
Abstract
Neurogenesis occurs in discrete regions of normal brains of adult mammals including humans, and is induced in response to brain injury and neurodegenerative disease. Whether intracerebral hemorrhage can also induce neurogenesis in human brain is unknown. Specimens were obtained from patients with primary intracerebral hemorrhage undergoing surgical evacuation of an intracerebral hematoma, and evaluated by two-photon laser scanning confocal microscopy. We found that neural stem/progenitor cell-specific protein markers were expressed in cells located in the perihematomal regions of the basal ganglia and parietal lobe of the adult human brain after primary intracerebral hemorrhage (
Introduction
Primary intracerebral hemorrhage (ICH) is a devastating neurologic disease with the highest mortality among all stroke subtypes, although it comprises only 10% to 15% of all cases of strokes (Broderick et al, 1999; Heiskanen, 1993; Qureshi et al, 2001). Management of patients with ICH is generally limited to supportive care or evacuation of the hematoma, although the efficacy of surgical removal is variable and controversial (Broderick et al, 1999; Heiskanen, 1993; Qureshi et al, 2001).
One way in which the brain appears to respond to diverse forms of injury is through an increase in the production of endogenous neural cells from resident neural stem/progenitor cells (NSCs), which are present in discrete brain regions—subventricular zone (SVZ) and subgranular zone of the hippocampal dentate gyrus (SGZ)—of adult mammals, including mice (Alvarez-Buylla and Lois, 1995), rats (Jin et al, 2003), non-human primates (McDermott and Lantos, 1991), and humans (Eriksson et al, 1998). These cells are induced to proliferate in response to ischemic stroke in animals and humans (Jin et al, 2003; Nakatomi et al, 2002; Raber et al, 2004); in the former, their progeny can also migrate into areas of brain damage (Jin et al, 2003), where they express phenotypic markers of mature neurons (neuronal nuclear antigen (NeuN), microtubule-associated protein-2 (MAP-2)) as well as regionally specific neuronal markers (calbindin, dopamine, and cAMP-regulated phosphoprotein-32; Liu et al, 1998; Nakatomi et al, 2002; Teramoto et al, 2003), and form synapses (Yamashita et al, 2006). Despite an age-related reduction in basal levels of neuroproliferation (Kuhn et al, 1996), NSCs in aged rats (Jin et al, 2004b) and humans (Jin et al, 2006) retain the capacity for proliferation, lesion-directed migration, or both in response to cerebral ischemia. Evidence for functional neuronal replacement through endogenous neurogenesis has been reported in a model of global cerebral ischemia that affects the hippocampus predominantly. Infusion of fibroblast growth factor 2 and epidermal growth factor led to regeneration of hippocampal CA1 neurons derived from the SGZ, which integrated into existing brain circuitry and are thought to have ameliorated neurologic deficits (Nakatomi et al, 2002). In another study, whole-brain ionizing radiation was used to ablate dividing NSCs in the SGZ of guinea-pigs before global ischemia (Raber et al, 2004). One month after ischemia, the number of newborn cells of neuronal lineage in the dentate granule cell layer was reduced by ~80% in irradiated animals. These animals showed impaired performance in a water-maze task, suggesting that ischemia-induced neurogenesis enhanced outcome in nonirradiated subjects.
Whether NSCs in the adult human brain respond to ICH in a similar manner is unknown, although increased neurogenesis has been reported after ICH in rats (Masuda et al, 2007) and subarachnoid hemorrhage in humans (Sgubin et al, 2007). After subarachnoid hemorrhage in mice, a more complex effect on neurogenesis has been reported: an acute (1 to 3 days) reduction in (5-bromodeoxyuridine-labeled) cell proliferation in both SGZ and SVZ, followed by an increase in the number of 5-bromodeoxyuridine- and NeuN-positive cells migrating into the granule cell layer (Mino et al, 2003). In this study, we investigated whether neurogenesis is induced in the human brain after ICH, using tissue obtained at surgery within 72 h of the event and evaluated by two-photon laser scanning confocal microscopy. Our data suggest that ICH induces proliferation of NSCs in the human brain.
Materials and methods
Human Brain Specimens
Specimens of perihematomal regions were obtained from patients with primary ICH (
Clinical features of patients with ICH
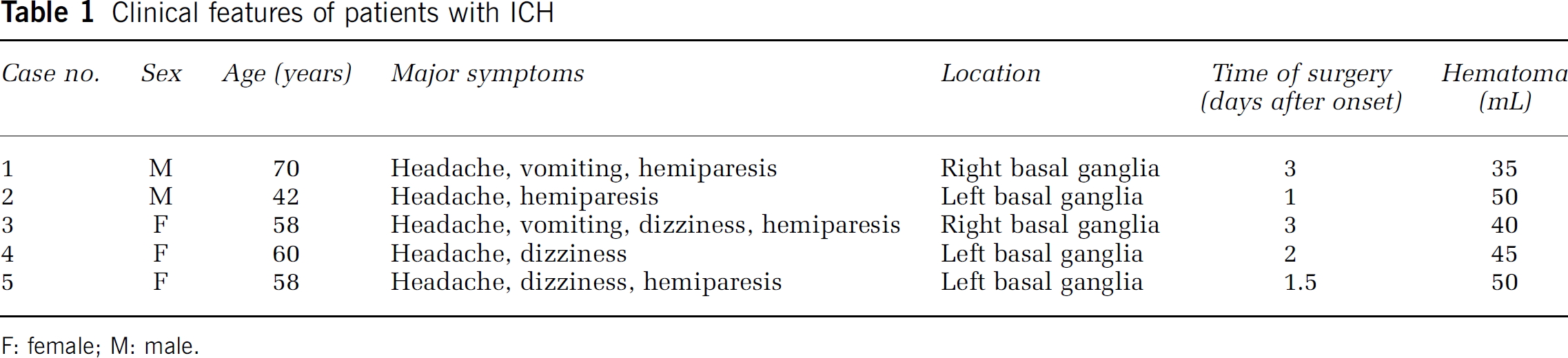
F: female; M: male.
Immunohistochemistry
Brain tissue was postfixed in paraformaldehyde for 24 h, incubated with 30% sucrose for 3 days, and embedded in paraffin; 6-μm sections were cut on a microtome and stored at room temperature. Sections were deparaffinized with xylene and rehydrated with ethanol. To obtain more efficient immunostaining, brain sections were subjected to an antigen retrieval procedure according to the manufacturer's instructions (Vector Laboratories, Burlingame, CA, USA). After heating under pressure for 2 mins, samples were washed extensively with phosphate-buffered saline. Endogenous peroxidase activity was blocked by 30 mins incubation at room temperature in 1% H2O2. After several washes with phosphate-buffered saline, sections were incubated in blocking solution (2% goat serum, 0.1% Triton X-100, 1% bovine serum albumin in phosphate-buffered saline) for 1 h at room temperature. Primary antibodies used (Table 2) were mouse monoclonal anti-Ki67 antigen (1:50; Novocastra, Newcastle upon Tyne, UK), rabbit anti-Ki67 antigen (1:100; Zymed, South San Francisco, CA, USA), goat anti-minichromosome maintenance 2 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-proliferating cell nuclear antigen (1:200; Chemicon, Temecula, CA, USA), rabbit anti-cleaved caspase-3 (1:200; Cell Signaling Technology, Boston, MA, USA), affinity-purified goat anti-doublecortin (DCX; 1:200; Santa Cruz Biotechnology), rabbit anti-glial fibrillary acidic protein (GFAP; 1:1,000; Sigma, St Louis, MO, USA), mouse monoclonal anti-βIII-tubulin (TUJ-1, 1:250; Covance, Berkeley, CA, USA), rabbit polyclonal anti-TUC-4 (turned on after division/Ulip-1/CRMP-4; 1:250; Chemicon), rabbit anti-Musashi1 (1:500; Chemicon), and mouse monoclonal anti-human-specific nestin (1:200; Chemicon). These antibodies crossreact with proteins of the appropriate molecular weight in human brain (Jin et al, 2004c). Primary antibodies were added in blocking buffer and incubated with sections at 4°C overnight. Sections were then washed with phosphate-buffered saline and incubated with biotinylated goat anti-rabbit or anti-goat antibody (1:200) (for polyclonal antibodies) or biotinylated horse anti-mouse antibody (1:200) (for monoclonal antibodies) for 1 h at room temperature. Avidin—biotin complex (Vector Elite; Vector Laboratories) and a diaminobenzidine or nickel solution (Vector Laboratories) were used to obtain a visible reaction product. Controls for immunohistochemistry included preabsorption and coincubation of the antibodies with the corresponding antigens. Sections were dehydrated, sealed, and coverslipped. A Nikon microscope and a Magnifire digital color camera were used for examination and photography of the slides, respectively.
Antibodies used for immunohistochemistry
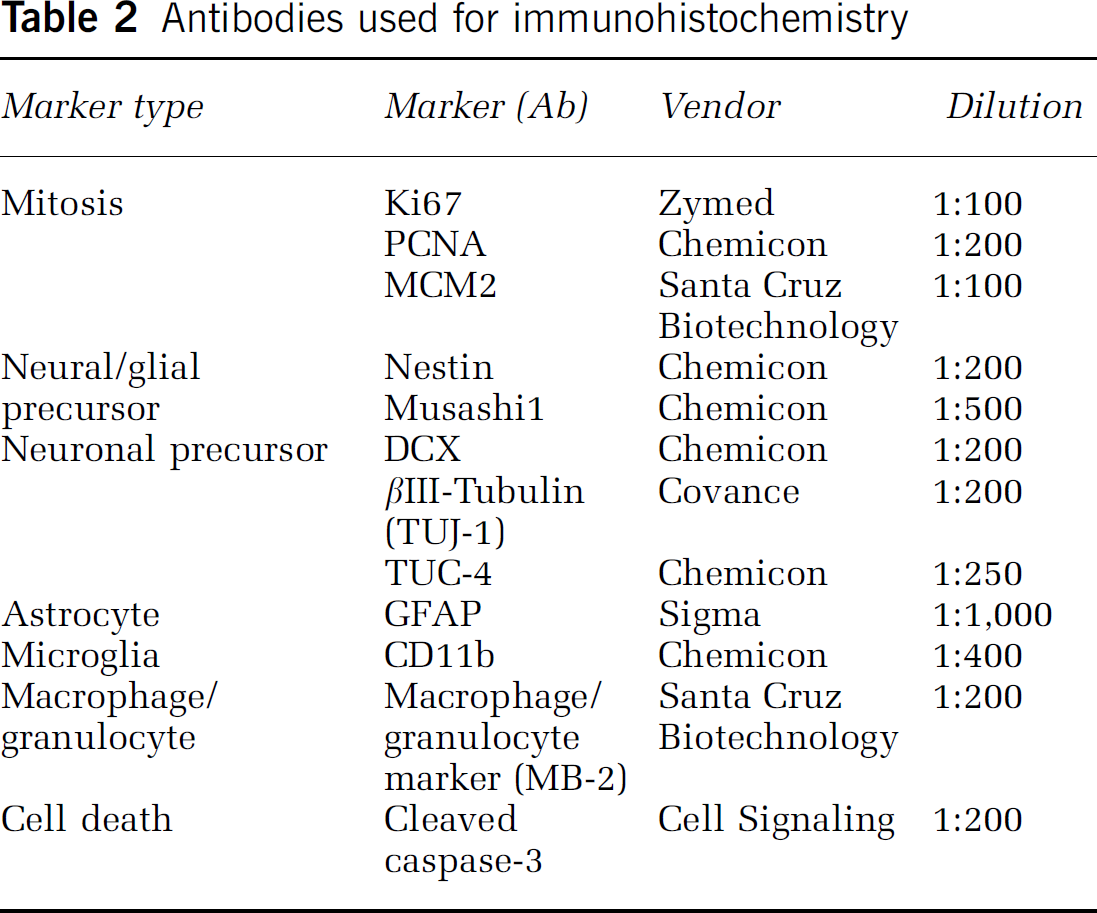
Double Immunostaining
Double immunostaining was performed on brain sections as previously described (Jin et al, 2003). The primary antibodies used, in addition to those listed above, were rabbit anti-CD11b (1:400; Chemicon) and mouse anti-macrophage/granulocyte (1:200; Santa Cruz Biotechnology). The secondary antibodies were Alexa Fluor 488-, 594-, or 647-conjugated donkey anti-mouse, anti-goat, or anti-rabbit IgG (1:200 to 500; Molecular Probes, Carlsbad, CA, USA). Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI) using proLong Gold antifade reagent (Molecular Probes). Fluorescence signals were detected using an LSM 510 NLO Confocal Scanning System mounted on an Axiovert 200 inverted microscope (Carl Zeiss Ltd) equipped with a two-photon Chameleon laser (Coherent Inc.), and images were acquired using LSM 510 Imaging Software (Carl Zeiss Ltd). Two-, three-, or four-color images were scanned using argon, 543 HeNe, 633 HeNe, and Chameleon (750 to 780 nm for DAPI) lasers. Selected images were viewed at high magnification, and three-dimensional images were constructed using Imars software. Controls included omitting either the primary or secondary antibody or preabsorbing the primary antibody.
Results
We reported recently that cells expressing NSC marker proteins are present in the cortical ischemic penumbra of patients with ischemic stroke, but not in equivalent regions of control brains, suggesting that ischemia-induced neurogenesis occurs in humans (Jin et al, 2006). To investigate whether NSCs in the adult human brain also respond to ICH, we performed immunocytochemistry for NSC protein markers in sections of perihematomal regions from five patients with primary ICH who underwent surgical evacuation (Table 1). βIII-Tubulin, an early marker of neuronal lineage
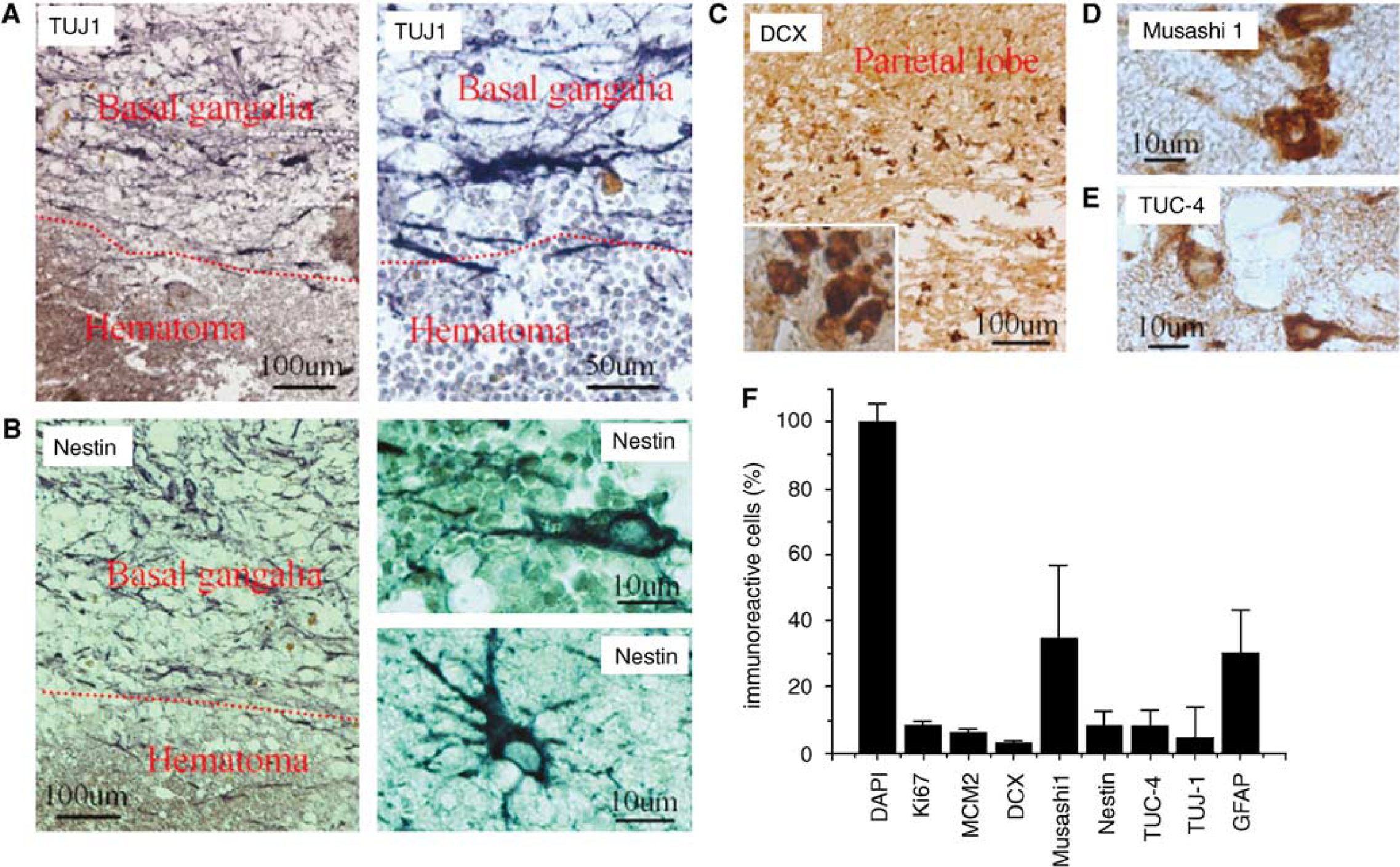
Expression of NSC marker proteins in the perihematomal region after ICH in adult human brain. (
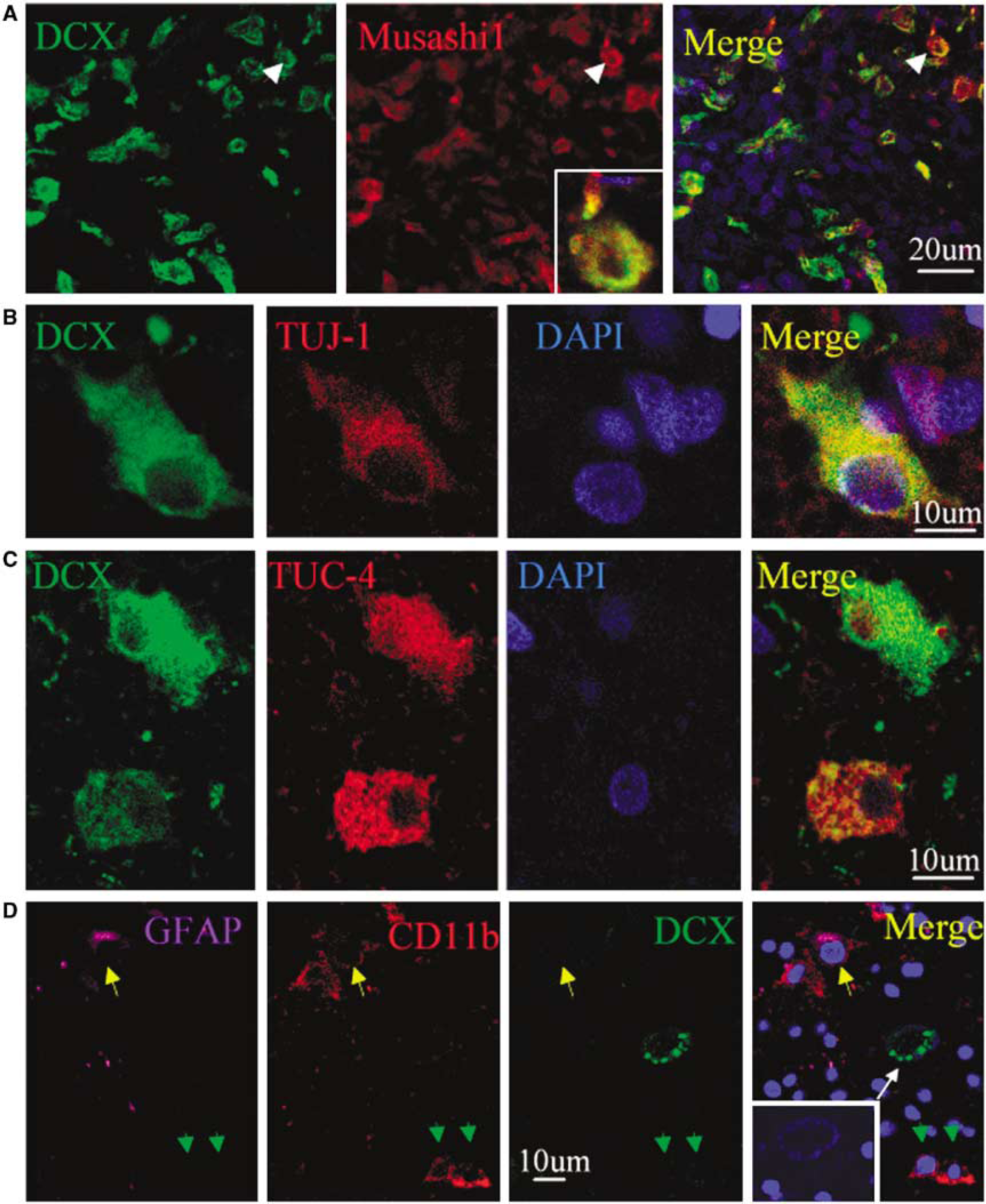
Coexpression of NSC and related markers in the perihematomal region after ICH. (
We next performed immunostaining using an antibody against the cell proliferation marker Ki67 antigen (Gerdes et al, 1984) to determine if cells expressing NSC marker proteins seen after ICH showed evidence of a proliferative state. As shown in Figure 3, Ki67-immunopositive cells were observed in perihematomal regions. Moreover, double-label immunostaining showed that these cells expressed other proliferation markers as well, including minichromosome maintenance 2 and proliferating cell nuclear antigen. Only some Ki67-positive cells showed evidence of caspase-3 cleavage or abnormal nuclear morphology (Figure 4), so it is unlikely that all simply represent dying cells attempting to enter the cell cycle. To determine if Ki67-positive cells were of neuronal lineage, we examined the coexpression of Ki67 antigen and NSC marker proteins using two-photon laser scanning confocal microscopy and Imars software. As shown in Figure 5, cells that expressed NSC protein markers such as TUJ-1, TUC-4, and DCX were also reactive for Ki67, and most Ki67-immunopositive cells did not express the astrocyte marker GFAP, microglia marker CD11b (Figure 5D), and macrophage marker (data not shown), indicating that most Ki67-positive cells in the perihematomal regions were NSCs.
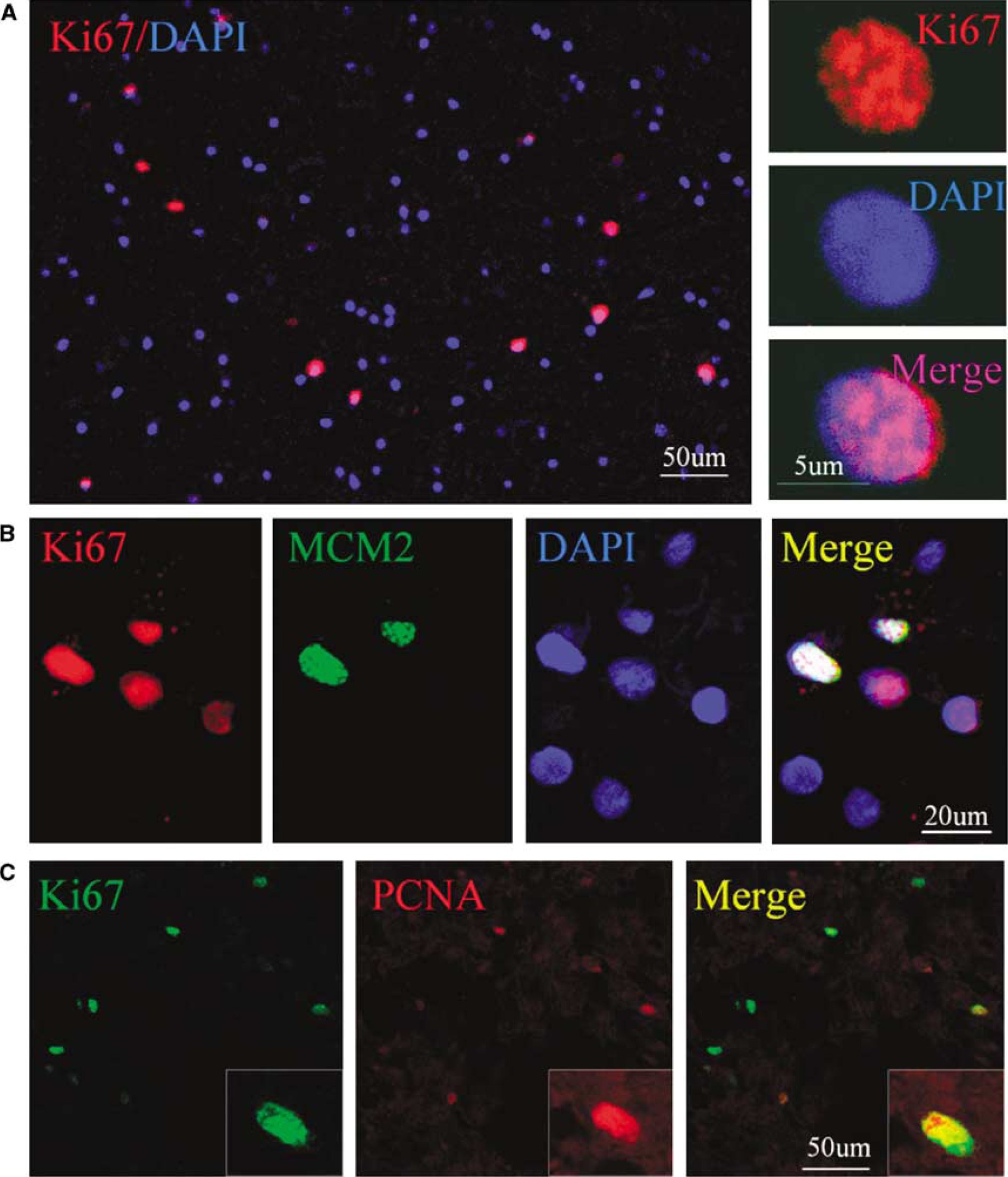
Presence of proliferative cells in perihematomal region after ICH. (
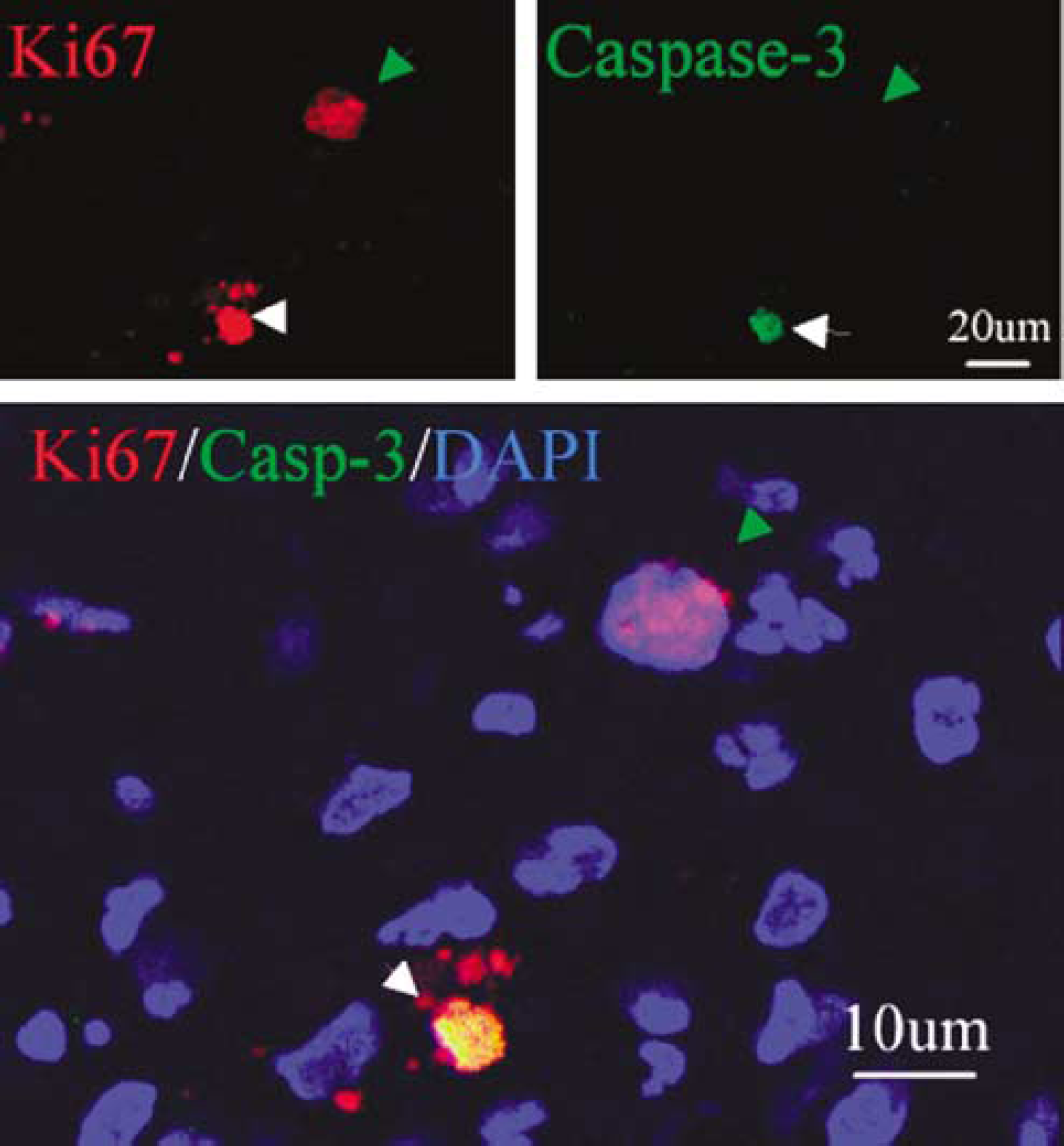
Relationship between Ki67 reactivity and caspase-3 activation. Ki67 (red) is expressed in the perihematomal region after ICH. Ki67 colocalizes with a cell death marker, the 17 to 20 kDa cleavage product of caspase-3 (green), in some (misshapen nuclei, white arrows) but not all (normal nuclei, green arrows) cells. Nuclei are counterstained with DAPI (blue).
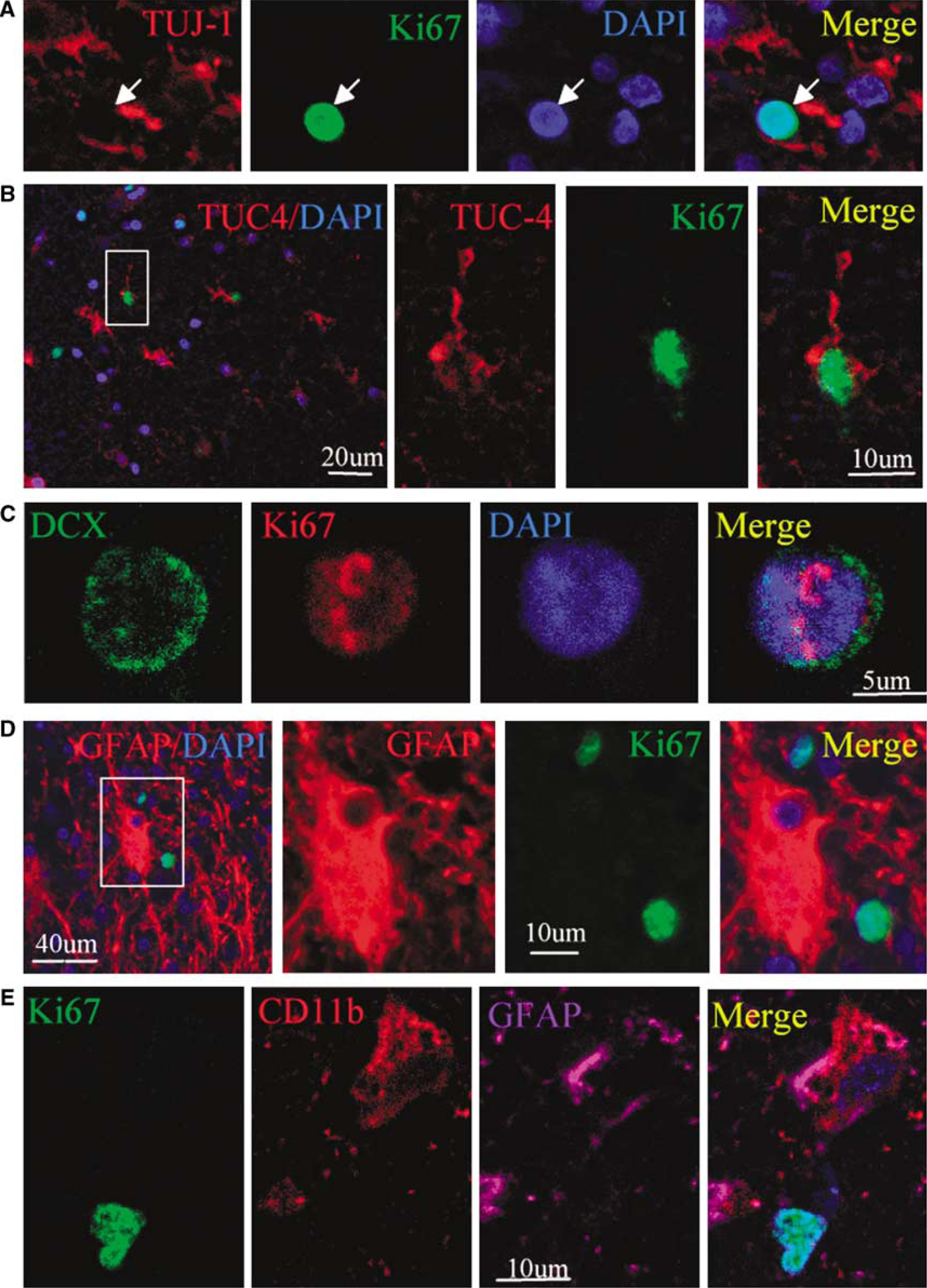
Coexpression of neuronal lineage and cell proliferation markers in perihematomal regions after ICH. (
Discussion
The main finding of this study is that evidence for neurogenesis can be detected in perihematomal brain regions after ICH in humans. We found such evidence in five out of five samples taken from patients who underwent surgical evacuation of a hematoma, consisting of expression of NSC markers and markers of cell proliferation, which could be observed together in the same cell. As reported previously, such coexpression is not seen outside canonical neuroproliferative zones, such as SVZ and SGZ, in the normal human adult brain (Jin et al, 2006). Injury-induced neurogenesis has been reported previously in a number of neurologic disorders in humans, including Huntington's disease, ischemic stroke, Alzheimer's disease, epilepsy, and aneurysmal subarachnoid hemorrhage. Increased SVZ neurogenesis with subsequent migration of newborn neurons into the region surrounding the hemorrhage has also been described in a rat model of ICH induced by injection of collagenase into the internal capsule (Masuda et al, 2007). The time course for the appearance of newborn cells of neuronal lineage in the present series (within 1 to 3 days after hemorrhage) is consistent with the short latency for the same phenomenon in rodent brain after stroke (Jin et al, 2001).
Some of the questions that arise in this context concern the stimulus that incites increased neurogenesis, the site of origin of newborn cells, and the basis for cell migration to the region of pathology. In contrast to ischemic stroke, the number of cells lost after ICH is not large, arguing that cell loss is less likely to provide the signal for increased neurogenesis. This is consistent with findings in transgenic mouse models of Alzheimer's disease (Jin et al, 2004a) and Huntington's disease (Jin et al, 2005), where neurogenesis is enhanced in the absence of substantial cell death. One possibility is that neurogenesis increases after ICH (and perhaps also after cerebral infarction) in response to increased intracranial pressure, which could be detectable at sites, such as SVZ or SGZ, that are remote from the lesion. Although the effect of increased intracranial pressure on adult neurogenesis has not been examined directly, neurogenesis is enhanced in experimental models of traumatic brain injury (Urrea et al, 2007), which also elevates intracranial pressure.
The site of origin of NSCs detected in the perihematomal region after ICH in humans is uncertain. As in a previous study of stroke-induced neurogenesis in human brain that used tissue samples obtained at biopsy, we were unable to investigate possible paths of NSC migration from distant sites, such as SVZ or SGZ, in our surgical specimens. In rats, however, enhanced migration of DCX-immunopositive cells from SVZ to the perihematomal region was observed (Masuda et al, 2007), which is reminiscent of what is seen in rodent models of stroke (Arvidsson et al, 2002; Jin et al, 2003; Nakatomi et al, 2002). Nevertheless, the greater migration distances that would be required in human, compared with rodent, brain raise the possibility that NSCs might arise locally.
If NSCs do migrate to sites of ICH in the human brain, as observed in rodents, what attracts them there? A variety of factors, including growth factors and chemokines, have been proposed as mediators of NSC migration in rodents. It has also been suggested that NSCs use blood vessels to guide them to appropriate destinations (Ohab et al, 2006). In biopsy specimens from the ischemic border zone of human stroke patients, DCX-positive cells appeared to cluster in the vicinity of such vessels, which could be because of such a pattern of migration. Alternatively, if NSCs arise locally in relation to human brain lesions, their association with blood vessels might reflect the existence of a vascular niche akin to that observed in SGZ (Palmer et al, 2000) and SVZ (Gotts and Chesselet, 2005).
Because cell loss after ICH is not extensive, ICH-induced neurogenesis is unlikely to have evolved as a mechanism for cell replacement. As a corollary, the potential therapeutic implications of further stimulating neurogenesis in this setting seem limited. However, ICH-induced neurogenesis (and gliogenesis) could serve purposes other than cell replacement, such as providing cytoprotective or trophic factors to cells surrounding a hematoma. In support of this notion, transplantation of a variety of cell types, including umbilical cord blood cells (Nan et al, 2005), mesenchymal stem cells (Zhang et al, 2006), adipose stem cells (Kim et al, 2007), and immortalized neural cells (Lee et al, 2007), improves outcome in rodent stroke models.
In conclusion, our results extend the known range of disorders associated with increased neurogenesis in human brain. The functional significance of injury-induced neurogenesis remains unclear, but neurogenesis may contribute to brain repair and functional recovery, either through cell replacement or by indirect mechanisms. In either case, increased understanding of the mechanisms that underlie injury-induced neurogenesis may lead to new treatments for neurologic disease.
Footnotes
Acknowledgements
This work was supported by National Institutes of Health Grants AG21980 (to KJ) and NS44921 (to DAG), and Zhejiang province Nature Science foundation Y206089 (to JS) and Y205164 (to RZ).