15 min prior to injection in 9 scans, at a dose of 0.01, 0.03, 0.1, 0.3 or 1 mg/kg (n=2 for all doses except 1.0 mg/kg). In 1 scan, 1.0 mg/kg Preladenant was administered as an IV bolus 93 min post injection (p.i.) of [123I]MNI420. Immediately following injection of [123I]MNI420, animals were scanned on a MollyQ SPECT camera (NeuroPhysics Corp., Shirley, MA, USA) for up to 4 hours (scan length: 3.8h±0.26 h). Volumes-of-interest (VOIs) were drawn on averaged SPECT images, to include the whole brain, caudate, putamen, striatum, brainstem, occipital cortex and cerebellum. All scans with preladenant were co-registered to the animal's baseline scan. To optimize imaging outcomes, several non-invasive pharmacokinetic models (SRTM, MRTM, Logan reference method) were compared to outcome measures derived from the time activity curves (specific binding, SUVr, BPnd apparent). As SRTM and MRTM failed to produce robust solutions for all scans, the Logan reference method was used to calculate BPnd. Receptor occupancy was defined as (Baseline BPnd – Blockade BPnd)/Baseline BPnd, expressed as a percent. Cerebellum was used as a reference region.
Results
Peak SUVs were ∼1.0, and at equilibrium, striatal SUVrs were 3–4. SUVrs stabilized by ∼90 min p.i., and remained stable up to 4 h p.i. Using the Logan reference method, the rank order for binding of [123I]MNI420 was caudate>putamen>>brainstem>occipital. In striatum, the EC50 was calculated to be 0.05 mg/kg, using either the Logan reference method, or using BPnd apparent, derived from the TAC 90–150 min p.i. Maximum displacements with preladenant were observed for doses of 0.1–1 mg/kg (70%–80% displacement in the striatum across all tested doses in this range). Preladenant displaced [123I]MNI420 in a dose-dependent fashion.
Conclusions
These data suggest that [123I]MNI420 binds selectively to A2a in vivo. BPnd apparent, calculated 90–150 min p.i. is highly correlated with noninvasive PK outcomes. The relatively longer half-life of 123I relative to commonly used PET isotopes may be advantageous in distributing this radioligand for additional research assessing the utility of [123I]MNI420 in human populations.
P082. Synthesis, radiolabelling and validation of (20R)-4,5-α-epoxy-17-methyl-3-hydroxy-6-(2-[18F]fluoroethoxy)-α,17-dimethyl-α-(2-phenyleth-1-yl)-6,14-ethenomorphinan-7-methanol ([18F]FEPEO)
1University of Cambridge, UK; 2ABX Radeberg, Germany; 3TU Munich, Germany
Background
We have investigated (20R)-4,5-a-epoxy-17-methyl-3-hydroxy-6-(2-[18F]fluoroethoxy)-a,17-dimethyl-a-(2-phenyleth-1-yl)-6,14-ethenomorphinan-7-methanol ([18F]FEPEO) a novel 18F labelled agonist for opioid receptor (OR) imaging [1]. [18F]FEPEO is a full agonist active at μ-, δ-, and κ-ORs, with profound selectivity for the κ-OR and μ-OR. Cerebral distribution of ([18F]FEPEO) in Lister-Hooded rats was investigated and ROI data was quantified.
Methods
The non-radioactive reference compound FEPEO and a labelling precursor for direct nucleophilic radiofluorination were prepared. An automated process for the production of [18F]FEPEO using a GE Tracerlab FXFN radiosynthesis module was developed. The radiotracer was subjected to in vitro and ex vitro autoradiography studies in rat brain sections. Radiotracer uptake into OR containing brain regions was investigated using microPET. The amount of radioactive metabolites in plasma and brain tissue was determined. Radiotracer uptake into a variety of brain regions was quantified using RTM, sRTM, invasive and non-invasive Logan graphical analysis, 1-tissue compartment and 2-tissue compartment plasma input modelling [3].
Results
[18F]FEPEO was obtained in a radiochemical yield (RCY) of 30±25% (n =16) via nucleophilic substitution using a trityl protected labelling precursor and subsequent cleavage of an O-trityl protective group. Radiotracer distribution mirrored the distribution of ORs in the rat brain. Radiotracer binding was found to be reversible and saturable via antagonist treatment. The cerebellum was validated as a suitable reference region for reference tissue modelling.
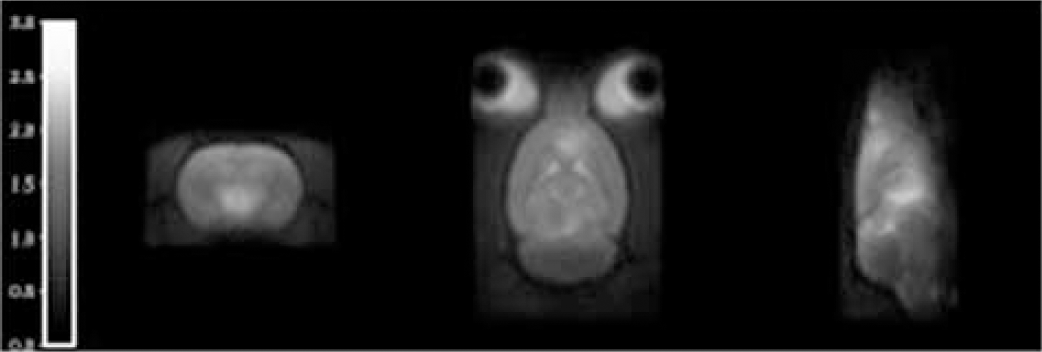
Non-displaceable binding potential (BPND) map from simplified reference tissue modelling, the cerebellum was used as a reference.
Conclusions
[18F]FEPEO was produced using an automated process using cGMP compliant equipment. This OR agonist showed selective and saturable uptake into OR containing brain regions with profound selectivity for the MOR in rats. This is the first validated 18F-labelled OR full agonist PET tracer available for PET. The cerebellar cortex is a suitable reference region for kinetic modelling of [18F]FEPEO-PET data.
Acknowledgements
This study was funded by the MRC ICCam grant (grant no.: G1000018).
P083. Characterization of RO5013853, a novel positron emission tomography radiotracer for the imaging of the glycine transporter type 1
1Hoffmann-La Roche Ltd., Basal, Switzerland; 2Johns Hopkins University, Baltimore, Maryland, USA
Background
The glycine transporter type 1 (GlyT1) is a promising target for the development of novel drugs for the treatment of schizophrenia and other CNS disorders. A specific positron emission tomography (PET) radiotracer for GlyT1 is a key tool for the clinical development of a GlyT1 inhibitor since it will allow assessment of brain penetration of drug candidates and enable quantification of transporter occupancy. In this study, we pharmacologically characterized RO5013853 and evaluated it as a PET ligand for GlyT1 in non-human primates and in healthy volunteers.
Methods
Affinity and kinetic properties of RO5013853 for human recombinant and rat native GlyT1 were determined using [3H]-RO5013853; autoradiographic studies were performed on rat, baboon and human brain sections. [11C]-RO5013853 radiosynthesis employed 11C methylation. Baboon and human PET included administration of [11C]-RO5013853 i.v., 90 minutes of scanning, separation of metabolites using high-performance liquid chromatography (HPLC), and determination of radial arterial input function. PET studies were performed using a high-resolution research tomography (HRRT) scanner (baboon) and GE Advance PET scanner (human). PET data were analyzed using a 2-tissue 5-parameter (2T5P) model, a 1-tissue 3-parameter model, and a graphic reference region method (Logan plot). The outcome variables were K1, VT and BPND.
Results
RO5013853 displays high affinity and reversible binding to both human recombinant and rat native GlyT1 (dissociation constant [Kd] was 2.4 and 2.0 nM for the human and the rat transporters, respectively). Autoradiographic studies showed that the distribution of [3H]-RO5013853 binding sites corresponded to the known distribution of GlyT1. The VT estimated (2T5P) obtained in baboon PET studies showed the following rank order of GlyT1 distribution: thalamus, pons and cerebellum > caudate, putamen and cortical regions. Pre-treatment with bitopertin, a selective GlyT1 reuptake inhibitor (GRI) presently in phase III clinical development, produced a plasma-concentration-dependent blockade of the tracer. Scans carried out in healthy male human volunteers (n=5; aged 23–56 years) confirmed the ability of [11C]-RO5013853 to image regions expressing GlyT1. Administration of [11C]-RO5013853 (∼30 mCi), with an average specific activity of 12905±7857 mCi/μmol, was shown to be well tolerated. HPLC metabolites were 50% at 90 minutes. A 2T5P model best described the human tracer tissue kinetics, which was consistent with primate imaging. VT was higher in the cerebellum, pons and thalamus (1.99 to 2.59 mL/mL) and lower in the putamen, caudate and cortical areas (0.86 to 1.13) with less than 10% difference between tests and retest scan. Tracer retention was effectively blocked after administration of the selective GRI, bitopertin.
Conclusions
[11C]-RO5013853 is a novel PET ligand for imaging of GlyT1 which can be used in occupancy studies of novel drugs targeting GlyT1 and to investigate the role of GlyT1 in CNS diseases.
P084. PET Imaging of VMAT2 with the novel radioligand [18F]FE-DTBZ-d4 in non-human primates: comparison with [11C]DTBZ and [18F]FE-DTBZ
Karolinska Institutet, Department of Clinical Neuroscience, Stockholm, Sweden
Background
The vescicular monoamine transporter type 2 (VMAT2) is responsible for the uptake of monoamines into the vesicles of the synaptic terminals. The VMAT2 radioligands [11C]DTBZ and [18F]FP-DTBZ have been used to assess the degree of nigrostriatal deficit in Parkinson's disease.1,2 We have recently developed [18F]FE-DTBZ-d4 as a potential radioligand for imaging VMAT2 in insulin-secreting beta-cells. [18F]FE-DTBZ-d4 has similar imaging properties as the non-deuterated analogue [18F]FE-DTBZ, with increased stability against defluorination. 3 The usefulness of [18F]FE-DTBZ-d4 as an imaging marker for VMAT2 in the brain remains to be investigated. The aim of this study was to investigate the brain uptake, kinetics, and metabolism of [18F]FE-DTBZ-d4 in non-human primates, in comparison with [11C]DTBZ and [18F]FE-DTBZ.
Methods
Three female cynomolgus monkeys (weight: 5.8–6.95 kg) were studied in five experimental days. PET measurements were conducted using the HRRT system. Three monkeys underwent two PET measurements. The first PET measurement (123 min) with [11C]DTBZ (181±7 MBq), followed by the second PET measurement (183 min) with [18F]FE-DTBZ-d4 (187±14 MBq), 3 h later. In two monkeys, the third PET measurement of 183 min was conducted 2-to-3 months later with [18F]FE-DTBZ (180±2 MBq). Venous blood samples were drawn 5 min before and 2.5, 15, 30, 45, 60, 90, and 120 min (180 min only for [18F]FE-DTBZ-d4 and [18F]FE-DTBZ) after tracer injection, for measurement of free fraction (fP) and metabolite analysis. Regions-of-interest were drawn on caudate, putamen, midbrain, and cerebellum. Quantification was performed with the Logan graphical analysis, using the cerebellum as reference region. The outcome measure was the binding potential (BPND).
Results
[18F]FE-DTBZ-d4 displayed higher brain uptake than [11C]DTBZ (%SUV: 428±101% vs. 306±32%, p=0.1) and faster wash-out (peak-to-120 min ratio: 2.8±0.5 vs. 2.3–0.5, p=0.07) (Figure). BPND values of [18F]FE-DTBZ-d4 (caudate: 4.4±1.1; putamen: 5.5±1.4; midbrain: 1.4±0.4) were higher than [11C]DTBZ (caudate: 3.0±0.6, p=0.04; putamen: 3.7±0.6, p=0.07; midbrain: 0.9±0.3, p=0.02). Both radioligands had similar fP (52±1% vs. 47±5%) and metabolism. At 90 min after injection, the percent of unchanged [18F]FE-DTBZ-d4 and [11C]DTBZ (retention time-Rt- 8 min) in plasma were 30±17% and 22±3%, respectively. The HPLC analysis showed the presence of two major radiometabolite peaks. At 90 min after injection, the first peak (Rt: 2.3 min) accounted for 51±24% ([18F]FE-DTBZ-d4) and 71±2% ([11C]DTBZ) of total plasma radioactivity, whereas the second peak (Rt: 5.8 min) accounted for 10±4% ([18F]FE-DTBZ-d4) and <1% ([11C]DTBZ). In the two monkeys studied, [18F]FE-DTBZ showed similar BPND values (caudate: 3.8±0.5; putamen: 4.7±0.3; midbrain: 1.1±0.1) and fP (56±0%), as compared with [18F]FE-DTBZ-d4, with a slightly faster metabolism (unchanged parent at 90 min: 13±1%).
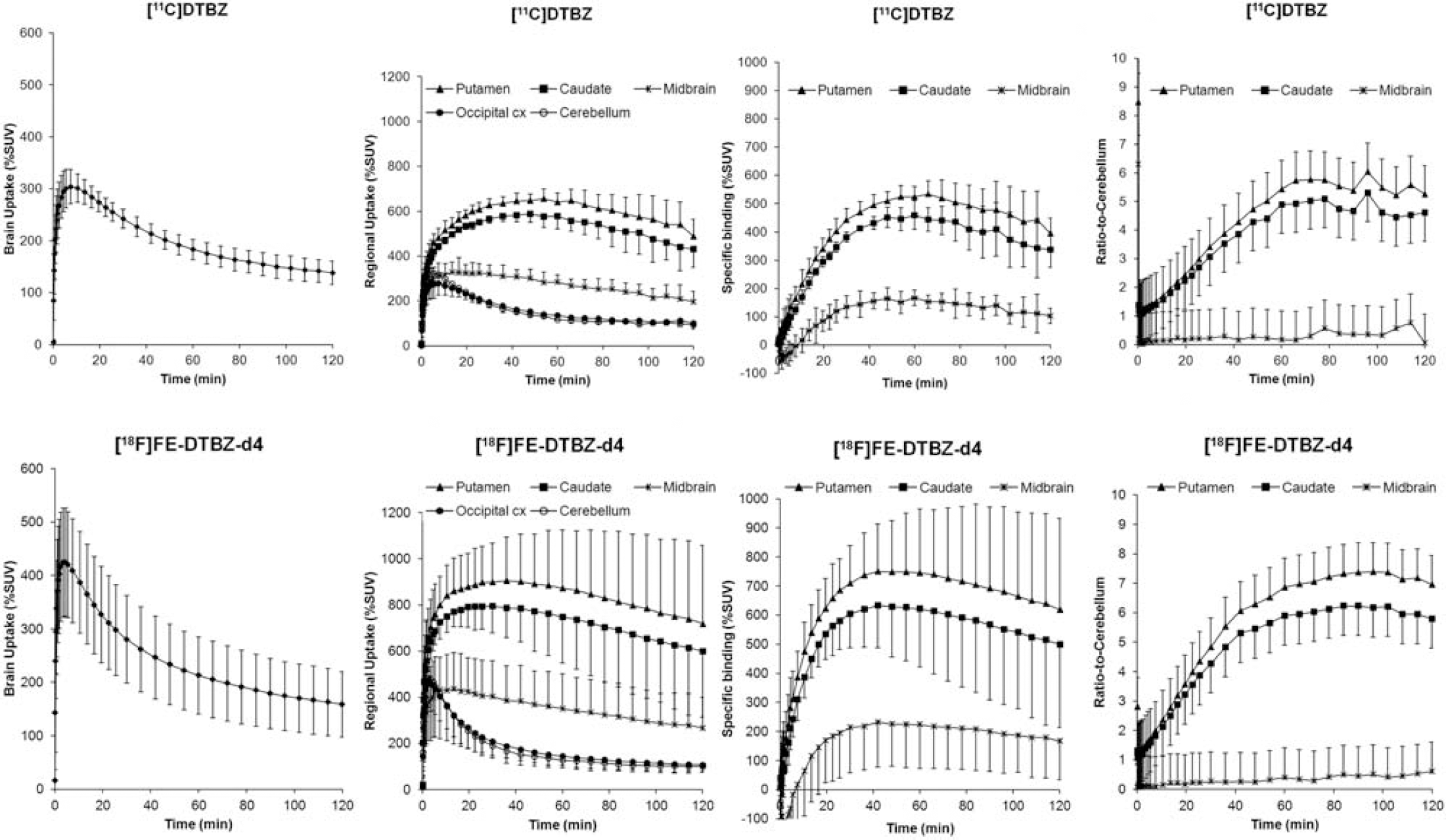
Average time-activity curves of [11C]DTBZ and [18F]FE-DTBZ-d4 in 3 monkeys examined (bar=1 SD).
Conclusions
[18F]FE-DTBZ-d4 is a suitable radioligand for quantification of VMAT2 in non-human primates, with better imaging properties than [11C]DTBZ. A preliminary comparison suggests that [18F]FE-DTBZ has similar characteristics as its non-deuterated analogue.
P085. Sensitive and specific amyloid-β PET imaging using the three novel radioligands [18F]AZD4694, [11C]AZD2184 and [11C]AZD2995 in Alzheimer's disease patients
1Karolinska Institutet, Center for Psychiatric Research, Department of Clinical Neuroscience, Stockholm, Sweden; 2AstraZeneca Research & Development, Neuroscience Research & Therapy Area, Södertälje, Sweden; 3Clinical Geriatrics, Dept. Neurobiology, Caring Sciences and Society, Karolinska Institutet, Stockholm, Sweden
Background
Several novel radioligands for Positron Emission Tomography (PET) imaging of amyloid-β have been developed in a collaboration between Karolinska Institutet and AstraZeneca. The ligands have been developed to minimize non-specific binding by reducing the lipophilicity when compared with previous radioligands. Here we present data with the three novel radioligands [18F]AZD4694 [1], [11C]AZD2184 [2] and [11C]AZD2995 and a comparison of outcome measures obtained by full kinetic modeling and a metabolite corrected arterial input function as well as simplified reference region based methods.
Methods
Healthy control subjects (CS) and patients with Alzheimer's Disease (AD) underwent PET imaging with either [18F]AZD4694 [11C]AZD2184 and/or [11C]AZD2995 using the High Resolution Research Tomograph. All subjects also underwent a T1 weighted MR scan for delineation of regions of interest.
Results
All three ligands showed favorable properties for PET imaging with sufficient brain exposure, reversible binding with rapid binding kinetics, as well as high specific and low non-specific binding. The time-activity curves could be described by a 2-tissue compartment model as well as be fitted using the Logan plot. The radioligands could be quantified using simplified reference regions methods which were validated against full kinetic modeling (Figure 1). [18F]AZD4694, [11C]AZD2184 and [11C]AZD2995 showed significant increase of amyloid-β in grey matter of AD patients (DVR of 1.6, 1.7 1.5 respectively) compared to controls (1.0, 1.1, 1.0 respectively). The white matter binding was low for all radioligands with DVR values of 1.4 for [18F]AZD4694; 1.3 for [11C]AZD2184 and 1.0 for [11C]AZD2995. The rank order was consistent with the previously determined determined lipophilicity values, for [11C]AZD4694 having the highest (3.2), followed by [11C]AZD2184 (1.8) and then [18F]AZD2995 (1.3). All three radioligands showed high levels of specific binding and could discriminate between healthy individuals and AD patients with [11C]AZD2184 and [18F]AZD4694 having the largest discrimanate power in cortical regions.
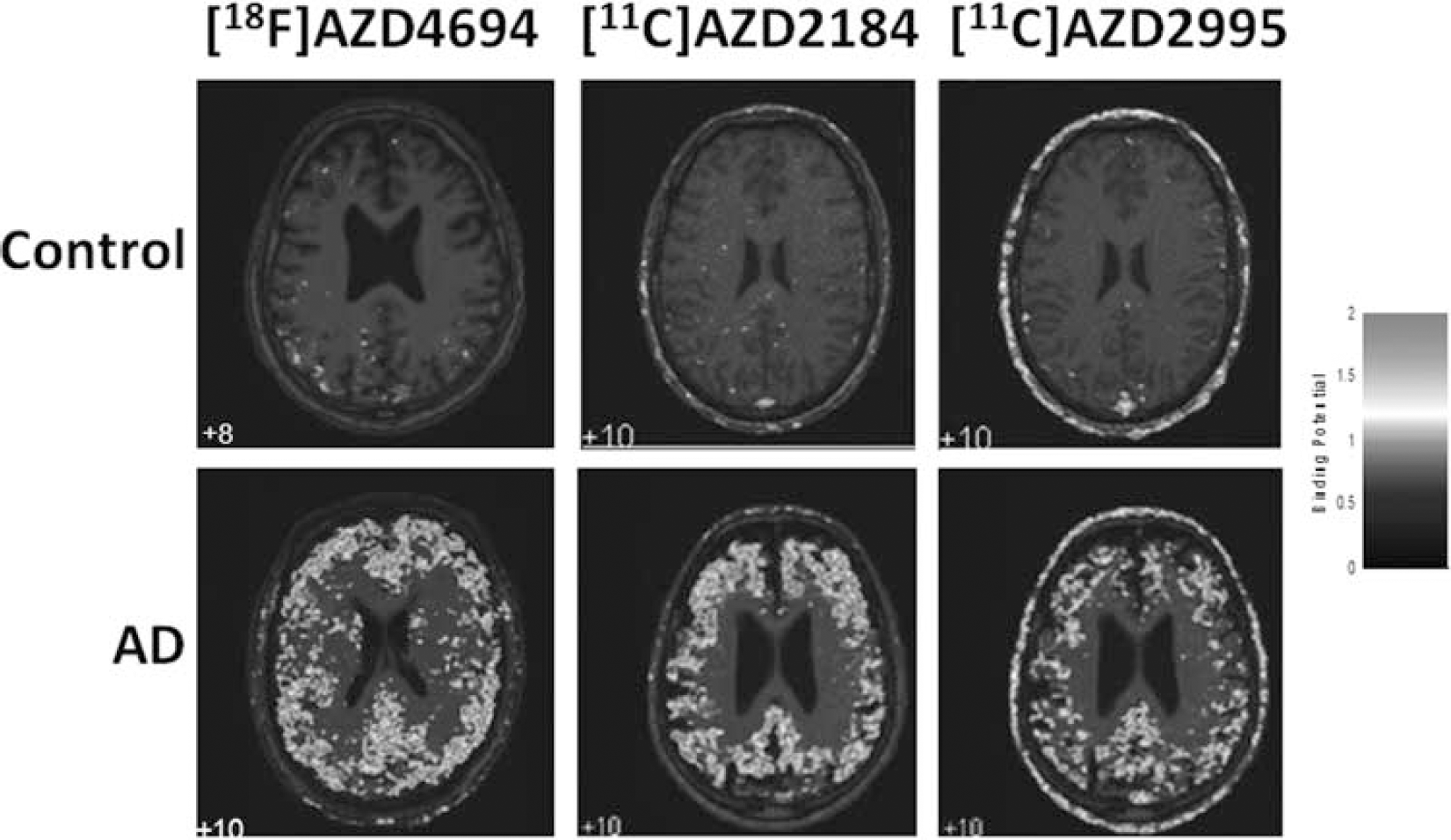
Parametric images created with wavelet aided parametric imaging, showing Binding Potential values calculated using reference Logan. PET images overlaid on respective subjects MR image.
Conclusions
The individual characteristics of the three radioligands may translate into advantages for different imaging purposes. For multi-tracer approaches an carbon-11 labeled ligand, such as [11C]AZD2184, is advantageous due to the potential to perform several PET measurements in the same subject on the same day. [11C]AZD2184 having high discriminate power can serve as a tool to study discrete levels of amyloid-β, which may be important in early prodromal stages of AD or in longitudinal studies of disease modifying therapies. Being labeled with the more long-lived radionuclide Fluor-18, allowing shipment to PET-facilities without their own cyclotrone, [18F]AZD4694 has potential as a biomarker to assist the clinical diagnosis of Alzheimer's Disease. [11C]AZD2995, with its very low non-specific binding, may provide advantages for micro-PET studies on transgenic rodents. We conclude that the present development of three novel radioligands for amyloid-β PET imaging have the potential to serve research and clinical applications in several ways.
P086. Labeling of Tubastatin A with Carbon-11 in the hydroxamic acid carbonyl position for PET imaging of histone deacetylase 6 in brain
1Molecular Imaging Branch, National Institute of Mental Health, NIH, Bethesda, Maryland, USA; 2Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago, USA
Background
Many neurological disorders are associated with imbalances in protein acetylation and transcriptional dysfunction for which histone deacetylases (HDACs) play an important role. 1 Aberrant acetylation of α-tubulin, a major substrate of HDAC6, leads to defects in intracellular transport that have been linked to neurodegenerative disorders, such as Alzheimer's disease 2 and Huntington's disease. 1 Tubastatin A is a selective HDAC6 inhibitor which has been shown to induce elevated levels of acetylated α-tubulin, confer dose-dependent protection against glutathione depletion-induced oxidative stress in primary cortical neuron cultures and restore defects in axonal transport.3,4 In this work, we aimed to develop a generic method for labeling hydroxamic acids with carbon-11 that could be used to prepare [11C]tubastatin A as a candidate PET radioligand for imaging HDAC6 in the brain.
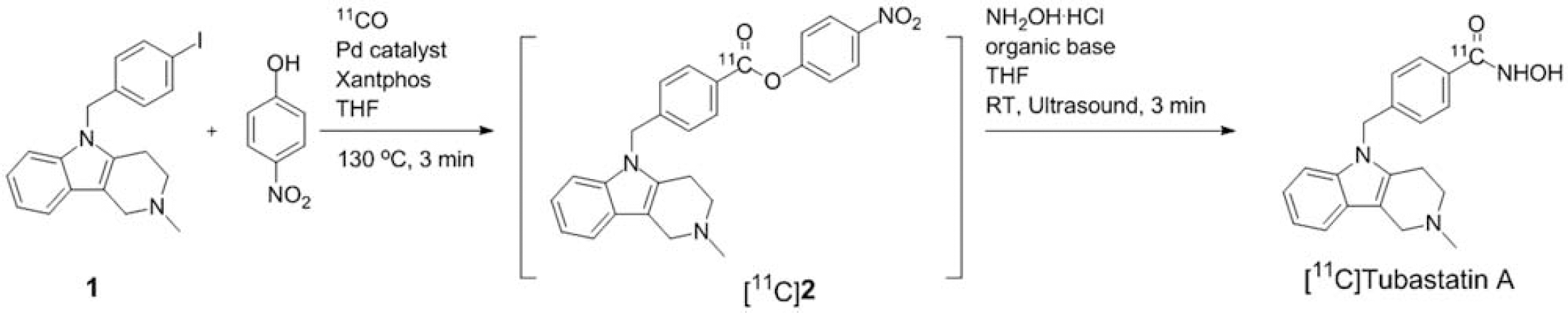
Radiosynthesis of [11C]tubastatin A via the aminolysis of [11C]
Methods
Radiosynthesis was performed with a modified Synthia [11C]CO module.
5
Tris(dibenzylidene-acetone)dipalladium(0) (0.6 μmol), Xantphos (1.2 μmol), 5-(4-iodobenzyl)-2-methyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indole (
Results
The intermediate [11C]nitrophenol ester ([11C]
Conclusions
A method for producing [11C]tubastatin A as a candidate PET radioligand for imaging HDAC6 in the brain was developed. Further optimization of the radiosynthesis and purification are in progress. This method should also be applicable to labeling other hydroxamic acids as potential PET radioligands.
Acknowledgements
This research was supported by the Intramural Research Program of the NIMH.
P087. Preclinical characterization of [11C]JNJ42491293, a PET tracer for the mGluR2 positive allosteric site
1Janssen Research & Development, Beerse Belgium; 2Laboratory for Radiopharmacy & Nuclear Medicine, KUL Leuven, Belgium; 3Janssen Research & Development, Toledo, Spain
Background
The metabotropic glutamate subtype 2 receptor (mGluR2) is a presynaptic membrane receptor distributed widely in brain that provides feedback inhibitory control of glutamate release. Augmenting mGluR2 function with a positive allosteric modulator (PAM) is expected to reduce activity dependent glutamate release which may be of therapeutic benefit for a range of neurological and psychiatric disorders. To confirm target engagement and define exposure-occupancy relationships of clinical candidates targeting this site, a site-specific positron emission tomography (PET) ligand has been developed. Here we report the preclinical evaluation of [11C]JNJ42491293, a PET ligand for in vivo imaging of mGluR2 in brain.
Methods
The chloro-triazolopyridine JNJ42491293 was selected based on affinity and selectivity for an mGluR2 allosteric site and acceptable physicochemical properties. [11C]JNJ42491293 was obtained by alkylation of the phenolic precursor with [11C]MeI followed by RP-HPLC purification. Biodistribution and μPET imaging were performed in rats. Radiometabolites were quantified in rat plasma and brain homogenates using HPLC. Blocking and displacement experiments were done using compounds with high affinity and selectivity for mGluR2.
Results
In vitro radioligand binding studies showed that JNJ42491293 binds with high affinity (IC50=9.6 nM) to the cloned human mGlu2 receptor. Further studies demonstrated that JNJ-42491293 acted as a PAM with a potency of 7.6 nM. JNJ42491293 showed >250-fold selectivity versus other mGlu receptors or neurotransmitter targets. [11C]JNJ42491293 was obtained with a radiochemical yield of 65% and a specific activity of 145 GBq/μmol. Rat biodistribution studies showed moderate brain uptake (0.88% ID at 2 min p.i.) and accumulation of radioactivity in all studied brain regions (>1% ID at 30 min p.i.) such as cortex, striatum, and cerebellum, consistent with the known distribution of the mGlu2 receptor. Plasma radiometabolite analysis showed the presence of one polar metabolite which was not detected in brain. Baseline μPET imaging showed that the maximum radioactivity concentration (SUV 1.7) in brain was reached at 12 min p.i., remained constant until 27 min, followed by slow wash-out. Blocking resulted in fast wash-out and chasing in displacement of radioactivity in all studied brain regions.
Conclusions
μPET blocking and displacement experiments in rats indicate that [11C]JNJ42491293 binds specifically and reversibly to an mGluR2 allosteric site, strongly suggesting that it is a promising candidate for in vivo imaging of mGluR2 using PET.
P088. 11C-TAZA and 11C-Dalene: new PET imaging agents for human brain senile plaques also bind to norepinephrine transporter
University of California, Irvine, USA
Background
Alzheimer's disease (AD) is a neurodegenerative disease characterized by β-amyloid senile plaques (SPs) that reside mainly in the frontal cortex and hippocampus regions of the brain. The aim of this study is to evaluate the effectiveness of two new tracers, 4-11C-methylamino-4′-dimethylaminoazobenzene (11C-TAZA) and 4-methylamino-4′-(N-11C-methyl-N-methylamino)stilbene (11C-Dalene) shown in Figure 1. Binding of11C-TAZA, 11C-Dalene and 11C-PIB was evaluated to the SP sites in post-mortem human brain hippocampus (AD and control) and binding to norepinephrine transporters (NET) were evaluated in normal rats using PET/CT.
Methods
Radiosynthesis was carried out by reacting 4-amino-4′-dimethylaminoazobenzene (1 mg/0.5 cc acetone for 11C-TAZA) and 4-methylamino-4′-(N-methylamino)stilbene (1 mg/0.5 cc acetone for 11C-Dalene) with 11C-methyltriflate prepared in the GE FXCPro synthesis unit. 11C-Methyltriflate was trapped at −20°C, subsequently heated for 5 mins at 80°C and purified using HPLC. In vitro autoradiography studies were performed on 7 micron slices of hippocampus of AD patients and control brains. Slices were incubated with the radiotracer (10–25 μCi/cc, 11C-TAZA, 11C-Dalene or 11C-PIB) in 40% ethanol-water for 1/2 hour. Non-specific binding was assessed using 10 μM PIB for the 3 tracers. Slices were then washed and exposed on to phosphor screens and slides were analysed by Optiquant image analysis program for SP binding Digital light units/mm2 (DLU/mm2). Sprague-Dawley rats were scanned after IV injection of 11C-TAZA and 11C-Dalene in Inveon PET/CT and displacement studies were done with atomoxetine (1 mg/kg). PET images were analysed using PMOD.
Results
11C-TAZA, 11C-Dalene and 11C-PIB were obtained in 25 to 100 mCi yields in specific activities >1000 Ci/mmol. The radiosynthesis was clean with >95% radiochemical product for each of them. 11C-TAZA and 11C-Dalene bound specifically to SP present in AD brains compared to the normal controls as show in the Figure 1. AD/Control hippocampus ratios were: 11C-TAZA>30; 11C-Dalene>5 and 11C-PIB>5. Presence of SP in the AD brains were confirmed using 4G8 antibody immunostaining for Aβ-amyloid. The control brains exhibited little or no SP. Unlabeled PIB dispaced 11C-TAZA and 11C-Dalene—suggesting similar binding sites. 11C-TAZA and 11C-Dalene also exhibited selective binding in NET brain regions such as brain stem and thalamus. Ratios of brain stem to cingulate gyrus (used as reference region) was >2. Atomoxetine reduced the binding of both 11C-TAZA and 11C-Dalene with the former showing greater reduction.
Conclusions
Our studies suggest that 11C-TAZA and 11C-Dalene are effective imaging agents for human SP in AD patients. Of the 3 radiotracers, 11C-TAZA showed the highest binding to SP. Our studies also suggest that 11C-TAZA and 11C-Dalene bind to NET. Further studies are underway to determine if there is overlap in the binding sites of SP and NET.
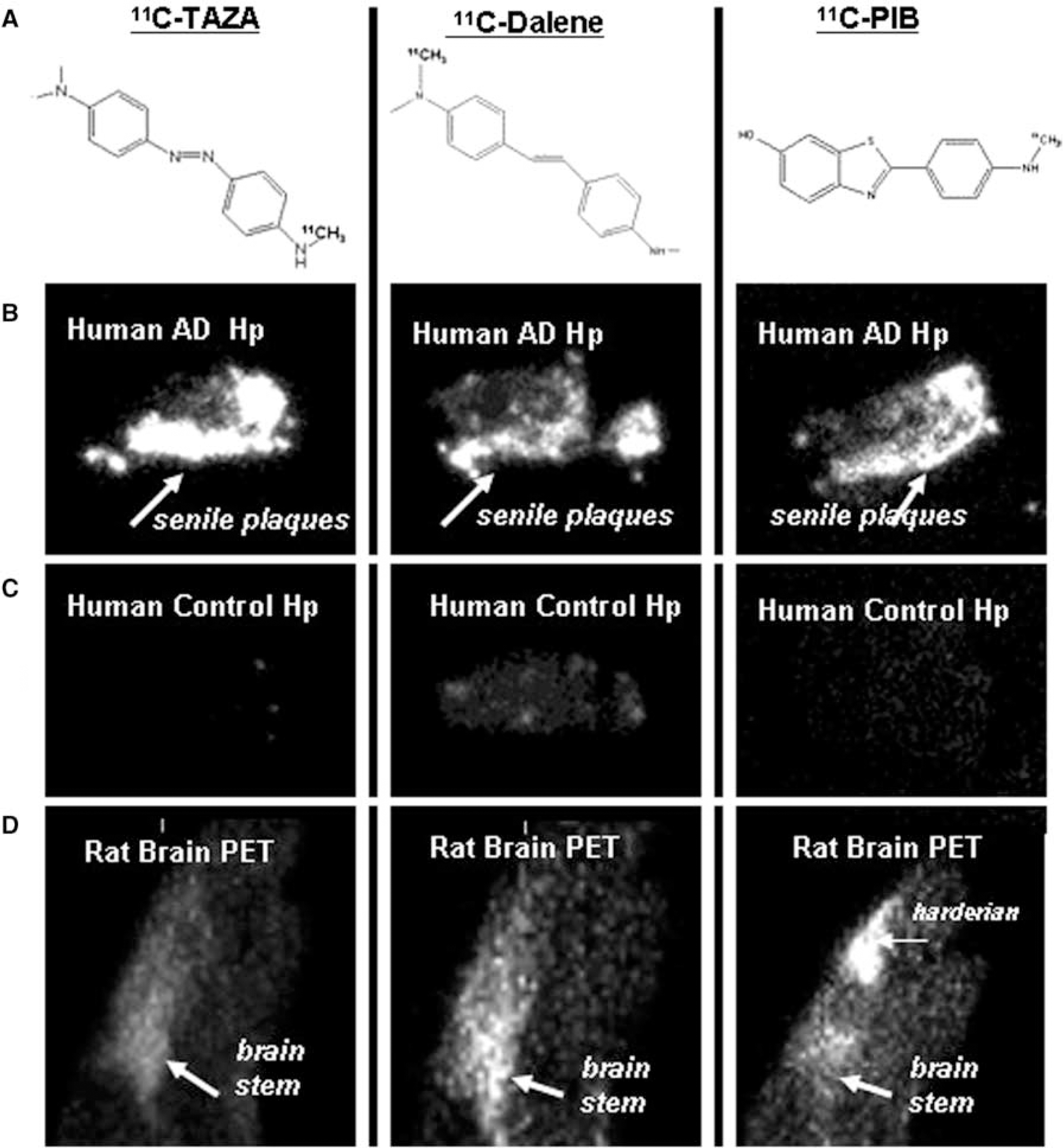
11C-TAZA, 11C-Dalene, 11C-PIB: Chemical structures (A); Binding to postmortem human AD brain hippocampal sections, in vitro autoradiographs (B); Binding to postmortem human control brain hippocampal sections, in vitro autoradiographs (C); Binding to normal rat brain sagittal sections, in vivo PET (D).
Acknowledgements
NIH/NIA R01AG029479, NIH/NIA R33AG030524, NIH/NIA P50 AG16573.
P089. Imaging norepinephrine transporter and glucose metabolism in rat brown adipose tissue
M. Reza Mirbolooki, Min-Liang Pan, Bhavin Patel, Christopher Liang, Himika Patel, Meenakshi Mukherjee, Cristian Constantinescu and
University of California, Irvine, USA
Background
Innervation of brown adipose tissue (BAT) with neurotransmitter, norepinephrine is essential for regulation of thermogenesis and thus has a role in obesity and diabetes. Norepinephrine acts by activating β3-adrenoceptor to increase BAT metabolic activity measurable using 18F-FDG PET. Norepinephrine transporter (NET) inhibitors such as atomoxetine play an important role in BAT activity by increasing norepinephrine levels. Therefore imaging NET along with glucose metabolism in BAT will provide useful tools to measure mass and activity of BAT.
Methods
Male Sprague-Dawley rats were fasted for 24 hrs prior to 18F-FDG administration. Rats were administered iv ∼0.3 mCi 18F-FDG under 2% isoflurane anesthesia. The same rats were treated with atomoxetine 1 mg/kg, 30 mins before 18F-FDG administration. Rats were awake for 60 mins and subsequently anesthetized for upper-body Inveon MicroPET/CT scan. To evaluate whether enhanced 18F-FDG uptake in activated BAT could be reduced by pharmacologic interventions, propranolol (β3-adrenorecptor inhibitor) 5 mg/kg was given intraperitoneally in anesthetized rats, 30 minutes prior to tomoxetine administration. For NET imaging,11C-TAZA (4-11C-methylamino-4′-dimethylaminoazobenzene) and 11C-Dalene (4-methylamino-4′-(N-11C-methyl-N-methylamino)stilbene) were administered iv (∼0.5–3 mCi) and scanned in Inveon PET/CT for 90 mins. Competition studies were done with atomoxetine (preinjection, 1–2 mg/kg). PET/CT data was analysed using IRW and PMOD.
Results
Tomoxetine increased the average 18F-FDG uptake of IBAT significantly, >15 times, compared to controls (365±170 vs. 23.9±8.7 kBq/cc) (Figure 1). This is consistent with blocking of NET by atomoxetine and increasing norepinephrine. Propranolol (adrenergic receptor inhibitor) reduced the average 18F-FDG uptake of IBAT significantly. Interscapular BAT (IBAT) was clearly visualized with both 11C-TAZA and 11C-Dalene with IBAT to reference muscle ratios >4. Atomoxetine reduced IBAT binding of both 11C-TAZA and 11C-Dalene by 60% and 35%, respectively. Incomplete displacement may be due to possible internalization of the radiotracers. Although there was similarity in the different BAT regions visualized by atomoxetine stimulated 18F-FDG uptake and NET agents 11C-TAZA and 11C-Dalene, 18F-FDG uptake appeared more prominent. Autoradiography of IBAT and white adipose tissue (WAT) confirmed the data obtained by PET.
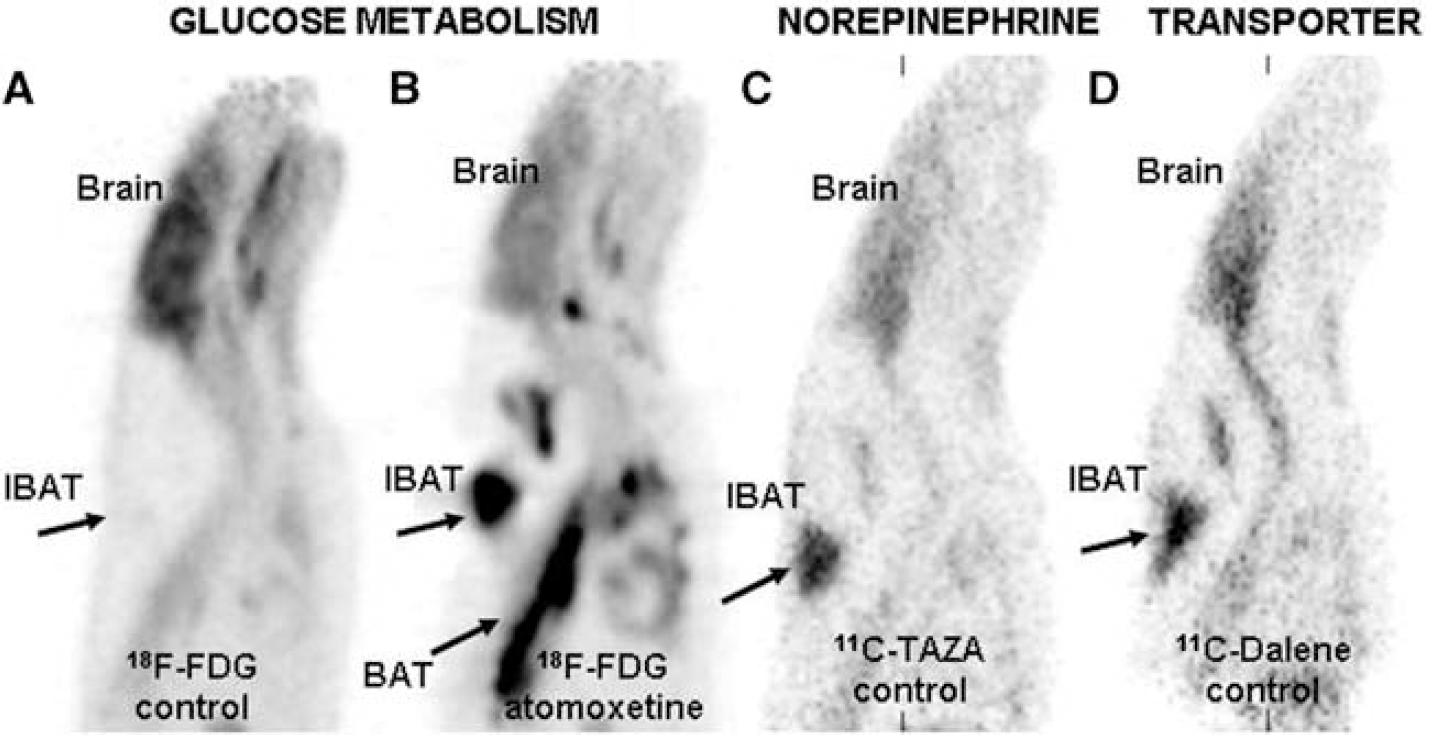
Rat PET studies showing brown adipose tissue (BAT): 18F-FDG uptake in control rat (A); 18F-FDG uptake in rat after preinjection of atomoxetine showing activation of BAT (B, arrows showing interscapular BAT);11C-TAZA showing NET binding in BAT (C, arrows showing interscapular BAT); 11C-Dalene showing NET binding in BAT (D, arrows showing interscapular BAT).
Conclusions
Our studies suggest that 11C-TAZA and 11C-Dalene are effective imaging agents for NET in BAT. Atomoxetine increases norepinephrine in BAT resulting in hypermetabolic effects measurable by 18F-FDG uptake at ambient temperature in the rodent model. Correlations of NET binding of 11C-TAZA and 11C-Dalene and 18F-FDG uptake in various BAT regions is underway in order to assess the relationship of NET concentration with metabolic activity.
Acknowledgements
NIH/NIDDK R21DK092917, RC1DK087352.
P090. [18F]-MNI654 and [18F]-MNI659: novel PET imaging tracers to quantify PDE10A
1Institute for Neurodegenerative Disorders, New Haven, Connecticut, USA; 2Pfizer Inc., Groton, Connecticut, USA
Background
Preclinical and early clinical evaluation of the PDE10A occupancy and displacement by candidate drugs is an important part of in vivo characterization in the clinical development of potential new treatments for schizophrenia and other neuropsychiatric disorders. Such studies aid in prioritization among competing drug candidates and provide rational dosing strategies for Phase 2. The aim of this research is to validate the two structurally related PDE10A (phosphodiesterase-10A) tracers [18F]MNI654 and [18F]MNI659 in primates (non-human and human).
Methods
Positron emission tomography (PET) studies were carried out in rhesus macaque (Macaca mulatta) and baboon (Papio anubis). Eleven PET studies (four baselines and seven blockade studies using MP10, a selective PDE10A inhibitor) were conducted with [18F]MNI654 (dose 4.9±0.2 mCi) and five PET studies (three baselines and two blockade studies) were conducted with [18F]MNI659 (dose 4.5±0.8 mCi). MP10 doses of 1.8, 0.6, 0.2 and 0.07 mg/kg were administered i.v. over 30 min prior to tracer injection. PET studies were also carried out on a Siemens HR+ camera over up to 5.5 hours in healthy human volunteers post injection of ∼5.0 mCi of [18F]MNI654 or [18F]MNI659 to evaluate an optimal outcome measure and its test/retest variability.
Results
In non-human primate, [18F]MNI654 and [18F]MNI659 displayed regional brain uptake in accordance with expected PDE10A distribution. Baseline and blockade studies were modeled with a two-tissue compartment model to estimate the distribution volume. The binding potential BPnd was derived using the occipital as a reference region. The occupancy was estimated from BPnd at baseline and following blockade with MP10. [18F]MNI654 and [18F]MNI659 PET imaging estimated a dose-dependent occupancy of PDE10A by MP10 similar to published results with [11C]MP10.
In humans, similar uptake was observed. Kinetic modeling was applied (two-tissue compartment, occipital as reference region). BPnd in the putamen was ∼12.0 for [18F]-MNI654 using 330 min of data and ∼3.5 for [18F]-MNI659 using 90 min of data.
Conclusions
Preliminary data suggest that both [18F]MNI654 and [18F]MNI659 may be useful for measuring PDE10A in human, albeit with different pharmacokinetics.
P091. Characterization of the opioid receptor-like 1 (ORL1) PET tracer [18F]MK-0911 in rhesus monkey and human brain
1Merck, West Point, Pennsylvania, USA; 2Tsukuba Research Institute, Japan; 3KU Leuven, Belgium; 4MSD Brussels, Belgium
Background
Opioid receptor-like 1 (ORL1), also known as nociceptin opioid peptide (NOP) receptor, is a G-protein coupled receptor with homology to classical opioid receptors (δ, κ, and μ). ORL1 has been implicated in a number of neuropsychiatric disorders such as pain, obesity, substance abuse, anxiety, and cognitive impairment. Therefore, ORL1 antagonists are of interest as potential drug therapies. An ORL1-specific PET tracer would be useful in clinical development by ensuring sufficient target engagement is achieved by well-tolerated doses of an ORL1 antagonist therapeutic candidate. Evaluation of the ORL1 PET tracer [11C]NOP-1a in monkey was recently reported [1,2]. Presented here is [18F]MK-0911 (Figure 1), a PET tracer for imaging ORL1 in rhesus monkey and human.
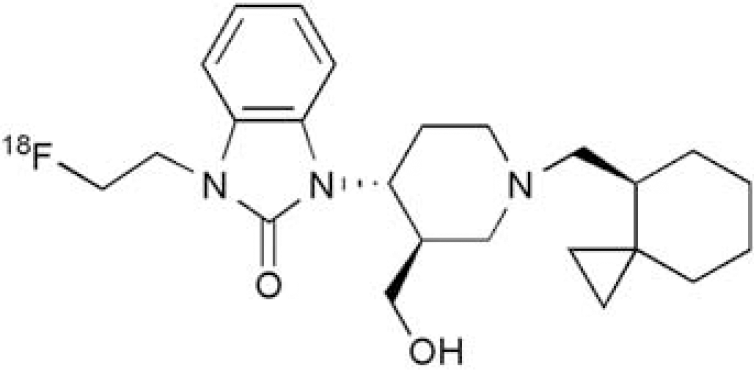
The structure of [18F]MK-0911.
Methods
[18F]MK-0911 (IC50=0.56 nM, log D=2.9) was synthesized with high specific activity (>1 Ci/umol) by reacting the appropriate precursor with [18F]fluoroethylbromide. Drug occupancy studies were performed by IV bolus + infusion (monkey) or PO administration (human) of an ORL1 antagonist prior to IV administration of [18F]MK-0911. Monkey PET data was analyzed by an area-under-the-curve (AUC) method using the cerebellum AUC at full blockade as a surrogate for nondisplaceable binding. Human PET data was analyzed by kinetic modeling using metabolite-corrected arterial input functions and by an AUC method similar to that used in monkey.
Results
The kinetics and distribution of [18F]MK-0911 in monkey and human brain were similar, with rapid distribution into all grey matter regions including cortex, striatum, and thalamus followed by moderate clearance. Lowest distribution was observed in the cerebellum and white matter, consistent with the known distribution of ORL1 [3]. [18F]MK-0911 distribution in the grey matter of both species was dose-dependently blocked in vivo by pre-treatment with a potent, selective ORL1 antagonist. At the highest drug plasma levels, [18F]MK-0911 uptake was homogeneous throughout the brain, revealing a specific signal and confirming the specificity of [18F]MK-0911 for ORL1. Data from [18F]MK-0911 PET studies with ORL1 antagonists was used to determine plasma concentration/ORL1 receptor occupancy relationships in rhesus monkey and human.
Conclusions
[18F]MK-0911 is a suitable PET tracer for use in occupancy studies with ORL1 antagonists in rhesus monkey and human.
P092. Development of candidate 18F-labeled PET imaging agents for β-amyloid plaque in Alzheimer's disease
Molecular Imaging Branch, National Institute of Mental Health, Bethesda, Maryland, USA
Background
Neurofibrillary tangles and β-amyloid plaques are the two hallmarks of Alzheimer's Disease (AD) [1]. [11C]PIB is currently the foremost useful PET radioligand for imaging β-amyloid plaques [2]. There remains a need for radioligands with a greater dynamic imaging range coupled with a longer-lived fluorine-18 label. To meet this need, we sought to design radioligands with higher binding affinity and lower lipophilicity than [11C]PIB, and with amenability to labeling with fluorine-18. Here we report the synthesis, radiolabeling and PET imaging in monkey of three candidate 18F-labeled ligands for imaging β-amyloid plaque.
Methods
Ligands (
Synthesis of [18F]
Results
Decay-corrected radiochemical yields of the purified and isolated 18F-labeled ligands were 3050%. The retention times of
Conclusions
Although [18F]
Acknowledgements
We thank Dr. Mary Herman (CBDB/NIMH) for providing human brain slices, and the Intramural Research Program of NIH (NIMH) for support.
P093. Test-retest evaluation of [18F]FPEB, a PET tracer for the mGluR5 receptors in humans
Jenna Sullivan1, Beata Planeta-Wilson1, Keunpoong Lim1, Shu-Fei Lin1, Soheila Najafzadeh1, Timothy McCarthy2, Yu-Shin Ding3, Richard E. Carson1, Evan D. Morris1,4 and
1Yale PET Center, Department of Diagnostic Radiology, Yale University School of Medicine, New Haven, Connecticut, USA; 2Pfizer Global Research and Development, Groton, Connecticut, USA; 3Department of Radiology, New York University School of Medicine, New York, New York, USA, 4Department of Psychiatry, Yale University School of Medicine, New Haven, Connecticut, USA
Background
[18F]FPEB displays high specificity and selectivity toward metabotropic glutamate receptor 5 (mGluR5) (Patel, 2005; Wang, 2007). It possesses the potential to be used in human studies to evaluate mGluR5 function, and possibly glutamate changes in various neurological disorders with PET. The goal of this study was to evaluate the reproducibility of receptor binding parameters measured with [18F]FPEB in humans.
Methods
All human PET imaging was conducted on the HRRT scanner following bolus plus constant infusion (Kbol=190) of [18F]FPEB. The primary outcome measures were regional brain volume of distribution (V T ), and binding potential (BPND). Five healthy control subjects were each scanned twice on separate days in a Test-Retest paradigm, to assess the variability and reproducibility of binding parameters. Arterial and venous blood samples were collected, and parent fraction was measured by HPLC to generate the metabolite-corrected arterial input function. Time activity curves (TACs) were extracted from 13 regions of interest (ROIs). Regional TACs were fitted with 2-tissue compartment model (2TC) and multilinear analysis (MA1) as well as with MRTM2 to estimate VT and BPND using the cerebellum white matter [CWM] as the reference region. Test-retest variability (TRV) and the intra-class correlation coefficient (ICC) were calculated to evaluate reliability of VT andBPND. Specific activity (SA) at end-of-synthesis (t: 5.20±1.12 Ci/μmol, rt: 5.95±0.70 Ci/μmol); injected dose (t: 4.28±1.01 mCi/nmol, rt: 4.27±0.93 mCi/nmol); and, injected mass (t: 0.27±0.09 μg, rt: 0.28±0.23 μg), were not significantly different between test and retest scans (n=5) (p>0.1).
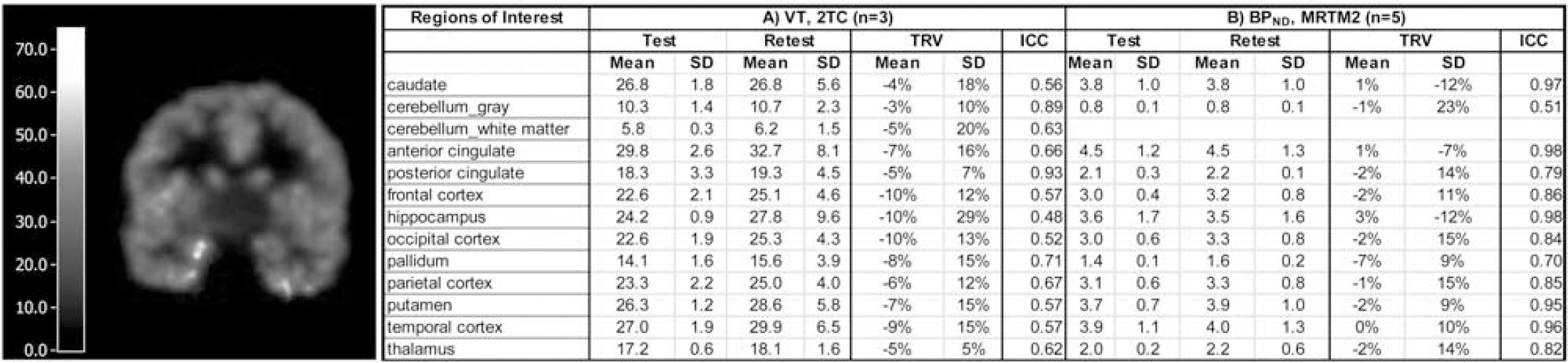
(Left) VT Image from one subject showing region-specific uptake in brain. (Right) [18F]FPEB TRV and ICC by region.
Results
Average [18F]FPEB plasma free fractions were 6%±1% (test), and 5% ±1% (retest), respectively. Metabolism of [18F]FPEB was rapid, with parent fraction representing 61.4±11.7%, 28.2±3.3%, and 27.4±4.3% of total radioactivity in the plasma at 8, 30, and 60 min post-injection (n=5). [18F]FPEB displayed the highest VT values in the caudate, putamen, and anterior cingulate; frontal-, occipital-, parietal- and temporal-cortices; and hippocampus. Intermediate VT values were observed in the thalamus, posterior cingulate, and palladium. Lowest VT values were observed in the cerebellum gray matter and CWM. Ultimately, blocking studies will be needed to validate CWM as a reference region. Test-retest reproducibility was high. The overall mean BPND value was 3.0. In subcortical regions: thalamus, putamen, caudate, and hippocampus were 2.0, 4.0, 4.0, 4.0, respectively. Cortical [18F]FPEB BPND values were 5.0, 4.0, 3.0, and 3.1 in the anterior cingulate, temporal, frontal, and occipital cortex, respectively. Figure 1 (Right) shows mean % TRV and SD and ICC, the latter of which is an index of parameter measurement reliability.
Conclusions
The novel PET tracer [18F]FPEB displays appropriate uptake kinetics and reasonable test-retest variability in the human brain, further promoting its potential use as a PET imaging agent to visualize mGluR5.
P094. In vivo selectivity of new kappa opioid receptor ligand [11C]LY2795050 in rhesus monkey
Yale PET Center, Yale University, New Haven, Connecticut, USA
Background
Radioligand competition assays using cloned human kappa opioid receptors (KOR) and mu opioid receptors (MOR) to assess the in vitro binding affinities of [11C]LY2795050 indicate high affinity and selectivity of the tracer to KOR in vitro (Ki=1.37 & 87.3 nM for kappa & mu, respectively) (Zheng, submitted). Our goal in the present study was to determine the in vivo selectivity of this new KOR antagonist ligand by examining the binding curves for [11C]LY2795050 and [11C]carfentanil in the presence of unlabeled LY2795050.
Methods
[11C]LY2795050 and [11C]carfentanil scans were performed for 120 min with varying doses of LY2795050 (1.6, 5, 10, 44, 395 ug/kg for scans with [11C]LY2795050; 45, 135, 405 ug/kg for scans with [11C]carfentanil) in multiple rhesus monkeys. Volume of distribution (VT) of [11C]LY2795050 was calculated using MA1 (t∗: 40 min) (Ichise, 2002) with metabolite corrected plasma activity curve. Simplified reference tissue model (Gunn, 1997) was used to estimate binding potential (BPND) of both [11C]carfentanil and [11C]LY2795050. Lassen plots (Cunningham, 2010) were applied to calculate occupancy. Occipital and cerebellum regions were used as reference regions for [11C]carfentanil and [11C]LY2795050, respectively. ED50MOR of LY2795050 was estimated from a binding curve of [11C]carfentanil data. Carfentanil was assumed to be highly selective for MOR. BPND of [11C]LY2795050 in the presence of unlabeled LY2795050 was modeled as the sum of specific binding at KOR (BPNDMOR) and MOR (BPNDKOR) (i.e., a two site model). A one-site model was also used which attributed all blockade to action at KOR. To estimate ED50KOR, models were fitted to total specific binding in 8 regions and monkeys, simultaneously. ED50KOR of LY2795050 was assumed to be invariant across all regions and monkeys. Goodness of fits to one vs two-site models was determined by F-test.
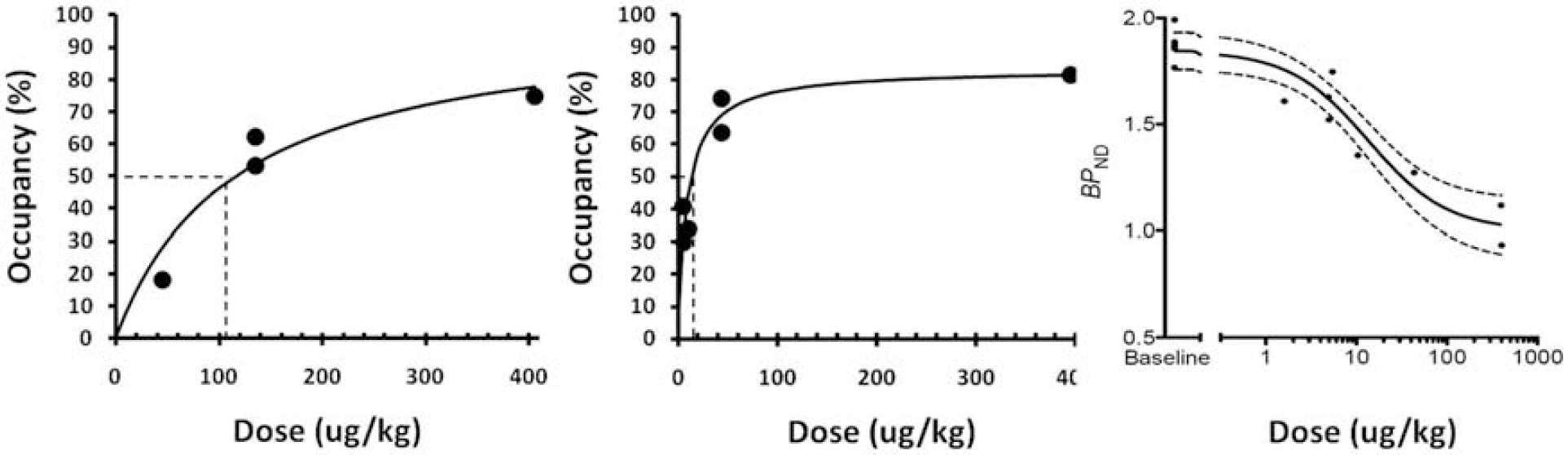
(Left) Binding curve of [11C]carfentanil and (middle) [11C]LY2795050 at varying doses of LY2795050. (Right) Fit of one-site model of [11C]LY2795050 BPND in the cingulate vs dose of LY2795050. Dotted lines show ED50 for each receptor, dotted curves are 95% confidence intervals.
Results
BPND of [11C]carfentanil showed a dose-dependent decline in most regions. Estimated ED50MOR was 115.8 ug/kg. There were no changes in VT for [11C]LY2795050 in the cerebellum at different doses of LY2795050. BPND of [11C]LY2795050 (cerebellum as the reference) also showed a dose-dependent decline. Estimated ED50KOR was 9.15 ug/kg and maximum occupancy was 83%. A one site model produced a better fit than two-site to describe the blocking of [11C]LY2795050 by unlabeled LY2795050. Estimated ED50KOR from one site model was 13.5 ug/kg.
Conclusions
The in vivo selectivity of [11C]LY2795050 is lower than the selectivity measured in vitro. If the prevalence of KOR and MOR are approximately equal in a region, the specific binding fraction of [11C]LY2795050 will be (ED50MOR/(ED50KOR+ED50MOR))=115.8/(115.8+13.5). Thus, 90% of the specific binding in an image due to a tracer injection of [11C]LY2795050 will be attributable to KOR binding. [11C]LY2795050 appears to be a promising tracer for selective imaging of KOR in vivo.
P095. Quantification of activated NMDA receptors with [18F]GE-179 PET
C.J. McGinnity1,2,
1Centre for Neuroscience, Department of Medicine, Imperial College London, London, UK; 2MRC Clinical Sciences Centre, London, UK; 3The Neurodis Foundation, CERMEP Imagerie du Vivant, Lyon, France; 4Department of Clinical and Experimental Epilepsy, UCL Institute of Neurology, London, and Epilepsy Society, Chalfont St. Peter, UK; 5GE Healthcare Ltd., The Grove Centre, Amersham, UK
Background
N-methyl D-aspartate (NMDA) receptor ion channels are overactive following acute cerebral insults and in chronic neurological and psychiatric disorders. To date, there have not been suitable positron emission tomography (PET) radiotracers for NMDA receptors. [18F]GE-179 is a novel radioligand that selectively binds to the open/active state of the NMDA receptor ion channel (Robins et al., 2010). Pre-clinical studies demonstrated high affinity for the intra-channel site of the rat NMDA receptor (KD=2.35± 0.2 nM) with acceptable lipophilicity (LogD7.4=2.49±0.1; GE Healthcare, unpublished data). Focal increases in uptake are seen in patients with frequent epileptiform discharges (McGinnity et al., 29th IEC 2011). Here, we describe the kinetic behaviour of the radioligand in vivo in humans.
Methods
Ten healthy participants without history of neurological or psychiatric illness (seven male, median age 46 (25–62) years) each had a 90-minute PET scan using a Siemens/CTI ECAT EXACT3D 962 HR+ PET camera following a median injection of 186 (173–192) MBq [18F]GE-179. Continuous arterial blood sampling over the first 15 minutes was complemented by discrete blood sampling over the duration of the scan. Metabolite-corrected parent plasma input functions were generated using CLICKFIT (v1.7) software. Dynamic images were motion-corrected. Weighted summation images (KBq/ml) were created with Receptor Parametric Mapping (RPM6), and co-registered with corresponding 3T T1-weighted MR images. Global uptake was calculated, as was uptake in six bilateral grey matter regions-of-interest (ROIs) obtained via anatomical segmentation of the MRI (MAPER; Heckemann et al., 2011). Volume-of-distribution (VT) was quantified in those ROIs with one-, two- and three compartment(s) models in CLICKFIT. Parametric VT images were generated by graphical analysis (Logan, 2003) using RPM6 (Figure 1).
Left - Mean time-activity curves for the putamena (solid line), parahippocampal gyri (dashed line with triangles), and white matter (dotted line with Xs; all decay-corrected). Right - [18F]GE-179 volume-of-distribution (VT) for 58-year-old healthy male.
Results
[18F]GE-179 accounted for a mean of 37% of plasma radioactivity at 30 minutes post-injection. The mean grey-matter radioactivity concentration peaked at 9 KBq/ml at t=7.5 minutes, and remained higher than that of white matter over 90 minutes. The highest median ratio of grey:white matter radioactivity concentration (1.8) was seen in early (t=5–30 minutes) summation images. The radioactivity concentration was highest in putamina, occipital lobes, and thalami, and lowest in the temporal lobes. The two-compartment, four rate constants model best described the radioligand's kinetics in grey matter, as assessed by the Akaike Information Criterion (AIC), with acceptable (<17%) regional between-subject co-efficients of variation. There was no correlation between VT and k1 in any ROI.
Conclusions
[18F]GE-179 exhibits high brain penetration with a heterogeneous distribution, moderately-paced kinetics in grey matter, acceptable between-subject variability, and rapid metabolism. Reliable quantification appears feasible using a two-compartment model. The lower uptake in the temporal lobes might relate to higher expression of NR2B than NR2A subunits with lower channel open probability. Further evaluation of [18F]GE-179 should facilitate investigation of activated NMDA receptor availability in a variety of conditions.
P096. Consortium to develop an alpha-synuclein imaging agent
1Michael J. Fox Foundation, New York, New York, USA; 2BioFocus, Cambridge, UK; 3Massachusetts General Hospital, Boston, Massachusetts, USA; 4University of Pittsburgh, Pennsylvania, USA; 5Washington University, St. Louis, Missouri, USA
Background
The Michael J. Fox Foundation (MJFF) is supporting a consortium with the goal of developing a PET radiotracer to image the distribution of alpha-synuclein in the brain. The accumulation of aggregated alpha-synuclein in the brain is the pathological hallmark of Parkinson's disease and is a frequent target for drugs being developed to treat PD. The ability to visualize alpha-synuclein in the brain could be useful both as a biomarker of the presence of disease and disease progression and as a tool for drug development.
Methods
Four groups have been selected by MJFF to participate in the consortium including three academic groups and a contract research organization (BioFocus). These groups are working to identify and test compounds that can ultimately be radiolabeled and used as PET imaging agents.
Results
Two approaches were pursued to identify compounds that bind to aggregated alpha-synuclein. The first approach utilized a competition binding assay using either the fluorescent ligand Thioflavin T or the radioligand [3H]-Chrysamine G. Following assay optimization, approximately 100,000 compounds selected from the BioFocus libraries were screened in both assay formats at a single concentration of 10 micromolar. From the Thioflavin T screen, approximately 2,000 compounds showing >45% inhibition of binding were selected for hit confirmation at 10 micromolar in duplicate, and 263 of these compounds proceeded to IC50 potency determination. Selectivity of these compounds was determined against aggregated beta-amyloid and tau using Thioflavin T and Thioflavin S assays respectively. The second approach utilized a Surface Plasmon Resonance (SPR) based Biacore assay developed to investigate the direct binding of compounds to monomeric alpha-synuclein, aggregated alpha-synuclein and aggregated beta-amyloid, immobilized to Biacore CM5 chips via amine coupling reactions. Following the screening of approximately 3,500 compounds against these three protein targets, 120 compounds were selected for KD determination.
Conclusions
Further characterization of interesting compounds identified from these approaches is ongoing, including binding studies in PD brains with Lewy body pathology.
P097. In vivo PET imaging of cerebral 5-HT7 receptors with novel radioligands
1Neurobiology Research Unit, Copenhagen University Hospital, Rigshospitalet, Copenhagen; 2PET and Cyclotron Unit, Rigshospitalet, Copenhagen; 3Institute for Medicinal Chemistry, Faculty of Pharmaceutical Sciences, University of Copenhagen, Denmarik
Background
Because of its possible involvement in brain disorders such as schizophrenia, depression, and circadian rhythms the 5-HT7 receptor is an interesting drug target. So far, no well-validated radiotracer for PET-imaging of the 5-HT7 receptor exists. We identified two compounds with high affinity and selectivity for the 5-HT7 receptor and investigate here their in vivo behaviour in the pig brain before and after pre-treatment with a structurally different 5-HT7 receptor antagonist.
Methods
We synthesized two PET radioligands [11C]Cimbi-717 and the ethyl derivative [11C]Cimbi-775 and studied the in vivo brain distribution of the tracers in female Danish Landrace pigs. After intravenous injection of the radiotracers, data were acquired by a HRRT PET scanner for 90 minutes. Arterial input measurements including corrections for radiolabeled metabolites were done and regional time-activity curves were retrieved by co-registration to an MRI-based atlas of the pig brain. In a total of 3 pigs, the 5-HT7 receptor selective antagonist SB-269970 (1 mg/kg/h or 4.2 mg/kg/h) was given intravenously prior to the second scan to investigate if cerebral [11C]Cimbi-717 binding was blocked. Furthermore, in vitro receptor autoradiography was performed with [11C]Cimbi-717 and [11C]Cimbi-775 on pig brain sections.
Results
Time-activity curves of [11C]Cimbi-717 and [11C]Cimbi-775 showed reversible tracer kinetics and high brain uptake. [11C]Cimbi-775 showed a more uniform distribution pattern across regions of interest, which is not consistent with the 5-HT7 receptor distribution (Varnäs et al., 2004). On the basis of the Akaike information criterion, the two-tissue compartment model was chosen for modelling. Distribution volumes confirmed the higher uptake of [11C]Cimbi-717 and a BPND of 4.8±1.16 (n=4) was calculated in the thalamus – a region with a high 5-HT7 receptor density. Pretreatment with SB-269970 decreased the cerebral binding of [11C]Cimbi-717 by up to 77% supporting that [11C]Cimbi-717 is selective for the 5-HT7 receptor. Radiometabolite analysis of the two tracers showed the presence of two metabolites for both tracers and a faster metabolism of [11C]Cimbi-775 than of [11C]Cimbi-717, consistent with a previous study comparing unsubstituted vs. 3-ethyl substituted oxindole moities (Volk et al., 2011). In vitro autoradiography on pig sections confirmed the higher levels of specific binding of [11C]Cimbi-717 over [11C]Cimbi-775.
Conclusions
Based on both in vitro and in vivo data, [11C]Cimbi-717 was superior to [11C]Cimbi-775 with regards to specific brain binding. [11C]Cimbi-717 is a promising radiotracer for in vivo imaging of the 5-HT7 receptor due to a good brain uptake and since it can be selectively blocked by SB-269970.
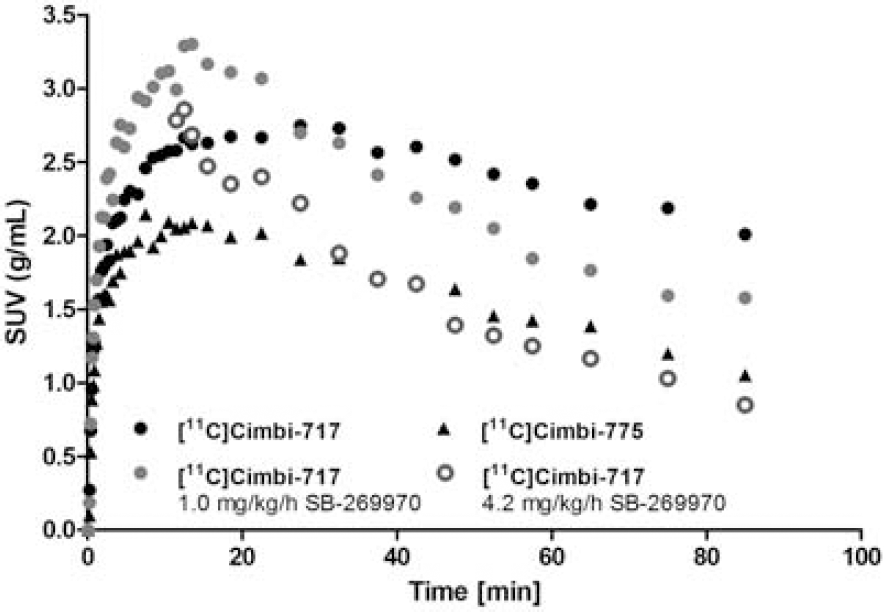
Time activity curves for [11C]Cimbi-717 and [11C]Cimbi-775 in the thalamus at baseline. For [11C]Cimbi-717, time activity curves in the thalamus is also shown after intravenous administration of two different doses of the 5-HT7 receptor specific antagonist SB-269970. Data is presented as standard uptake value (SUV).
P098. Optimized bolus/infusion scheme and determination of in vivo Kd for [11C]AZ10419369 in non-human primates
1Neurobiology Research Unit, Copenhagen University Hospital, Rigshospitalet, Copenhagen; 2A.A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital, Charlestown, MA, United States
Background
Positron emission tomography (PET) can be used to assess alterations in neurotransmitter levels in vivo in a non-invasive manner, as convincingly demonstrated for the dopamine system. The prospects for imaging endogenous serotonin (5-HT) release are improved by the introduction of 5-HT1B receptor partial agonist radioligands, e.g., [11C]AZ10419369, where binding is reduced in a dose-dependent manner by intravenous fenfluramine adminstration. The aim of this study was first to optimize a bolus/infusion scheme for [11C]AZ10419369 in the rhesus monkey to allow for a within-scan challenge and secondly, to calculate the AZ10419369 in vivo dissociation constant (Kd).
Methods
Anesthetized rhesus macaques underwent PET- and MR- scanning with a 3T Tim Trio with a BrainPET insert inside the bore of the scanner; data were acquired continuously for up to 150 min. The 5-HT1B receptor radioligand [11C]AZ10419369 was given as different bolus plus infusion schedules with Kbol ranging from 1.3 h to 3 h. Regional time-activity curves were retrieved by delineating brain regions on the anatomical MRI-image. Sixty-five min after bolus injection, either 10 μg of AZ10419369 was given as an i.v. bolus injection or the 5-HT releaser fenfluramine (5 mg/kg) was infused i.v. over 5 min. Cerebral 5-HT1B receptor binding BPND was quantified at steady-state with cerebellum as the reference region. The occupancy was calculated based on fitting of the displacement curve assuming a koff=0.02 min−1 (1) and the free fraction in cerebellum (fND) was determined based on plasma water concentration of [11C]AZ10419369. In vivo Kd of the radiotracer was calculated as: Kd=−((F∗O)-F)/O), where O is the occupancy and F is the free fraction of AZ10419369 after the challenge injection.
Results
Time-activity curves of [11C]AZ10419369 showed that a Kbol of 1.8 h generated steady-state levels in both high and low binding regions as early as 35 min after injection whereas Kbol values of 2.6–3 h, resulted in steady state levels at 55–75 min, leading to difficulties in accurate quantification of within-scan challenge studies. The free fraction of [11C]AZ10419369 in cerebellum, fND, was determined to 0.008. I.v. injection of 10 μg AZ10419369 lead to a conservative estimate of 33 % occupancy of the 5-HT1B receptors and accordingly, a Kd was determined to 0.017 nM. Fenfluramine infusion led to a ∼30% reduction in occipital cortex BPND measured 30 min post infusion of fenfluramin. An immediate and transient increase in [11C]AZ10419369 was also observed after injection of fenfluramine similar to what has previously been reported (2).
Conclusions
A bolus/infusion scheme of 1.8 h is optimal for obtaining steady state in high and low binding regions as fast as possible. An in vivo Kd of 0.017 nM means that a maximum of 1.19 μg of AZ10419369 can be injected in order to keep occupancy below 5%.