Granulocyte colony-stimulating factor (G-CSF) is a candidate neuroprotective factor following cerebral ischemia. To determine whether G-CSF acts partly through the inhibition of nitric oxide synthase (NOS)-2 expression, we administered G-CSF to male NOS-2−/− mice after cerebral ischemia. Although male NOS-2−/− mice exhibit resistance to the gross effects of cerebral ischemia, they display neuronal loss and skilled motor deficits following cerebral ischemia. Administration of G-CSF during reperfusion reduced motor deficit and neuronal loss. Thus, G-CSF is still effective in NOS-2 gene-deficient mice, suggesting that part of the mechanism of action is independent of NOS-2.
Granulocyte colony-stimulating factor (G-CSF), a member of the cytokine family of growth factors, is a potential neuroprotective factor following cerebral ischemia (Gibson et al, 2005a,
b
; Schabitz et al, 2003; Schneider et al, 2005). Cerebral ischemia triggers a complex series of detrimental events including excitotoxicity, inflammation, and cell death along with increased nitric oxide production through the activation of nitric oxide synthase (NOS) enzymes. Nitric oxide produced through NOS-2 is detrimental following cerebral ischemia, because NOS-2 gene-deficient mice display smaller infarcts than do their wild-type counterparts (Iadecola et al, 1997; Loihl et al, 1999a).
Administration of G-CSF suppresses specific elements of the inflammatory response evoked by cerebral ischemia, including edema formation and increased interleukin-1β expression (Gibson et al, 2005a,
b
). In addition, G-CSF reduces the number of NOS-2-immunopositive cells within the peri-infarct region after cerebral ischemia (Komine-Kobayashi et al, 2006; Sehara et al, 2007). Although we did not previously (Gibson et al, 2005b) show reduced NOS-2 mRNA expression after G-CSF treatment, this may be because of studying the whole hemisphere rather than the peri-infarct region alone. Thus, it is unclear whether the neuroprotective effect of G-CSF may be attributed, at least in part, to a direct suppression of NOS-2 expression. In this study, we investigated the neuroprotective ability of G-CSF in the absence of a functional NOS-2 gene.
Materials and methods
Transient Ischemia
This study was conducted in accordance with the UK Animals (Scientific Procedures) Act, 1986 (Project Licence 40/2206). Mice were housed in a pathogen-free facility (University of Nottingham) on a 12-h light–dark cycle with ad libitum access to food and water. Mice with the NOS-2 gene deletion (−/−) were originally obtained from Carl Nathan, John MacMicking, and John Mudgett (MacMicking et al, 1995) and were out-crossed to C57 BL/6 wild-type (wt) mice to generate NOS-2 heterozygote (+/−) mice for breeding. The NOS-2 (−/−) mice were selected on the basis of genotyping, as described previously (Loihl et al, 1999b).
All mice were adult male C57 BL/6 NOS-2−/− weighing between 24 and 32 g. In total, 21 mice were used: 16 mice underwent transient middle cerebral artery occlusion (MCAO) (1 died and 1 was excluded because of inadequate reperfusion) and 5 were included as sham-operated controls. Focal cerebral ischemia was induced for 60 mins by occlusion of the right middle cerebral artery, and cerebral blood flow was monitored as described previously (Gibson et al, 2005a,
b
). Sham-operated mice underwent the same surgical procedure, except that the filament was not advanced far enough to occlude the middle cerebral artery. Mice subjected to MCAO were randomly assigned to receive either G-CSF (Amgen, Breda, The Netherlands, n = 7, 50 μg/kg dissolved in saline) or vehicle (n = 7, saline) injected subcutaneously at the onset of reperfusion, i.e., 1 h after ischemia. The experimenter was blinded to the treatment that the mice received before all subsequent testing/analyses.
Tests of Motor Function
An accelerating rotarod (Letica Scientific Instruments, Barcelona, Spain) was used to test the motor function as described previously (Gibson et al, 2005a,
b
). Data are expressed as the percentage of mean duration per day compared with presurgery control value. In addition, mice were subjected to the grid test as described previously (Gibson et al, 2005a,
b
). Foot faults are expressed as the number of errors made by the contralateral limbs as a percentage of total errors made.
Histology
At 7 days after MCAO, mice were anesthetized and transcardially perfused using 20 mL 0.9% saline, followed by 1 mL/g body weight buffered 4% paraformaldehyde fixative solution. Coronal cryostat sections (of 25-μm thickness) were processed for NeuN (neuronal nuclei) immunostaining (mouse monoclonal NeuN antibody; Chemicon, Hampshire, UK; 1:1,000) using Diaminobenzidine (Sigma, Welwyn Garden City, UK) as the chromogen. Digital photographs of the striatal and lateral cortex were taken from sections spaced 125 μm apart at × 20 magnification using Axiovision Software (Welwyn Garden City, UK). Neuronal loss was analyzed by counting immunopositive neurons within the striatum and lateral cortex on a restricted number of sections spaced evenly (125 μm) apart from allowing the mean number per animal to be calculated. Cell counts were then expressed as a ratio of immunopositive neurons ipsilateral/contralateral to the lesion to identify any cell loss in relation to the unilateral ischemia.
Statistical Analysis
All data were found to be normally distributed when tested using the Kolmogorov–Smirnov test (P > 0.10). All data are expressed as mean ± s.e.m. and analyzed using parametric tests. Cerebral blood flow data were analyzed using a two-tailed Student's t-test. Survival data were expressed by applying the Kaplan–Meier curve, followed by the Mantel–Haenszel log-rank test to identify differences between the curves. Experiments conducted over a series of days (i.e., weight gain, rotarod, foot fault) were analyzed by two-way ANOVA (analysis of variance). Grid test data were also analyzed using linear regression to compare the slopes of the curves. Immunocytochemical cell counts were analyzed by two-way ANOVA for differences according to treatment (or genotype) and location, i.e., the striatum or lateral cortex. Post hoc analyses were carried out with Bonferroni's test. All data were analyzed using GraphPad Prism Version 5.00 for Windows (GraphPad Software, San Diego, CA, USA). The criterion for statistical significance was set at P < 0.05.
Results
Cerebral Blood Flow
Doppler monitoring showed no significant differences in cerebral blood flow after MCAO induction following G-CSF (17.96 ± 2.36%) or vehicle treatment (18.63 ± 3.02%). After the onset of reperfusion, G-CSF or vehicle treatment did not significantly alter the increase in relative cerebral blood flow (G-CSF: 68.04 ± 7.64%; vehicle: 75.31 ± 10.62%), at least for the first 5 mins after withdrawal of the filament.
General ‘Well-Being’
All sham-operated mice survived for the 7 days after surgery, whereas 1 mouse that underwent transient MCAO died. Analysis using the Mantel–Haenszel log-rank test (χ2 = 1.714) showed that survival rates did not differ between the experimental groups (P = 0.42). All experimental groups lost weight over the first 2 days after surgery. Shams regained weight faster than did MCAO-subjected animals regardless of whether the MCAO animals received G-CSF (F1,12 = 114.3, P < 0.0001) or vehicle treatment (F1,12 = 33.83, P < 0.0001). Treatment with G-CSF in NOS-2−/− mice did not significantly alter the rate of weight gain after MCAO (P = 0.0503).
Functional Outcome
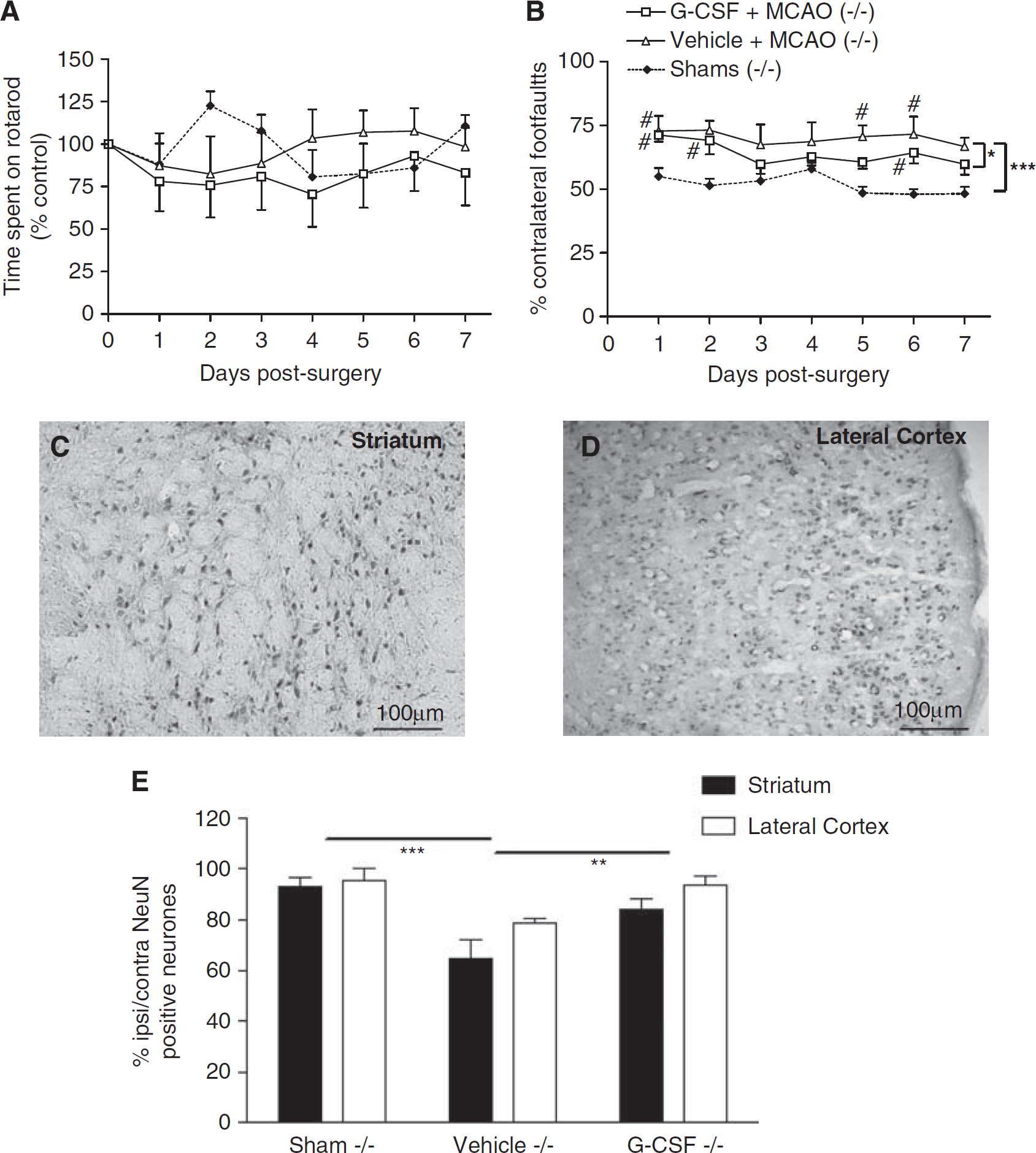
Motor function, assessed using the rotarod, showed the absence of a functional deficit in NOS-2−/− mice after MCAO compared with genotype-matched shams (P > 0.05, Figure 1A). Treatment with G-CSF did not affect rotarod performance in NOS-2−/− mice compared with vehicle treatment (P = 0.11, Figure 1A). On the grid test, there was a significant increase in the number of errors made following MCAO (regardless of treatment) compared with genotype-matched shams (F1,12 = 42.96, P < 0.0001, Figure 1B). However, G-CSF treatment reduced the functional deficit in NOS-2−/− mice compared with vehicle treatment (F1,14 = 5.47, P < 0.05, Figure 1B). In addition, G-CSF-treated animals showed a significant improvement in recovery over time (P = 0.0384) which was absent after vehicle treatment.
(A) Assessment of rotarod performance showed that after middle cerebral artery occlusion (MCAO), NOS-2−/− mice that received granulocyte colony-stimulating factor (G-CSF) or vehicle were indistinguishable from shams. The data are expressed as the percentage (%) of mean duration per day compared with the presurgery control value. (B) Skilled motor performance assessed using the grid test showed that unilateral deficits, measured by the number of contralateral foot faults (expressed as the % of total errors made), were significantly higher in NOS-2−/− mice after MCAO compared with shams, P < 0.0001. # represents a significant difference from sham animals on the day of testing. Treatment with G-CSF significantly reduced this deficit, *P < 0.05. After sham surgery, no unilateral functional deficit was observed. (C, D) Neuronal nuclei (NeuN)-positive neurons were identified in the striatal areas and the lateral cortex. (E) Cell counts of NeuN-positive neurons showed that NOS-2−/− mice exhibited neuronal loss 7 days after MCAO (***P < 0.001) which was significantly reduced after G-CSF treatment (**P < 0.01).
Neuronal Loss
Neuronal nuclei-positive neurons were counted in areas of the striatum (Figure 1C) and lateral cortex (Figure 1D). After MCAO and vehicle treatment, there was a significant decrease in the ratio of NeuN-positive compared with genotype-matched shams (F1,12 = 18.76, P < 0.001, Figure 1E). However, post hoc tests showed that the neuronal loss associated with NOS-2−/− genotype occurred in the striatal areas rather than in the lateral cortex (P < 0.01). Treatment with G-CSF after MCAO reduced the neuronal loss in NOS-2−/− mice compared with vehicle treatment (F1,14 = 13.71, P < 0.01, Figure 1E).
Discussion
The lack of a NOS-2 gene provided protection in terms of the severity of the general motor deficit that was present after cerebral ischemia. However, in terms of skilled motor coordination, as assessed using the grid test, a deficit was present after cerebral ischemia even in the absence of a functional NOS-2 gene. The severity of this skilled motor deficit was reduced over a period of 7 days after G-CSF treatment. Neuronal cell loss occurred in the striatum and lateral cortex after cerebral ischemia, even in mice lacking a functional NOS-2 gene, and the cell loss was reduced at 7 days after ischemia following G-CSF treatment. Thus, NOS-2 gene deletion in male mice does not completely protect them from the effects of transient cerebral ischemia in terms of skilled motor deficit and neuronal loss, and G-CSF is able to protect in the absence of a functional NOS-2 gene.
Transient MCAO has been shown to cause significant motor impairments as assessed using the rotarod and grid test in rats (Rogers et al, 1997) and mice (Gibson et al, 2005a). Although NOS-2 gene deletion prevented mice from exhibiting a deficit on the rotarod test, it did not prevent the deficit being identified on the grid test. This is presumably because the affected limb was compensated by the nonaffected limb on the rotarod, as shown previously following brain insults (Napieralski et al, 1998), which is not possible on the grid test, as it specifically assesses unilateral deficit and measures placement dysfunction of the affected limbs (Yager et al, 2006). Our previous study of traumatic brain injury also showed that only the grid test, and not the rotarod, is sensitive enough to identify a motor deficit in NOS-2-deficient mice (Jones et al, 2004). In the current study, in which a deficit was present after NOS-2 gene deletion, this was improved after G-CSF treatment, suggesting that G-CSF exerted a beneficial effect independent of NOS-2. However, the improvement in the grid test after G-CSF treatment was not observed immediately after MCAO (i.e., 24 h) but was observed over a period of 7 days.
The effectiveness of G-CSF in mice lacking a functional NOS-2 gene was also shown by reduced neuronal loss in the striatal and cortical areas at 7 days after ischemia following G-CSF administration in NOS-2−/− mice. The G-CSF suppresses specific elements of the inflammatory response following cerebral ischemia, in particular the injury-induced expression of interleukin-1β and edema formation (Gibson et al, 2005a,
b
). Although G-CSF has been reported to have no effect on NOS-2 mRNA expression at 72 h after ischemia (Gibson et al, 2005b), other reports indicate a suppression of NOS-2 protein at 72 h after ischemia in response to G-CSF treatment (Komine-Kobayashi et al, 2006). The lack of any earlier effects of G-CSF on NOS-2 protein expression suggests that this may be in response to other factors, such as decreased interleukin-1β expression, rather than as a direct effect of G-CSF. The demonstration of an effect of G-CSF in mice with a NOS-2 gene deletion supports our previous observation (Gibson et al, 2005b) that G-CSF does not act through NOS-2 suppression
Footnotes
Acknowledgements
The authors thank the staff of the BMSU, University of Nottingham, for their care of experimental animals and Dr Blair Grubb (Department of Cell Physiology and Pharmacology, University of Leicester) for provision of histology facilities. PMWB is Stroke Association Professor of Stroke Medicine. CLG conducted all experiments and the majority of the analysis. PMWB participated in study design. SPM conceived the study, participated in study design, and provided the coding system for blinding. All authors contributed to, read, and approved the final manuscript.
The authors declare no conflict of interest.
References
1.
GibsonCLBathPMWMurphySP (2005a) G-CSF reduces infarct volume and improves functional outcome after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab25:431–9
2.
GibsonCLJonesNCPriorMJWBathPMWMurphyDP (2005b) G-CSF suppresses edema formation and reduces interleukin-1β expression after cerebral ischemia in mice. J Neuropath Exp Neurol64:1–7
3.
IadecolaCZhangFCaseyRNagayamaMRossME (1997) Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci17:9157–64
4.
JonesNCConstantinDGibsonCLPriorMJWMorrisPGMarsdenCAMurphySP (2004) A detrimental role for nitric oxide synthase-2 in the pathology resulting from acute cerebral injury. J Neuropath Exp Neurol63:708–20
5.
Komine-KobayashiMZhangNLiuMTanakaRHaraHOsakaAMochizukiHMizunoYUrabeT (2006) Neuroprotective effect of recombinant human granulocyte-colony stimulating factor in transient focal ischemia of mice. J Cereb Blood Flow Metab26:402–13
6.
LoihlAKAsensioVCampbellILMurphySP (1999a) Expression of nitric oxide synthase (NOS)-2 following permanent focal ischemia and the role of nitric oxide in infarct generation in male, female and NOS-2 gene deficient mice. Brain Res830:155–64
7.
LoihlAKWhalenSCampbellILMudgetJSMurphySP (1999b) Transcriptional activation following cerebral ischemia in mice of a promoter-deleted NOS-2 gene. J Biol Chem274:8844–9
8.
MacMickingJDNathanCHomGChartrainNFletcherDSTrumbauerMStevensKXieQWSokolKHutchinsonHChenHMudgettJS (1995) Altered responses to bacterial-infection and endotoxic-shock in mice lacking inducible nitric-oxide synthase. Cell81:641–50
9.
NapieralskiJABanksRJChesseletMF (1998) Motor and somatosensory deficits following uni- and bilateral lesions of the cortex induced by aspiration or thermocoagulation in the adult rat. Exp Neurol154:80–8
10.
RogersDCCampbellJAStrettonJLMackayKB (1997) Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke28:2060–6
11.
SchabitzW-RKollmarRSchwaningerMJuettlerEBardutzkyJScholzkeMNSommerCSchwabM (2003) Neuroprotective effect of granulocyte-stimulating factor after focal cerebral ischemia. Stroke34:745–51
12.
SchneiderAKrugerCSteiglederTWeberDPitzerCLaageRAronowskiJMaurerMHGasslerNMierWHasselblattMKollmarRSchwabSSommerCBachAKuhnH-GSchabitzW-R (2005) The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest115:2083–98
13.
SeharaYHayashiTDeguchiKZhangHTsuchiyaAYamashitaTLukicVNagaiMKamiyaTAbeK (2007) Decreased focal inflammatory response by G-CSF may improve stroke outcome after transient middle cerebral artery occlusion in rats. J Neurosci Res85:2167–74
14.
YagerJYWrightSArmstrongEAJahrausCMSaucierDM (2006) The influence of aging on recovery following ischemic brain damage. Behav Brain Res173:171–80