
Abstract
Breast cancer resistance protein (BCRP) is the most abundant multidrug efflux transporter at the human blood–brain barrier (BBB), restricting brain distribution of various drugs. In this study, we developed a positron emission tomography (PET) protocol to visualize Bcrp function at the murine BBB, based on the dual P-glycoprotein (P-gp)/Bcrp substrate radiotracer [11C]tariquidar in combination with the Bcrp inhibitor Ko143. To eliminate the contribution of P-gp efflux to [11C]tariquidar brain distribution, we studied mice in which P-gp was genetically knocked out (Mdri1a/b(−/−) mice) or chemically knocked out by pretreatment with cold tariquidar. We found that [11C]tariquidar brain uptake increased dose dependency after administration of escalating doses of Ko143, both in Mdr1a/b(−/−) mice and in tariquidar pretreated wild-type mice. After 15 mg/kg Ko143, the maximum increase in [11C]tariquidar brain uptake relative to baseline scans was 6.3-fold in Mdr1a/bf(−/−) mice with a half-maximum effect dose of 4.98 mg/kg and 3.6-fold in tariquidar (8 mg/kg) pretreated wild-type mice, suggesting that the presented protocol is sensitive to visualize a range of different functional Bcrp activities at the murine BBB. We expect that this protocol can be translated to the clinic, because tariquidar can be safely administered to humans at doses that completely inhibit cerebral P-gp.
Keywords
Introduction
The adenosine triphosphate-binding cassette (ABC) transporter breast cancer resistance protein (BCRP), which is encoded by the ABCG2 gene, is widely expressed in the intestine, liver, mammary gland, placenta, and the brain (Robey et al, 2009; Vlaming et al, 2009). It transports a broad range of structurally diverse chemicals, including several clinically used drugs, against concentration gradients across cellular membranes. It has, for instance, been documented that BCRP mediates intestinal, biliary, and mammary secretion of various substrate drugs (Hirano et al, 2005; Merino et al, 2006). At the blood–brain barrier (BBB), BCRP is expressed in the luminal membrane of brain capillary endothelial cells, where it impedes distribution of its substrates to brain parenchyma by active efflux transport back into the blood compartment (Agarwal et al, 2011). BCRP has been shown to be colocalized at the BBB with P-glycoprotein (P-gp), another important member of the ABC transporter family (Sisodiya et al, 2006). Contrary to initial assumptions that P-gp is the most abundant ABC transporter at the human BBB, recent quantitative proteomics data indicate that BCRP expression at the human BBB is 1.43-fold higher than that of P-gp (Uchida et al, 2011). In mice, however, the expression of Bcrp (i.e., the murine homolog of BCRP) at the BBB is 3.3-fold lower than that of P-gp (Kamiie et al, 2008). A growing body of literature shows that brain distribution of several drugs, such as the tyrosine kinase inhibitors gefitinib, imatinib, and sorafenib, is limited because they are dual substrates of P-gp and Bcrp (Agarwal et al, 2010; Bihorel et al, 2007; Lagas et al, 2010). These two transporters form a concerted defense mechanism at the BBB, in which one transporter substitutes the other when one is chemically or genetically disrupted (de Vries et al, 2007; Kodaira et al, 2010). For dual substrates of Bcrp and P-gp, however, the function of Bcrp in restricting drug brain penetration in rodents is often masked due to the more pronounced role played by P-gp at the BBB, likely due to the higher expression of P-gp at the rodent BBB (Kamiie et al, 2008). Whereas several lines of evidence suggest that changes in P-gp expression and/or function might have a key role in the causation and pathogenesis of certain neurologic disorders, such as epilepsy and Alzheimer's disease (Löscher and Potschka, 2005), the role of BCRP in neurologic disease is much less understood.
Positron emission tomography (PET) imaging with radiolabeled substrates of P-gp, such as racemic [11C]verapamil, (R)-[11C]verapamil, or [11C]-N-desmethyl-loperamide, has been very useful, particularly in combination with administration of unlabeled P-gp inhibitors such as cyclosporine A or tariquidar, in studying P-gp function in vivo, both in animals and in humans (Bankstahl et al, 2011; Bauer et al, 2010b, 2012; Kreisl et al, 2010; Kuntner et al, 2010; Liow et al, 2009; Sasongko et al, 2005). So far, no PET radiotracers suitable for visualization of BCRP have been described, which can be explained in part by the fact that there is currently a lack of BCRP selective substrate probes due to highly overlapping substrate recognition patterns of P-gp and BCRP (Kodaira et al, 2010). The availability of a PET imaging protocol that allows study of BCRP function would enable a better understanding of the role this important efflux transporter plays at the healthy and diseased BBB.
The potent third-generation P-gp/Bcrp inhibitor tariquidar (Fox and Bates, 2007), at tracer concentrations as used in PET experiments, was shown to be a dual substrate of P-gp and Bcrp (Bauer et al, 2010a; Kannan et al, 2011b; Kawamura et al, 2010; Römermann et al, 2011). In the present study, we demonstrate that PET imaging with [11C]tariquidar can be used to assess Bcrp function at the murine BBB when P-gp is either genetically or chemically knocked out (i.e., by pretreatment with cold tariquidar). Because recent data show that tariquidar can be safely administered to humans at doses which completely inhibit P-gp at the BBB (Bauer et al, 2012), the presented PET protocol appears promising for future translation to the clinic.
Materials and methods
General
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich Chemie (Schnelldorf, Germany) or Merck (Darmstadt, Germany) and were of analytical grade and used without further purification. Isoflurane was obtained from Baxter Vertriebs Gmbh (Vienna, Austria). The Bcrp inhibitor Ko143 (Allen et al, 2002) was purchased from Axon Medchem BV (Groningen, The Netherlands). For intravenous injection, Ko143 was freshly dissolved in dimethyl sulfoxide and diluted with a solution containing 10% (v/v) Tween 80, 25% (v/v) PEG300 and 65% (v/v) sterile water to a final dimethyl sulfoxide concentration of 5% (v/v). Formulated Ko143 solution was injected into mice at a volume of 4 to 6 mL/kg. The dual P-gp/Bcrp inhibitor tariquidar dimesylate (chemical purity >98%) (Fox and Bates, 2007) was synthesized at the Department of Medicinal Chemistry (University of Vienna, Austria), freshly dissolved in 2.5% (w/v) aqueous dextrose solution before each administration and injected into mice at a volume of 4 mL/kg.
Radiotracer Synthesis and Formulation
[11C]Tariquidar was synthesized as described previously (Bauer et al, 2010a). For intravenous injection, [11C]tariquidar was formulated in 0.9% aqueous saline containing 75 μL Tween 80 to an approximate concentration of 370 MBq/mL. Radiochemical purity, as determined by radio-HPLC, was greater than 98%, and specific activity at the end of synthesis was >100 GBq/μmol.
Animals
Female Mdr1a/b(−/−) and Bcrp1(−/−) mice with an FVB genetic background were obtained from Taconic (Germantown, NY, USA). Female FVB wild-type mice were either purchased from Taconic or from Charles River (Sulzfeld, Germany). All animals were housed in Makrolon type 2 cages under controlled environmental conditions (22 ± 3°C, 40% to 70% humidity, 12-hour light/dark cycle) with free access to standard laboratory animal diet (ssniff R/M-H, ssniff Spezialdiäten GmbH, Soest, Germany) and water. An acclimatization period of at least 1 week was observed before the animals were used in experiments. The study was approved by the local animal welfare committee (Amt der Niederösterreichischen Landesregierung) and all study procedures were performed in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC). All efforts were made to minimize both the suffering and the number of animals used in this study.
Experimental Design
Two different experimental settings were used in this study. In the first set of experiments, female FVB wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) mice aged 6 to 9 months and weighing 25.4 ± 4.4 g were assigned to six groups (two groups per strain with n = 4 to 5 each). Each group received an intravenous injection of either vehicle solution or 10 mg/kg of the selective Bcrp inhibitor Ko143 (Allen et al, 2002) at 1 hour before start of the [11C]tariquidar PET scan. The dose and time point of Ko143 pretreatment were chosen according to the literature (Zhang et al, 2011). For dose–response measurements, female Mdr1a/b(−/−) mice (29.2 ± 7.7 g) were randomized into four additional groups (n = 3 to 4 per group) and pretreated with different doses of Ko143 (1, 3, 5, or 15 mg/kg) at 1 hour before start of the [11C]tariquidar PET scan.
For the second set of experiments, female FVB wild-type mice aged 6 to 12 weeks and weighing 23.2 + 2.1 g were used. Two hours before start of the PET scan, mice were pretreated by intravenous injection of cold tariquidar (15 or 8 mg/kg). One hour before start of the PET scan, mice received an additional intravenous injection of Ko143 (15 mg/kg) or Ko143 vehicle solution (n = 4 per dose group except for 15 mg/kg tariquidar/15 mg/kg Ko143 dose group where n = 7). Further groups of female FVB wild-type mice (n = 4 per dose group) were pretreated with cold tariquidar (8 mg/kg) and two additional doses of Ko143 (3 or 6 mg/kg) at 2 hours and 1 hour, respectively, before start of the [11C]tariquidar PET scan.
Positron Emission Tomography Imaging
Before each experiment, the animals were placed in an induction box and anesthetized with 2.5% isoflurane. During the imaging period, anesthesia was maintained with 1% to 2% isoflurane administered via a cone mask and the isoflurane level was adjusted depending on the depth of anesthesia. Animal respiratory rate and body temperature were constantly monitored during the data acquisition period (SA Instruments Inc, Stony Brook, NY, USA). The animals were kept warm throughout the experiment at ~38°C. Mice were positioned in a custom-made imaging chamber and the lateral tail vein was cannulated for intravenous administration. For PET imaging, a microPET Focus220 (Siemens Medical Solutions, Knoxville, TN, USA) was used. A 60-minute dynamic emission scan was recorded after intravenous injection of 27 ± 11 MBq [11C]tariquidar in a volume of 0.1 mL.
Postimaging Procedures
After completion of the imaging procedure, blood was withdrawn under isoflurane anesthesia from the orbital sinus vein into preweighed micropipettes. Blood samples were weighed and measured for radioactivity in a gamma counter (Perkin-Elmer Instruments, Wellesley, MA, USA). Blood radioactivity data were corrected for radioactive decay and expressed as standardized uptake value (SUV = (radioactivity per cubic centimeter/injected radioactivity) × body weight).
Positron Emission Tomography Data Analysis
The dynamic emission PET data were sorted into 23 frames, which incrementally increased in time length from 5 seconds to 10 minutes. Images were reconstructed using Fourier rebinning of the 3D sinograms followed by two-dimensional filtered backprojection with a ramp filter, resulting in a voxel size of 0.4 × 0.4 × 0.796 mm3. The standard data correction protocol (normalization, decay correction, and injection decay correction) was applied to the data. For attenuation correction, a corresponding transmission scan using a rotating 57Co point source, recorded before the PET scan, was used. A calibration factor for converting PET units of the recorded images into radioactivity concentration units was derived by imaging a phantom cylinder filled with a known 11C-radioactivity concentration. Using the image analysis software Amide, regions of interest were manually outlined on each plane of the PET summation image (0 to 60 minutes) covering the whole brain area. The regions of interest of the planes were then summed to generate volumes of interest of comparable size. The volumes of interest were then transferred to the PET images of the individual time frames to derive time-activity curves, expressed as SUV. Brain uptake of [11C]tariquidar was expressed as the brain-to-blood radioactivity concentration ratio (Kb,brain) at 60 minutes after radiotracer injection, which can be considered as an approximation of volume of distribution (VT) derived from kinetic modeling. Kb,brain was calculated by dividing the brain radioactivity concentration in the last PET frame (50 to 60 minutes after start of scan) by the blood radioactivity concentration as determined by the gamma counter measurements.
For assessment of the Ko143 dose–response relationship in Mdr1a/b(−/−) mice, the mean vehicle Kb,brain was subtracted from individual Kb,brain values measured after different Ko143 doses and normalized to the mean Kb,brain after maximum inhibition (relative response). Log dose–response relationship was analyzed by nonlinear regression analysis based on a log-sigmoidal model with variable slope using Prism 5.0 software (GraphPad Software, La Jolla, CA, USA).
Statistical Analysis
Differences between the groups were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparison test using Prism 5.0 software. The level of statistical significance was set to P < 0.05.
Results
Bcrp Inhibition with Ko143 Leads to Increased Brain Uptake of [11C]Tariquidar in Mdr1a/b(−/−) Mice but not in Wild-Type and Bcrp1(−/−) Mice
Groups of wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) mice (n = 4 to 5 per group) underwent [11C]tariquidar PET scans after intravenous administration of vehicle or Ko143 (10 mg/kg) (Figure 1). In all three mouse genotypes, mean Kb,brain was low after administration of vehicle with the following rank order: wild-type (1.0 ± 0.10) < Bcrp1(−/−) (2.0 ± 0.3) < Mdr1a/b(−/−) (2.4 ± 0.1). Pretreatment with Ko143 had no statistically significant effect on Kb,brain in wild-type and Bcrp1(−/−) mice (Figure 1D). In Mdr1a/b(−/−) mice, a marked 5.2-fold increase in Kb,brain to 12.6 ± 2.4 was observed after Ko143 pretreatment relative to vehicle group (P < 0.001; Figure 1D). Blood radioactivity concentrations at the end of the PET scan ranged from 0.10 to 0.16 (SUV) and were not significantly different between groups.
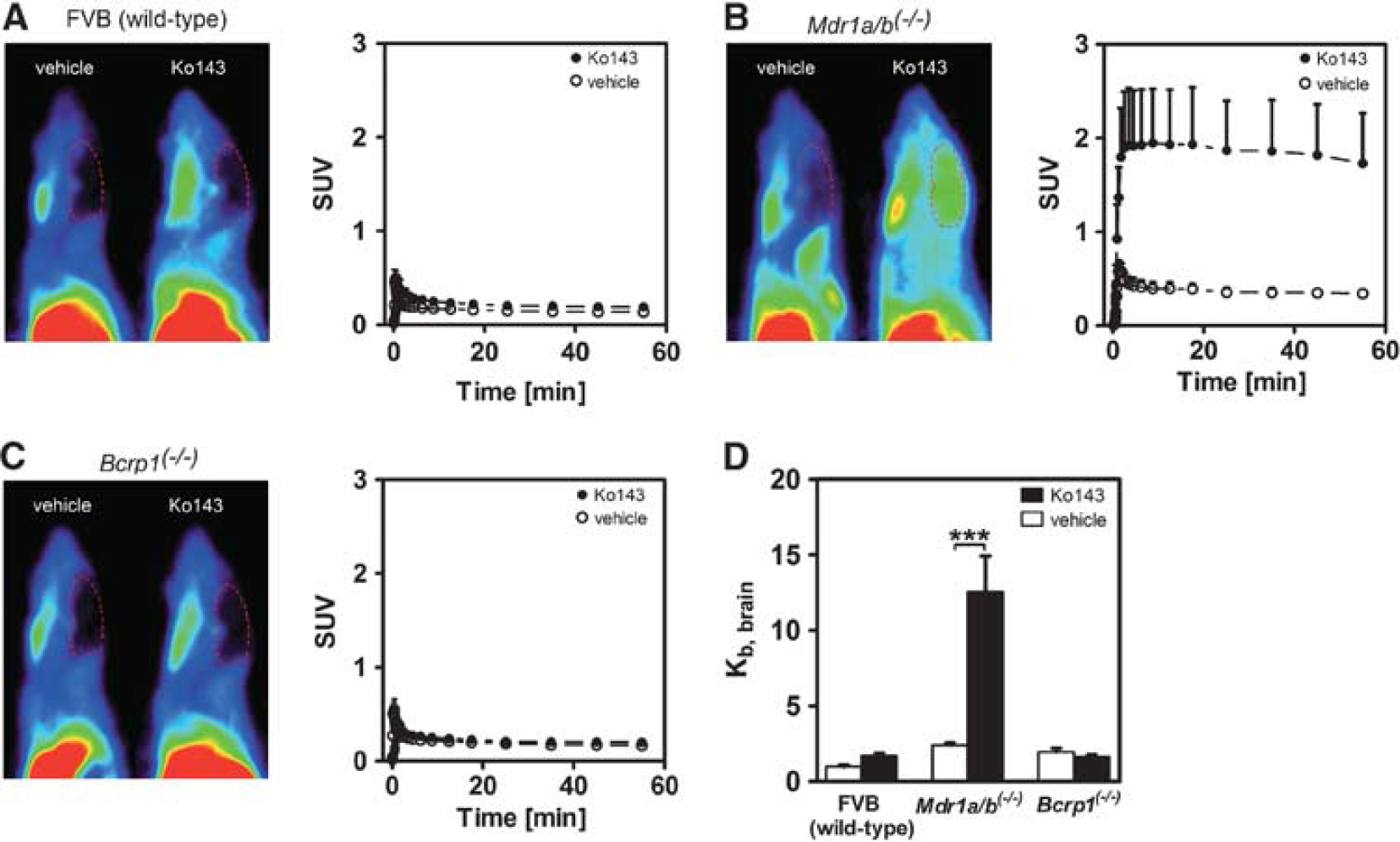
Sagittal positron emission tomography (PET) summation images (0 to 60 minutes) and mean (+ standard deviation, s.d.) time-activity curves of [11C]tariquidar in wild-type (
Brain Uptake of [11C)Tariquidar Is Dependent on Administered Ko143 Dose in Mdr1a/b(−/−) Mice
Pretreatment of Mdria/b(−/−) mice with escalating doses of Ko143 (1, 3, 5, 10, or 15 mg/kg, n = 3 to 4 per dose group) led to a dose-dependent increase in brain radioactivity uptake of [11C)tariquidar, whereas no changes in blood radioactivity concentrations were observed (Figure 2). After the highest Ko143 dose (15 mg/kg) mean Kb,brain was 6.3-fold increased relative to the vehicle group [P < 0.001; Figure 2C). Mean Kb,brain after 15 mg/kg of Ko143 was 15.1 ± 2.5, which was similar to Kb,brain of [11C]tariquidar in Mdr1a/b(−/−)Bcrp1(−/−) mice (14.4 ± 1.7) (Bauer et al, 2010a), suggesting complete inhibition of Bcrp at the BBB by Ko143 (Figure 2C). A sigmoidal Hill function was fitted to log dose–response data of Ko143 in Mdr1a/b(−/−) mice, providing an estimated half-maximum effect dose (ED50) of 4.98 mg/kg (95% confidence interval, 3.86 to 6.42 mg/kg) and a Hill slope (n) of 2.29 (95% confidence interval, 1.20 to 3.39; Figure 3).
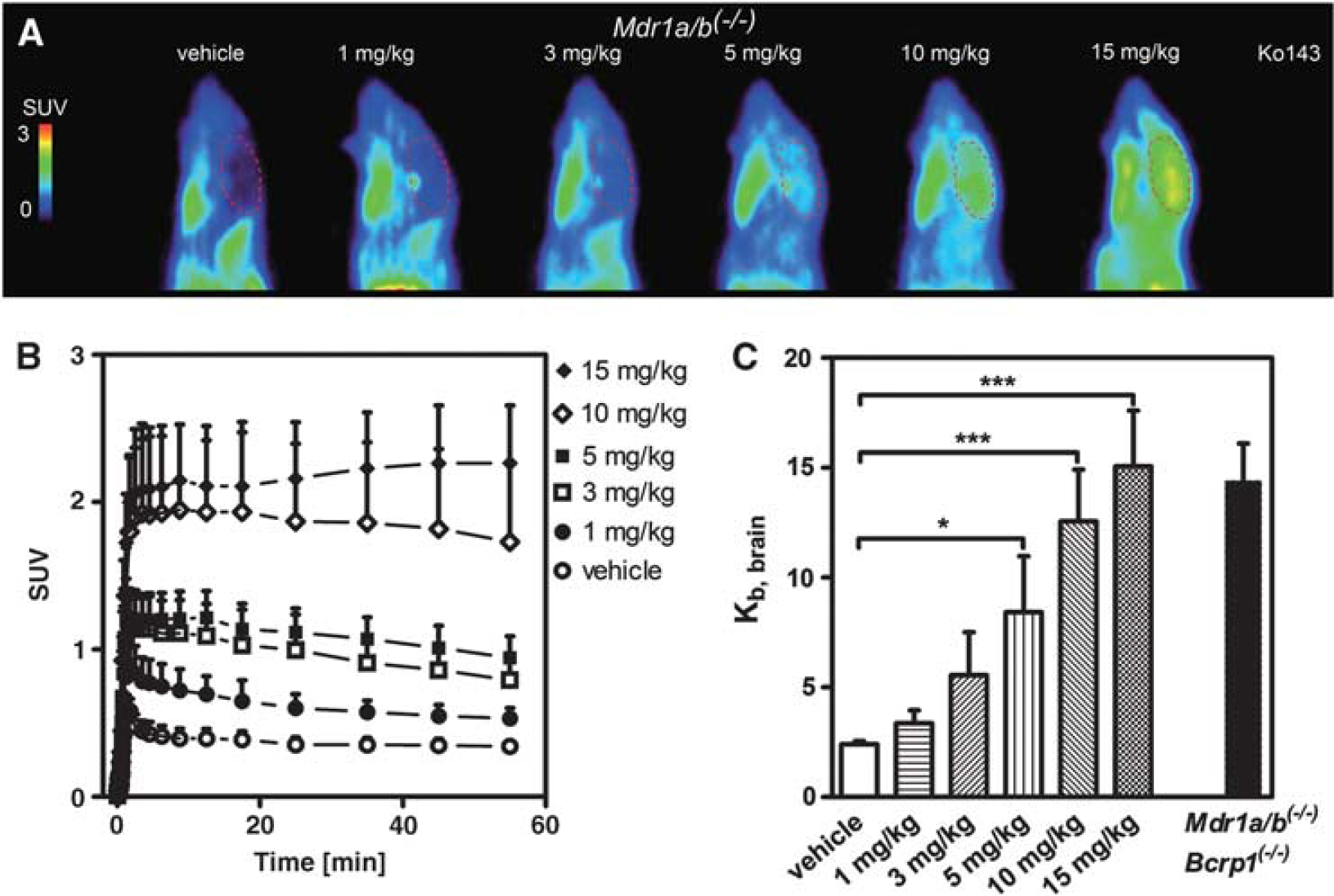
Sagittal positron emission tomography (PET) summation images (0 to 60 minutes) (
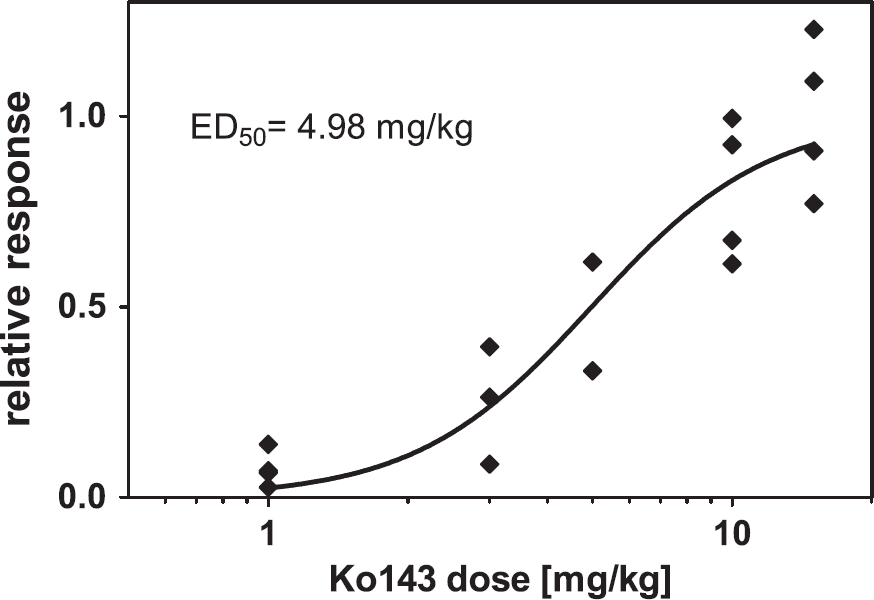
Relative response in terms of increase in brain-to-blood radioactivity concentration ratio at the end of the positron emission tomography (PET) scan (Kb,brain) in Mdr1a/b(−/−) mice plotted against Ko143 dose administered intravenously at 1 hour before start of PET scan. A sigmoidal Hill function was fitted to the data and gave an estimated half-maximum effect dose (ED50) value of 4.98 mg/kg (95% confidence interval, 3.86 to 6.42 mg/kg) and a Hill slope (n) of 2.29 (95% confidence interval, 1.20 to 3.39). For definition of relative response, see ‘Materials and methods’.
[11C]Tariquidar Brain Uptake is Dependent on Administered Ko143 Dose in Wild-Type Mice Pretreated with Cold Tariquidar
To identify a tariquidar dose that fully inhibits P-gp and minimally inhibits Bcrp at the BBB, groups of wild-type mice (n = 4 per dose group) were first pretreated with 15 or 8 mg/kg tariquidar followed by 15 mg/kg Ko143 or vehicle solution before performing [11C]tariquidar PET scans (Figure 4). In animals which had received 15 mg/kg tariquidar, baseline Kb,brain (i.e., for scans without Ko143 administration) was higher than in Mdr1a/b(−/−) mice (4.0 ± 0.9 versus 2.4 ± 0.1) whereas it was comparable to Mdr1a/b(−/−) mice in animals pretreated with 8 mg/kg tariquidar (Figure 4). This indicated that the 15 mg/kg tariquidar had partially inhibited Bcrp whereas the 8 mg/kg dose had not. After Ko143 administration, Kb,brain values were significantly, i.e., 1.8-fold (P < 0.001) and 3.6-fold (P < 0.001) increased relative to baseline scans for the 15 and 8 mg/kg tariquidar pretreatment group, respectively. Kb,brain values after 15 mg/kg Ko143 were 7.2 ± 0.5 for the 15 mg/kg and 8.7 ± 1.0 for the 8 mg/kg tariquidar dose group, respectively. For comparison, the ratio of Kb,brain Ko143-treated (15 mg/kg) Mdr1a/b(−/−) mice to that in vehicle-treated Mdr1a/b(−/−) mice was 6.3 (Figure 4). A further group of wild-type mice (n = 4 per dose group) underwent [11C]tariquidar PET scans after pretreatment with tariquidar (8 mg/kg) and two additional doses of Ko143 (3 or 6 mg/kg) (Figure 5). Comparable to Mdr1a/b(−/−) mice (see previous section), a Ko143 dose-dependent increase in brain radioactivity uptake was observed (Figure 5). Kb,brain was increased 1.7-fold in the 3 mg/kg Ko143 dose group (P < 0.05) and 2.3-fold in the 6 mg/kg Ko143 dose group (P < 0.001) relative to Ko143 vehicle-treated animals (Figure 5).
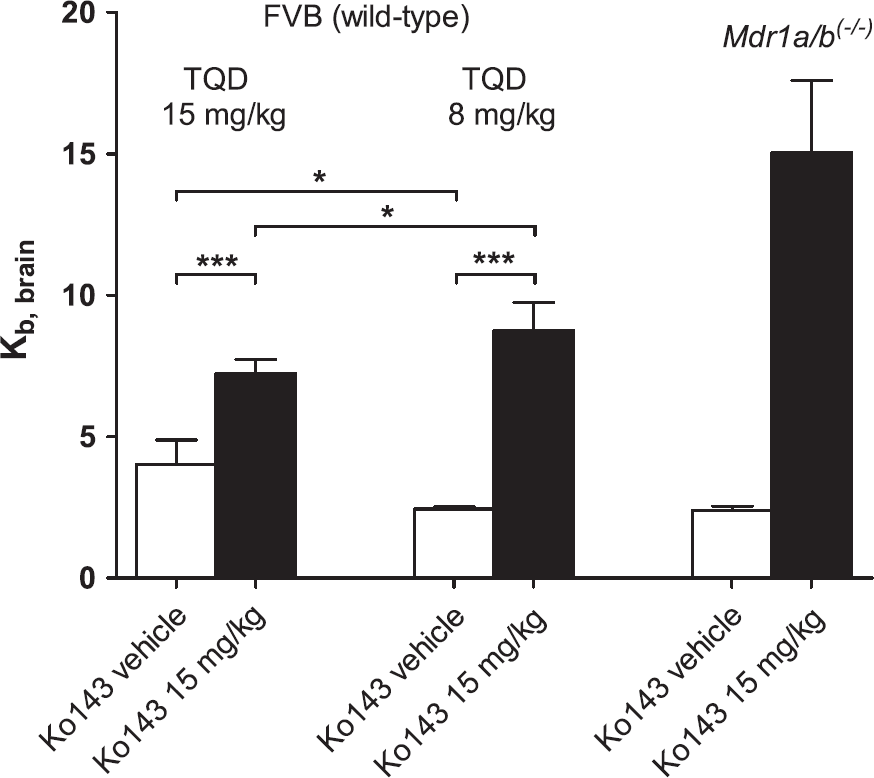
Mean (+ standard deviation, s.d.) brain-to-blood radioactivity concentration ratios of [11C]tariquidar at the end of the positron emission tomography (PET) scan (Kb,brain) for wild-type mice pretreated intravenously with 15 or 8 mg/kg tariquidar (TQD) followed by pretreatment with either Ko143 vehicle solution or 15 mg/kg Ko143 (n = 4 per dose group except for the 15 mg/kg tariquidar/15 mg/kg Ko143 dose group where n = 7; *P < 0.05, ***P < 0.001, one-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparison test). Tariquidar was administered at 2 hours and Ko143 at 1 hour before start of the PET scan. For comparison, Kb,brain of [11C]tariquidar in vehicle-treated and Ko143-treated (15 mg/kg) Mdr1a/b(−/−) mice is also shown.
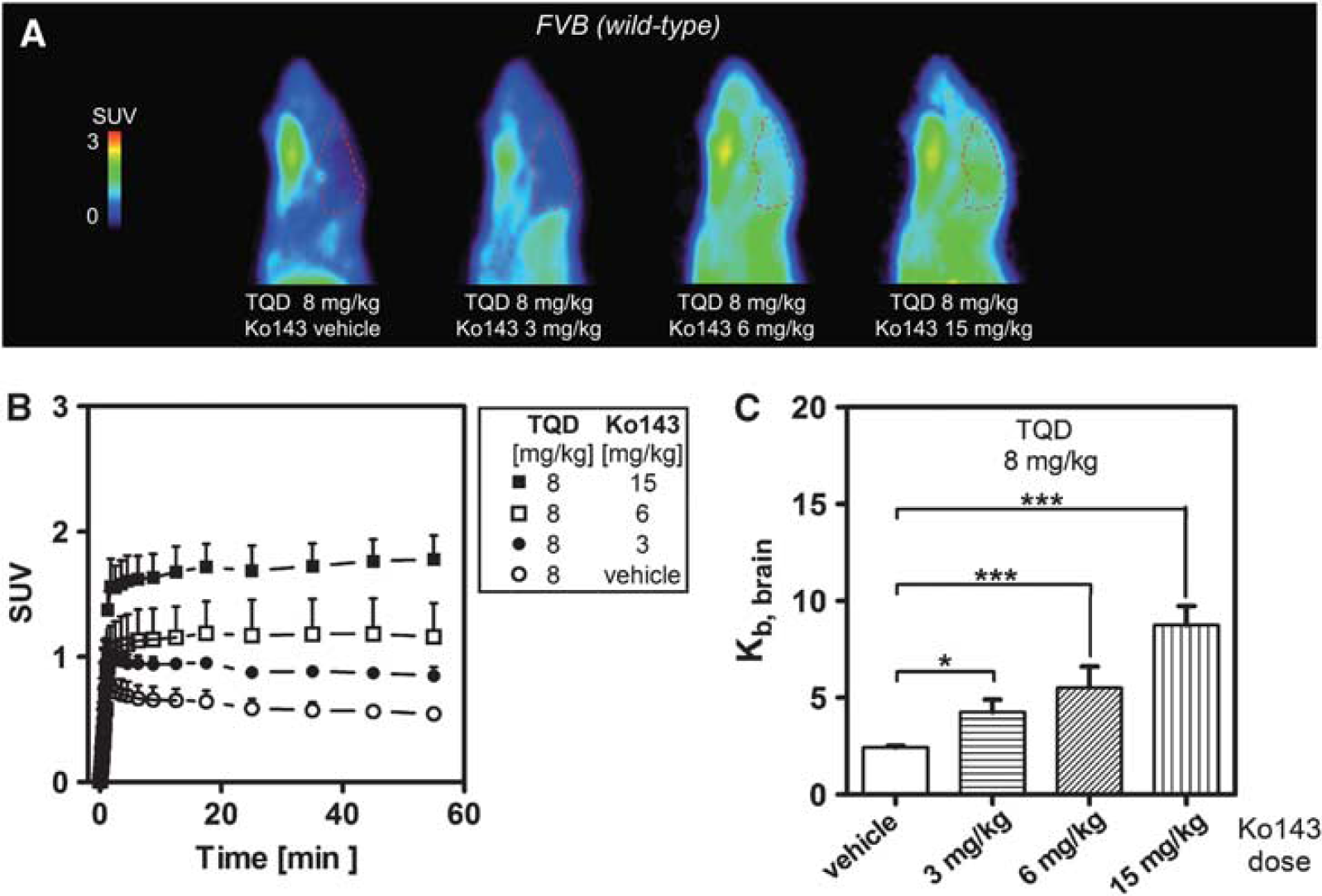
Sagittal positron emission tomography (PET) summation images (0 to 60 minutes) (
Discussion
We demonstrate in this study that [11C]tariquidar can be used to assess the functional activity of Bcrp at the murine BBB in vivo by modulating Bcrp activity with Ko143 as an inhibitor. Because [11C]tariquidar, at tracer concentrations, is a dual P-gp/Bcrp substrate [Bauer et al, 2010a; Kannan et al, 201lb; Kawamura et al, 2010; Römermann et al, 2011), we eliminated the influence of P-gp efflux on [11C]tariquidar brain distribution either by using animals in which P-gp was genetically knocked out (Mdr1a/b(−/−) mice), or by pretreating wild-type mice with cold tariquidar at a dose which completely inhibits cerebral P-gp but does not inhibit Bcrp. Besides showing that [11C]tariquidar PET at P-gp saturating tariquidar concentrations is sensitive to visualize a range of different cerebral Bcrp activities at the murine BBB, we also demonstrate that Ko143 can be used in vivo as a potent inhibitor of Bcrp which is selective for Bcrp over P-gp.
[11C]Tariquidar is based on the potent third-generation P-gp inhibitor tariquidar (Fox and Bates, 2007), which also inhibits Bcrp, but at several times higher concentrations than P-gp (Pick et al, 2008). The reported half-maximum effect concentrations (IC50) of tariquidar for inhibition of Hoechst 33342 transport in P-gp and BCRP overexpressing cells are 72 and 1,445 nmol/L, respectively (Pick et al, 2008). We initially developed [11C]tariquidar as a PET tracer to visualize cerebral P-gp expression levels (Bauer et al, 2010a), as opposed to substrates such as (R)-[11C]verapamil or [11C]-N-desmethyl-loperamide, which visualize P-gp function. However, contrary to our initial assumptions, we found that a tracer dose of [11C]tariquidar, as used in PET imaging, behaved as a dual P-gp and Bcrp substrate at the rodent BBB in vivo (Bauer et al, 2010a; Kawamura et al, 2010). Our findings were substantiated by in vitro experiments with [3H]tariquidar in cell lines transfected with human ABCG2, which showed that tariquidar is efficiently transported by BCRP (Kannan et al, 2011b). Also, our own data obtained from in vitro transport assays using P-gp- and Bcrp-overexpressing cells demonstrated that tariquidar was concentration dependently transported by P-gp and Bcrp and that saturation of P-gp transport was reached at a 5-time lower concentration than saturation of Bcrp transport (1 μmol/L for P-gp versus 5 μmol/L for Bcrp) (Römermann et al, 2011).
In the present study, we further confirmed that [11C]tariquidar is transported by Bcrp at the murine BBB by performing [11C]tariquidar PET scans in groups of wild-type, Mdr1a/b(−/−), and Bcrp1(−/−) mice, which were pretreated with Ko143 (10 mg/kg) (Figure 1). The fumitremorgin C derivative Ko143 has been developed as a potent Bcrp inhibitor that lacks affinity to P-gp (Allen et al, 2002; Matsson et al, 2009). Although Ko143 is commonly used for in vitro pharmacology, its in vivo use has rarely been reported (Giri et al, 2008; Xiao et al, 2012). Our observation that [11C]tariquidar brain uptake was not increased in wild-type (Figure 1A) and Bcrp1(−/−) mice (Figure 1C) after Ko143 treatment, but markedly increased in Mdr1a/b(−/−) mice (Figure 1B), confirms that Ko143 is selective for Bcrp over P-gp in vivo. If Ko143 had inhibited P-gp, then we would also have observed increases in brain uptake of the dual P-gp/Bcrp substrate [11C]tariquidar in wild-type and Bcrp1(−/−) mice, in which P-gp was functional.
As a next step, we assessed the dose–response relationship of Ko143 to increase cerebral uptake of [11C]tariquidar in Mdr1a/b(−/−) mice (Figure 2). Recent quantitative proteomics data suggest that expression levels of Bcrp in brain capillary endothelial cells of Mdr1a/b(−/−) mice are comparable to Bcrp expression levels in wild-type mice (Agarwal et al, 2012). Our experiments showed that Ko143 potently and dose dependently inhibited cerebral Bcrp in Mdr1a/b(−/−) mice with an ED50 of 4.98 mg/kg leading to increased brain exposure of [11C]tariquidar (Figures 2C and 3). We confirmed that the highest studied Ko143 dose (15 mg/kg) resulted in full inhibition of Bcrp by demonstrating that [11C]tariquidar brain uptake in Mdr1a/b(−/−) mice treated with 15 mg/kg Ko143 was similar to [11C]tariquidar brain uptake in Mdr1a/b(−/−)Bcrp1(−/−) mice (Bauer et al, 2010a; Figure 2C). Our findings are important as the dose–response relationship of Ko143 for inhibition of cerebral Bcrp was not known previously. Our data might therefore facilitate future use of Ko143 as an inhibitor of cerebral Bcrp for in vivo rodent studies.
It is remarkable that [11C]tariquidar brain uptake in Mdr1a/b(−/−) mice covered a 6.3-fold concentration range between baseline scans and scans where Bcrp was fully inhibited with Ko143 (Figure 2C). This amplitude is in fact similar to what has been observed for the P-gp substrate radiotracer (R)-[11C]verapamil (which is not a substrate of Bcrp) between conditions of fully functional and completely inhibited cerebral P-gp activity (Kuntner et al, 2010). As recently pointed out by Kannan and co-workers in their review article (Kannan et al, 2009), the suitability of a PET tracer to visualize a cerebral efflux transporter is determined by the magnitude of the difference in brain concentrations between baseline and blocked scans. If this difference is very small, then the radiotracer will in all likelihood not be sensitive enough to detect subtle changes in cerebral transporter activity, which are expected to occur in disease. Moreover, for a PET tracer to be useful for visualization of transporter function it is mandatory that brain uptake after transporter blockade be measurable. In other words, even if a radiotracer shows a great difference in brain uptake between baseline and blocked conditions it still might not be suitable for PET imaging when the magnitude of brain PET signal in the blocked condition is very small. For instance, the muscle relaxant dantrolene was shown to be transported by Bcrp and not by P-gp (Enokizono et al, 2008; Kodaira et al, 2010; Xiao et al, 2012) and was therefore proposed as a candidate for development of a Bcrp selective PET tracer (Takada et al, 2010). However, Kb,brain values of dantrolene were shown to increase from 0.05 in wild-type mice to only ~0.16 in Bcrp1(−/−) mice (Xiao et al, 2012). A Kb,brain of 0.16 may be too low to be useful for brain PET imaging. In fact, the Kb,brain of [11C]tariquidar in wild-type mice (1.0 ± 0.1) under baseline conditions (when Bcrp was fully functional) was already ~6 times greater than that of dantrolene under conditions when Bcrp was fully blocked (Bcrp1(−/−) mice) (Xiao et al, 2012). However, the better Bcrp over P-gp selectivity of dantrolene might pose an advantage of this probe over [11C]tariquidar.
Another important criterion for a PET tracer to be useful for visualization of transporter activity at the BBB is the absence of radiolabeled metabolites which are taken up into brain parenchyma and might thereby confound the PET measurements (Kannan et al, 2009). This has, for instance, been shown to be the case for (R)-[11C]verapamil (Lubberink et al, 2007). Our own data and data from Kawamura and colleagues suggest that [11C]tariquidar displays remarkable metabolic stability without any radiolabeled metabolites detectable in plasma and brain of rats or mice during the time course of a PET scan (Bauer et al, 2010a; Kawamura et al, 2010). The proposed [11C]tariquidar PET protocol therefore already meets two important criteria for a suitable transporter substrate tracer (high magnitude of signal and radiochemical purity of signal) (Kannan et al, 2009).
The third criterion put forward by Kannan is selectivity for the efflux transporter under investigation (Kannan et al, 2009). It is clear from our data that this criterion is not met because [11C]tariquidar is a dual P-gp/Bcrp substrate. However, we were able to demonstrate in this work that this limitation can be overcome by studying Bcrp function with [11C]tariquidar in mice in which P-gp has been genetically knocked out (Mdr1a/b(−/−) mice; Figure 2). How can this be translated to human subjects? An alternative to the use of Mdr1a/b(−/−) mice could be to induce chemical inhibition of P-gp by an appropriate inhibitor which can be safely used in humans, such as tariquidar. Because tariquidar also inhibits Bcrp (Pick et al, 2008), we sought, as a first step, to identify a tariquidar dose that fully inhibits P-gp and minimally inhibits Bcrp (Figure 4). For this purpose, groups of wild-type mice were pretreated with two different tariquidar doses (15 or 8 mg/kg) followed by [11C]tariquidar PET scans without or with preadministration of Ko143 at a fully Bcrp inhibiting dose (15 mg/kg). In scans without Ko143 administration, Kb,brain of [11C]tariquidar was higher in animals pretreated with 15 mg/kg tariquidar than in Mdr1a/b(−/−) mice (Figure 4), which indicated that the 15 mg/kg tariquidar dose had partially inhibited Bcrp. In contrast, Kb,brain was comparable in mice pretreated with 8 mg/kg tariquidar and in Mdr1a/b(−/−) mice (Figure 4), suggesting that the 8 mg/kg tariquidar dose had fully inhibited P-gp without affecting Bcrp. After administration of 15 mg/kg Ko143, Kb,brain values of [11C]tariquidar were higher in animals pretreated with 8 mg/kg than with 15 mg/kg tariquidar. Consequently, the ratio of Kb,brain after Ko143 administration to Kb,brain in baseline scans was twice as high for the 8 mg/kg than for the 15 mg/kg tariquidar dose group (3.6-fold versus 1.8-fold), suggesting that the lower tariquidar pretreatment dose was better suited to study Bcrp function at the murine BBB. We did not use tariquidar doses lower than 8 mg/kg because we had shown in a previous study in rats using (R)-[11C]verapamil as radiotracer that doses <8 mg/kg resulted in incomplete inhibition of P-gp at the BBB (Kuntner et al, 2010).
Why was brain uptake of [11C]tariquidar after administration of 15 mg/kg Ko143 lower in tariquidar pretreated wild-type mice than in Mdr1a/b(−/−) mice treated with 15 mg/kg Ko143 or in Mdr1a/b(−/−) Bcrp1(−/−) mice (Figure 4)? It seems unlikely that this was due to incomplete inhibition of Bcrp in wild-type mice because we had shown in Mdr1a/b(−/−) mice that the 15 mg/kg Ko143 dose fully inhibited Bcrp (Figure 2C). A possible explanation could be that tariquidar, which is a weak base, becomes ionically trapped in acidic lysosomes in the brain (Kannan et al, 2011a). It might be possible that cold tariquidar, which was administered before the PET scan, competed with the lysosomal accumulation of [11C]tariquidar, which could have resulted in an overall lower brain uptake of [11C]tariquidar after Bcrp inhibition in tariquidar pretreated wild-type mice than in Mdr1a/b(−/−) mice (which had not been pretreated with cold tariquidar) (Figure 4). In line with this assumption, Kb,brain of [11C]tariquidar after Ko143 administration was higher in animals pretreated with 8 mg/kg than in those treated with 15 mg/kg tariquidar (Figure 4), pointing to a lesser degree of competition for lysosomal trapping for the lower tariquidar dose. We had additionally observed in a previous study that Kb,brain of [11C]tariquidar was reduced by ~36% in Mdr1a/b(−/−)Bcrp1(−/−) mice after administration of 15 mg/kg tariquidar compared with untreated animals (Römermann et al, 2011), which further supports our hypothesis. It cannot be excluded, however, that other factors than competition for lysosomal accumulation, such as a pharmacokinetic interaction between tariquidar and Ko143 or the partial inhibition of an unknown influx transporter of [11C]tariquidar by cold tariquidar, accounted for the lower maximum brain uptake of [11C]tariquidar after Bcrp inhibition in tariquidar pretreated wild-type mice as compared with Mdr1a/b(−/−) mice.
After having identified the 8 mg/kg tariquidar pretreatment dose as optimal for studying Bcrp function, we showed a Ko143 dose-dependent increase in [11C]tariquidar brain uptake in two further groups of wild-type mice pretreated with 8 mg/kg tariquidar (Figure 5). This demonstrated that the used PET protocol is sensitive to visualize a range of different Bcrp activities at the BBB, not only in Mdr1a/b(−/−) mice but also in wild-type mice in which P-gp is chemically knocked out.
We expect that our method can be translated to the clinic as we have previously shown, by performing (R)-[11C]verapamil PET scans in healthy volunteers after administration of escalating tariquidar doses (up to 8 mg/kg), that tariquidar plasma levels which result in quantitative inhibition of cerebral P-gp (> 1,000 ng/mL) can be safely achieved in humans (Bauer et al, 2012). In contrast, only a modest inhibition of P-gp at the human BBB is achieved with the older generation of P-gp inhibitors such as cyclosporine A, even at a maximal allowable cyclosporine A blood concentration (Sasongko et al, 2005). We have not measured tariquidar concentrations in mouse plasma in the present study. However, in a previous study in which tariquidar was administered to Sprague-Dawley rats at a dose of 7.5 mg/kg, which was comparable to the tariquidar dose used in mice in the present study (8 mg/kg), tariquidar plasma concentrations were in the order of 3,000 ng/mL (Kuntner et al, 2010). Based on these data, it appears unlikely that the tariquidar concentrations needed to completely inhibit P-gp at the human BBB (~1,000 ng/mL) will cause significant inhibition of BCRP. Nevertheless, it cannot be excluded that species differences between mice and humans may affect the translatability of our PET protocol to humans.
Even though no potent BCRP selective inhibitor is currently available for human use, performance of [11C]tariquidar PET scans after administration of P-gp saturating tariquidar doses might be useful to selectively assess BCRP function at the human BBB. Such a PET protocol could be used, for instance, in studying the functional consequences of single-nucleotide polymorphisms in the ABCG2 gene (Kobayashi et al, 2005) or to study BCRP function in patients with Alzheimer's disease, in whom BCRP at the BBB may be upregulated as a protective mechanism to prevent entry of Aβ peptides from plasma into brain (Xiong et al, 2009).
In summary, we developed a PET protocol based on the dual P-gp/Bcrp substrate radiotracer [11C]tariquidar in combination with the potent Bcrp inhibitor Ko143 which allows for visualization of functional activity of Bcrp at the BBB of mice in which P-gp is either genetically knocked out (Mdr1a/b(−/−) mice) or chemically knocked out (by pretreatment with cold tariquidar). This study protocol is expected to be translatable to the clinic, because tariquidar can be safely administered to humans at doses that completely inhibit cerebral P-gp.
Disclosure/conflict of interest
The authors declare no conflict of interest.