
Abstract
The BcI-2 homology (BH) domain 3-only proteins are a proapoptotic subgroup of the BcI-2 gene family, which regulate cell death via effects on mitochondria. The BH3-only proteins react to various cell stressors and promote cell death by binding and inactivating antiapoptotic BcI-2 family members and direct activation of proapoptotic multi-BH domain proteins such as Bax. Here, we review the in vivo evidence for their involvement in the pathophysiology of status epilepticus and contrast it to ischemia and traumatic brain injury. Seizures in rodents activate three potent proapoptotic BH3-only proteins: Bid, Bim, and Puma. Analysis of damage after seizures in mice singly deficient for each BH3-only protein supports a causal role for Puma and to a lesser extent Bim but, surprisingly, not Bid. In ischemia and trauma, where core aspects of the pathophysiology of cell death overlap, multiple BH3-only proteins are also activated and Bid has been shown to be required for neuronal death. The findings suggest that while each neurologic insult activates multiple BH3-only proteins, there may be specificity in their functional contribution. Future challenges include evaluating the remaining BH3-only proteins, explaining different causal contributions, and, if possible, exploring neurologic outcomes in mouse models deficient for multiple BH3-only proteins.
Introduction
Neuronal death is a consequence of various acute neurologic insults, including status epilepticus (prolonged, continuous seizures), stroke, and traumatic brain injury (TBI). Neuronal death underlies the functional deficits associated with these injuries and may be causally involved in chronic processes such as epileptogenesis (Pitkanen et al, 2007). The cell death process in each form of injury shares some mechanisms in common, including glutamate-induced excitotoxicity and signaling pathways with the molecular features of apoptosis. Indeed, neurologic injury in each condition can be reduced by glutamate receptor antagonists or interrupting the function of apoptosis-associated proteins such as caspases, apoptosis-inducing factor, and proapoptotic genes such as p53 (Liou et al, 2003; Mehta et al, 2007). There are also pathophysiologically distinct components to each insult, for example increased rather than decreased blood supply in status epilepticus as compared with ischemia or TBI, and a direct mechanical component to injury in TBI (Bramlett and Dietrich, 2004).
The Bcl-2 gene family regulates mitochondrial release of apoptogenic factors such as cytochrome c and as such are a critical, apical network of cell death-regulatory molecules. Multiple proapoptotic and antiapoptotic members of this family have been explored for their possible influence on cell death after neurologic injuries. A subgroup of this family, the so-called Bcl-2 homology (BH) domain 3-only proteins, may function as the sentinels of cell stress and initiate cell death signaling. In the present review, we consider the evidence for involvement of the BH3-only proteins in neuronal death after status epilepticus, stroke, and TBI, drawing in particular from findings in BH3-only protein-deficient mice.
Neuronal Death After Status Epilepticus
Seizures are the result of abnormally synchronous discharges of groups of neurons in the brain and are the cardinal feature of epilepsy. In temporal lobe epilepsy, the most common form in adults, seizures may arise from the hippocampus, which often exhibits neuron loss and gliosis (hippocampal sclerosis) (Chang and Lowenstein, 2003). Epileptic seizures usually last no more a few minutes but when termination mechanisms fail status epilepticus may evolve. Such prolonged seizure discharges, whether or not they are accompanied by convulsive behavior, can cause neuronal death. Evidence for this comes from seminal papers by Ben-Ari et al, (1979); Meldrum et al, (1973); Olney et al, (1974); and Sloviter and Damiano (1981), among others. The principal mechanism underlying cell death after seizures is thought to be glutamate excitotoxicity (Fujikawa, 2005; Meldrum, 1991). Overactivation of glutamate receptors leads to pathologic entry of sodium and calcium through ligand- and voltage-gated channels in neurons. Obligate water entry causes cell swelling, there is harmful free radical production, cytoplasmic vacuolization, lysosomal release of proteases, activation of calpain (a calcium-dependent cysteine protease), nuclear pyknosis, and cell death with morphology consistent with necrosis (Fujikawa, 2006). Glutamate agonists such as kainic acid (KA) trigger seizures and model the pathology seen in humans after status epilepticus (Cendes et al, 1995; Fujikawa et al, 2000), and glutamate receptor antagonists curtail seizures and the associated damage (Meldrum, 1992, 2002). However, cell death signaling pathways associated with execution of apoptosis are also activated in several models of status epilepticus and appear to be regulated in human temporal lobe epilepsy (Henshall and Simon, 2005). Evidence comes from the observation that inhibition of protein synthesis inhibits a component of seizure-induced neuronal death, there is mitochondrial release of cytochrome c and apoptosis-inducing factor, activation of enzymes associated with effecting apoptosis such as caspases, neurons undergo nuclear DNA fragmentation and a portion of seizure-induced neuronal death can be blocked by targeting caspases, p53, and other signaling elements (for review see Engel and Henshall (2009)).
Neuronal Death After Cerebral Ischemia and Traumatic Brain Injury
Neuronal death following ischemia and TBI arise from different primary insults, but there are common aspects to their pathophysiology that are also shared with status epilepticus, including glutamate-induced excitotoxicity, oxidative stress, mitochondrial dysfunction, and inflammation (Bramlett and Dietrich, 2004; Liou et al, 2003). Focal cerebral ischemia arises when there is prolonged occlusion of a major artery, whereas in global ischemia cerebral blood flow reduction is brief but complete. The brain is exquisitely vulnerable to reduced substrate supply due to high metabolic demands and the excitotoxic properties of glutamate (Moskowitz et al, 2010). Within minutes, cellular adenosine triphosphate falls leading to cell depolarization, release of potassium, and increasing intracellular sodium and calcium levels through voltage-gated channels and other mechanisms. There is tissue swelling as water enters along with ionic failure. Depolarization and energy failure result in glutamate release (Moskowitz et al, 2010). Glutamate-mediated excitotoxicity is an important mediator of ischemic cell death in the early stages because damage can be attenuated by glutamate receptor antagonists (Simon et al, 1984). Activation of calcium-dependent proteases ensues. Glutamate-independent sodium and calcium entry also occurs via pH-sensitive channels that are activated by anaerobic metabolism-produced acidosis (Xiong et al, 2004). An hypoxia response is triggered, led in particular by hypoxia-inducible factor 1α, which mediates adaptive responses or promotes cell death (Semenza, 2000). Inflammatory cells enter brain parenchyma that may contribute to delayed cell death via free radical and cytokine production (Chopp and Zhang, 1996). Other pathologic changes include inhibition of protein synthesis and defects in protein degradation (Meller, 2009; Paschen, 2003a). A halo of partially perfused tissue commonly surrounds the dense ischemic core after stroke where incomplete metabolic collapse means an energy- and gene-dependent process contributes to cell death. Here, apoptosis-associated pathways may contribute significantly to expansion of the infarct zone and this watershed zone is partially salvageable by various antiapoptotic interventions (Liou et al, 2003; Mehta et al, 2007).
Traumatic brain injury results from mechanical or acceleration—deceleration injury to the brain and is usually divided into primary and secondary mechanisms. The pathophysiology specific to TBI includes a hemorrhagic component and differences in the degree of metabolic changes, extent of focal versus diffuse injury, nonneuronal cell contributions, and white matter damage (Bramlett and Dietrich, 2004; Loane and Faden, 2010; Royo et al, 2003). Secondary damage is delayed and features prominent activation of cell death pathways due to deinnervation (Liou et al, 2003; Loane and Faden, 2010; Raghupathi et al, 2000). Microscopic and biochemical evidence supports apoptosis in cells undergoing secondary cell death and inhibitors of caspases can prevent TBI-induced neuronal death (Raghupathi et al, 2000; Royo et al, 2003).
BcI-2 Family Proteins
Bcl-2 was discovered as a proto-oncogene, which functioned by inhibiting cell death rather than promoting cell proliferation (Vaux et al, 1988). Additional positive and negative regulators of cell death with homology to Bcl-2 followed with identification of Bax and Bcl-xL, respectively (Boise et al, 1993; Oltvai et al, 1993). The Bcl-2 family emerged on the basis of BH domains (Reed, 2006). Antiapoptotic members all share four BH domains and include Bcl-2, Bcl-xL, Bcl-w, and Mcl-1 (Youle and Strasser, 2008). Of these, Bcl-xL appears to be the most highly expressed in adult brain. Bcl-w is also present whereas Mcl-1 and Bcl-2 are not expressed at significant levels in the normal adult brain (Krajewski et al, 1995a; Merry et al, 1994). Bax-like proteins, which include Bak and Bok, possess three BH domains and are critical mediators of mitochondrial dysfunction during apoptosis. Bax is widely expressed in the brain (Krajewski et al, 1995b), while Bok is present only in CA3 neurons (Lein et al, 2004). Fulllength Bak is present only in nonneuronal cells in brain, whereas neurons express an unusual BH3-only splice variant of Bak which has antiapoptotic activity (Sun et al, 2001). The BH3-only proteins, which function to activate Bax/Bak, share only one BH domain in common (Strasser, 2005). Together, the Bcl-2 family of proteins constitute a complex network of molecules that regulate cell death by influencing mitochondrial release of apoptogenic proteins in response to physiologic or pathologic stimuli, with the BH3-only proteins the upstream initiators (Lomonosova and Chinnadurai, 2008).
Bcl-2 Homology Domain 3-only proteins
Origins
Vertebrate BH3-only proteins are related to the Caenorhabditis elegans gene Egl-1, which functions to inhibit the Bcl-2 homolog CED-9 (Conradt and Horvitz, 1998). The first two mammalian BH3-only proteins cloned were Bad (Yang et al, 1995) and Bik (Boyd et al, 1995). At least eight BH3-only proteins have been identified in mice and humans (Strasser, 2005). The BH3-only proteins derive their proapoptotic functions from their BH3 amphipathic helix and possibly other regions within their structure. The BH3 domain is short, comprising a 9 to 16 amino-acid sequence. Otherwise, these proteins bear no resemblance to one another. In fact, only Bid shares any structural similarity with the multidomain family members such as Bcl-2 (Youle and Strasser, 2008). Currently, the mammalian BH3-only protein subfamily includes, in order of published discovery after Bad and Bik; Bid (Wang et al, 1996), Hrk/DP5 (Imaizumi et al, 1997; Inohara et al, 1997), Bim/Bod (Hsu et al, 1998; O'Connor et al, 1998), Noxa (Oda et al, 2000), Puma/Bbc3 (Han et al, 2001; Nakano and Vousden, 2001; Yu et al, 2001), and Bmf (Puthalakath et al, 2001). BNIP3 and its homologs are another recognized BH3-only protein (Chinnadurai et al, 2008), and there are other, less closely related members including Bcl-Rambo (Kataoka et al, 2001) and Mule (Zhong et al, 2005). Several BH3-only protein homologs including Bad and Bid have been found in nonmammalian species; however, Puma, Bik/Blk, and Hrk/DP5 appear to be present only in mammals (Coultas et al, 2002).
Activation/Control
The BH3-only proteins display diverse tissue expression and activation mechanisms. Some, like Puma and Noxa, are under stringent transcriptional control, with minimal expression in healthy cells, including neurons. Several others are readily detectable in normal tissues, including Bad and Bid. For the constitutively expressed members, proapoptotic functions are restrained through phosphorylation, through proteasomal degradation, and by interaction with other molecules (Lomonosova and Chinnadurai, 2008). For example, Bad is bound to the 14-3-3 molecular chaperone protein (Zha et al, 1996). Others share a combination of mechanisms. Bim is sequestered to the cytoskeleton (Puthalakath et al, 1999) and its activity can be potentiated by phosphorylation by the c-Jun N-terminal kinase (JNK) (Putcha et al, 2003). Transcriptional control is also important, with Bim levels regulated by Forkhead box class O (FoxO) 3a (Dijkers et al, 2000), c-Jun downstream of JNK (Harris and Johnson, 2001; Whitfield et al, 2001), and C/EBP homologous protein CHOP/GADD153 (Puthalakath et al, 2007).
Downstream from Bcl-2 Homology Domain 3-only Proteins
Ultimately, interactions between Bcl-2 family proteins result in Bax/Bak oligomerization, conformational change, and insertion into the outer mitochondrial membrane, which triggers mitochondrial outer membrane permeabilization (MOMP) by a mechanism that is not fully understood (Chipuk et al, 2010). The importance of this Bax/Bak checkpoint is underscored by findings from mice doubly deficient for bax and bak, which display defects in developmental cell death and cellular resistance to most forms of stress-induced apoptosis (Lindsten et al, 2000; Wei et al, 2001). However, on the basis of brain expression of Bax and Bak (Krajewska et al, 2002; Krajewski et al, 1996; Sun et al, 2001) and data from mice singly deficient for Bax or Bak (Fannjiang et al, 2003; Lindsten et al, 2000; White et al, 1998; Xiang et al, 1998), only Bax appears to be required for neuronal death under most conditions. Following MOMP, there is release of intramitochondrial proteins such as cytochrome c, which binds the apoptotic protease activating factor 1. Apoptotic protease activating factor 1 recruits caspase-9 to a large complex that processes caspase-3, resulting in cell death by cleavage of various intracellular proteins. Other apoptogenic molecules are released from mitochondria including apoptosis-inducing factor (Susin et al, 1999), which appears to mediate nuclear pyknosis in neurons (Cheung et al, 2005; Landshamer et al, 2008).
Mechanism of Action and Relative Potency of Bcl-2 Homology Domain 3-only Proteins
The BH3-only proteins are critical for Bax/Bak-dependent MOMP and apoptosis. Indeed, Ren et al, (2010) recently generated triple knockout mice lacking BH3-only proteins Bid, Bim, and Puma, which were highly resistant to various apoptotic stimuli, including in neuronal injury models. Two competing theories have been proposed to explain their proapoptotic effects. In the first, so-called sensitizer/derepressor model, BH3-only proteins function by binding to and inhibiting antiapoptotic Bcl-2 family proteins via interaction of the BH3 domain with a cleft formed from BH1-3. That is, activation of BH3-only proteins inhibits the protective functions of Bcl-2-like antiapoptotic proteins enabling Bax/Bak-mediated MOMP (Youle and Strasser, 2008). There is, however, specificity in these interactions with certain BH3-only proteins capable of interacting with only a subset of anti-apoptotic proteins, whereas others avidly bind all. This gives rise to a hierarchy based on potency. Those with limited targeting are deemed ‘weak’ BH3-only proteins, and this includes Bad. Those that bind all, for example Bid, Bim, and Puma, are termed ‘potent’. Weak BH3-only proteins appear to require others (e.g., Bim) in order to execute apoptosis (Kim et al, 2006). In the second model, a subset of BH3-only proteins that includes Bid, Bim, and possibly Puma, directly bind and activate Bax/Bak (Chipuk et al, 2010). Further evidence in support of this model comes from recent binding experiments (Kim et al, 2009) and the finding that Bax does not undergo oligomerization in the absence of Bid, Bim, and Puma (Ren et al, 2010). However, some data are at odds with the direct activator model (Jabbour et al, 2009), and Bcl-2 mutants that bind BH3-only proteins but not Bax/Bak can still prevent apoptosis (Cheng et al, 1996; Youle and Strasser, 2008). A further mechanism may be to convert, on binding, antiapoptotic Bcl-2 family proteins into membrane-inserted versions that have a Bax/Bak-like effect on MOMP (Youle and Strasser, 2008). More likely, BH3-only protein-mediated membrane insertion of antiapoptotic proteins simply serves to inactive their antiapoptotic functions (Wilson-Annan et al, 2003). Figure 1 summarizes some of the main structural features of BH3-only proteins Bid, Bim, and Puma.
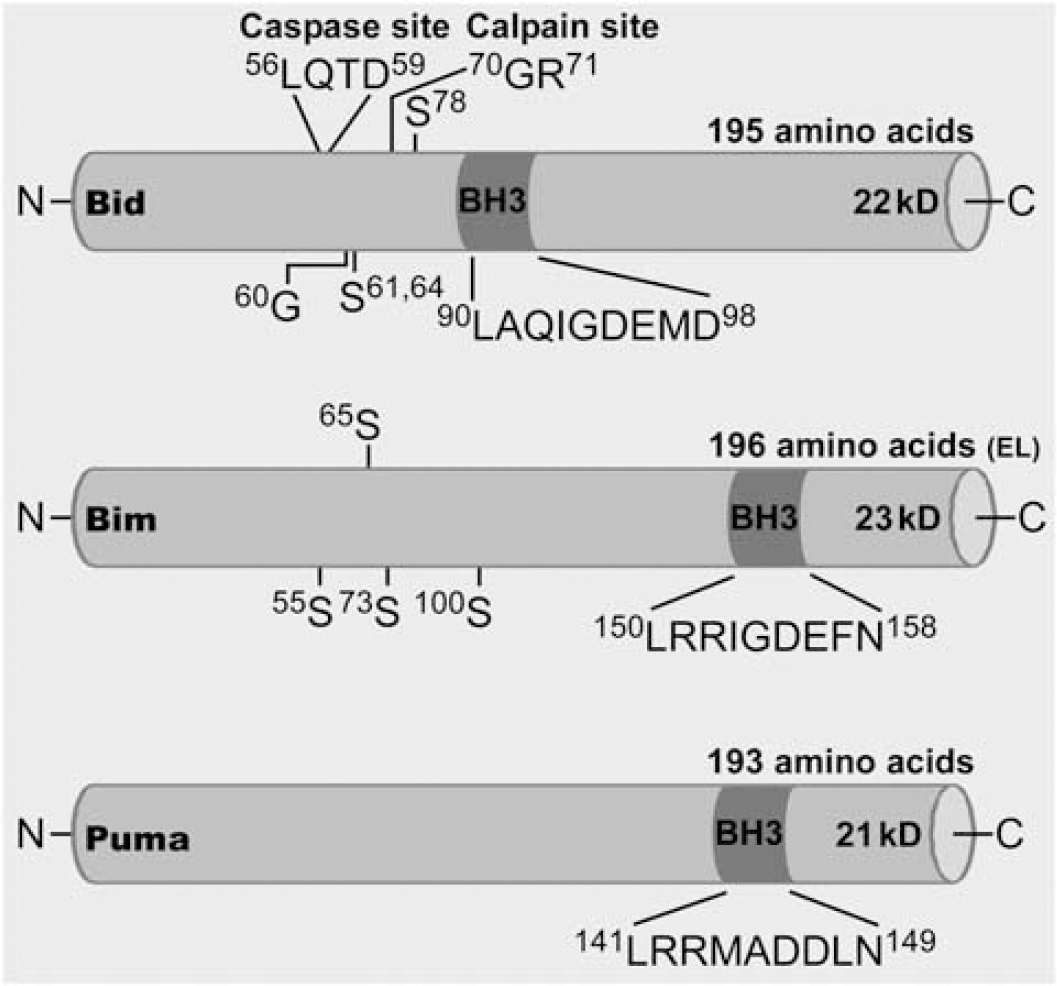
Molecular structures of Bid, Bim, and Puma. Cartoon showing mus musculus structural features of each Bcl-2 homology domain 3 (BH3)-only protein (Sources: NCBI and uniprot.org and the cited literature in this review). Included are sequence length, estimated molecular weights, and annotations for some of the proposed phosphorylation sites (S, serine residues), and for Bid, posttranslational modification by cleavage and N-myristoylation (G, glycine residue).
Linking Bcl-2 Homology Domain 3-only Proteins to Perturbed Neuronal Physiology After Seizures, Ischemia, and Trauma
What might link disturbances in cell physiology after neurologic injury to BH3-only protein activation? Calcium-dependent signaling pathways may be directly responsible. Bad activation is via dephosphorylation that may be mediated by the calcium-dependent phosphatase calcineurin (Wang et al, 1999). Calcineurin and the phosphatase and tensin homolog PTEN phosphatase have also been linked to control of Bim via regulating FoxO1/3a and Akt activity (Chang et al, 2007; Fukunaga and Shioda, 2009; Gary and Mattson, 2002; Li et al, 2009). Raised intracellular calcium levels could also be responsible for activating Bid since the calcium-dependent protease calpain has been shown to cleave and activate Bid (Chen et al, 2001), as well as other Bcl-2 family proteins. Indeed, while some studies have implied caspase-8 is involved in Bid activation after seizures and ischemia, reviewed below, whether sufficient levels of key components of the caspase pathway are present in adult brain remains controversial (Hu et al, 2000; Yakovlev et al, 2001). Translocation of full-length Bid to mitochondria may also be sufficient to cause neuronal death, although the mechanism is not fully understood (Konig et al, 2007).
Calcium influx in nonneuronal cells (Bouillet et al, 1999) and neuronal cells (Concannon et al, 2010) can induce Bim-dependent apoptosis. The energy sensor AMP kinase is activated when cellular adenosine triphosphate is depleted and this has been reported in models of calcium-dependent excitotoxicity (Lee et al, 2009; Weisova et al, 2009). AMP kinase was recently shown to mediate glutamate-induced neuronal death via Bim (Concannon et al, 2010). Also associated with perturbed calcium, oxidative stress, and excitotoxicity, JNK and FoxO3a are capable of upregulating Bim and Puma (Dijkers et al, 2000; Harris and Johnson, 2001; Whitfield et al, 2001; You et al, 2006). A direct effect of calcium influx on Puma induction is unlikely (Concannon et al, 2007; Villunger et al, 2003).
Acute neurologic insults are known to cause protein misfolding and endoplasmic reticulum stress, as well as proteasomal inhibition (Paschen, 2003b; Rao et al, 2004). Endoplasmic reticulum stress can result in Puma induction (Reimertz et al, 2003), and recent work suggests that this is mediated by activating transcription factor 4 and CHOP (Galehdar et al, 2010). N-methyl-
Finally, DNA damage is a potential trigger for BH3-only proteins in these models, in particular Puma but also Bid, which may be p53 mediated (Akhtar et al, 2006; Nakano and Vousden, 2001; Sax et al, 2002; Villunger et al, 2003; Wyttenbach and Tolkovsky, 2006). DNA damage does not appear to be an important activator of Bim-induced cell death (Bouillet et al, 1999; O'Connor et al, 1998). The schematic in Figure 2 summarizes some of the known molecular pathways that may be responsible for the activation of Bid, Bim, and Puma as they relate to the discussed neurologic injury models.
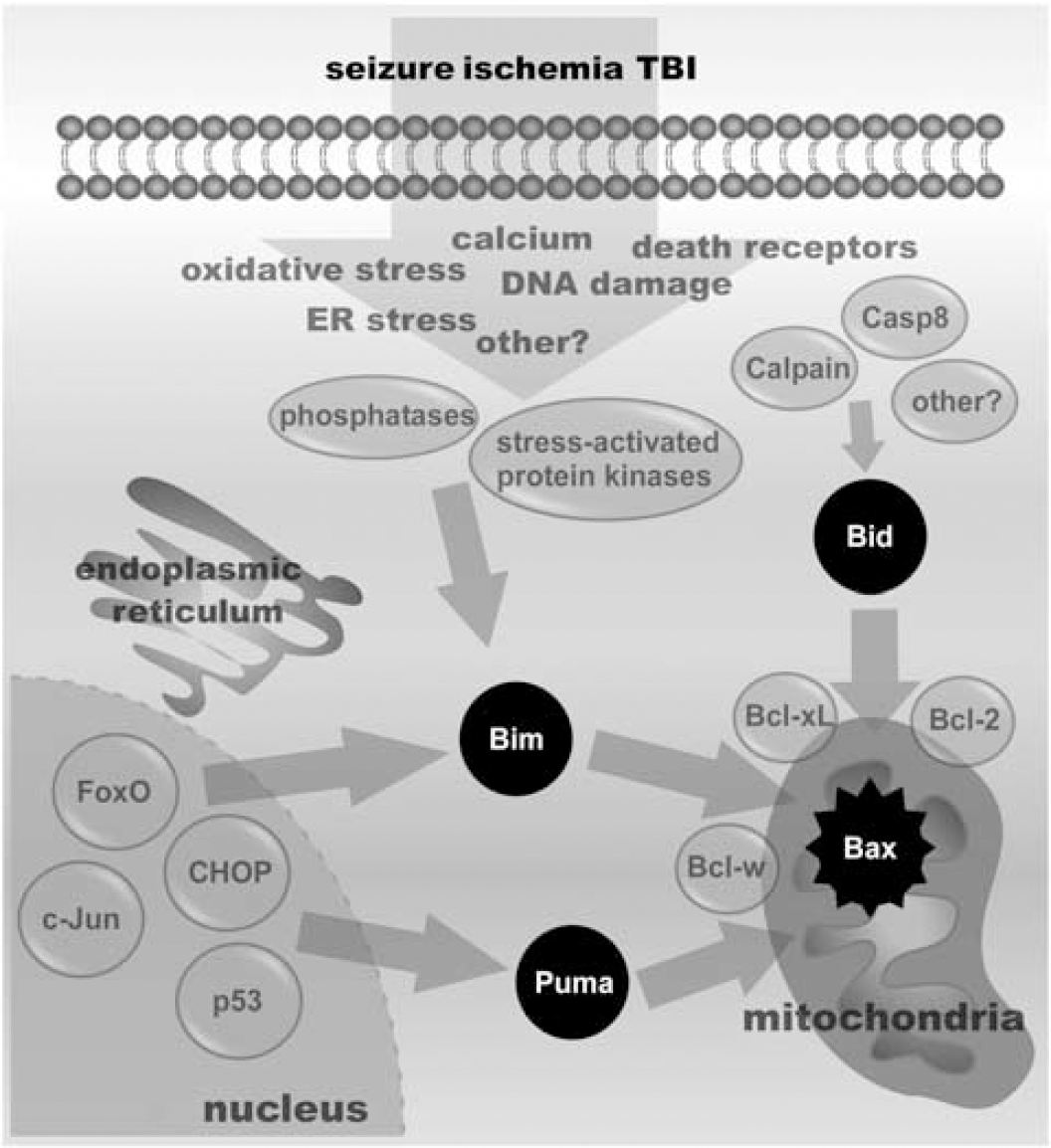
Schematic of the possible molecular pathways leading to BcI-2 homology domain 3 (BH3)-only protein activation. Cartoon showing some of the various factors triggered by seizures, ischemia, and traumatic brain injury (TBI) in neurons and how these may result in activation of Bid, Bim, and Puma. These include phosphatases such as PTEN and calcineurin and stress-activated protein kinases such as c-Jun N-terminal kinase (JNK), which can phosphorylate or upregulate Bim, proteases that can cleave and activate Bid, and various transcription factors that can upregulate Bim and/or Puma. Casp8, caspase-8; ER, endoplasmic reticulum.
BcI-2 Homology Domain 3-only Proteins in Status Epilepticus, Ischemia and Traumatic Brain Injury
Introduction
There is strong evidence that multi-BH domain proapoptotic Bax and several of the antiapoptotic Bcl-2 family proteins influence neuronal death after seizures, ischemia, and TBI. This includes in vivo studies in which these genes are knocked out or overexpressed (Cao et al, 2002; Gibson et al, 2001; Ju et al, 2008; Martinou et al, 1994; Murphy et al, 2007; Raghupathi et al, 1998; Sun et al, 2003; Tehranian et al, 2008; Xiang et al, 1998). What is the evidence for BH3-only protein activation in these models and what is the relative importance of individual members? The data come from biochemical studies on their expression, interaction (coimmunoprecipitation), and translocation events, and from studies in mice deficient in one or other BH3-only protein. Table 1 summarizes the findings on Bid, Bim, and Puma for which knockout mouse data are available in status epilepticus, ischemia, and TBI. Description of the non-central nervous system phenotypes of mice deficient in BH3-only proteins can be found elsewhere (Roset et al, 2007; Strasser, 2005; Youle and Strasser, 2008). Figure 3 summarizes the contributions of the three BH3-only proteins to neuronal death in each model.
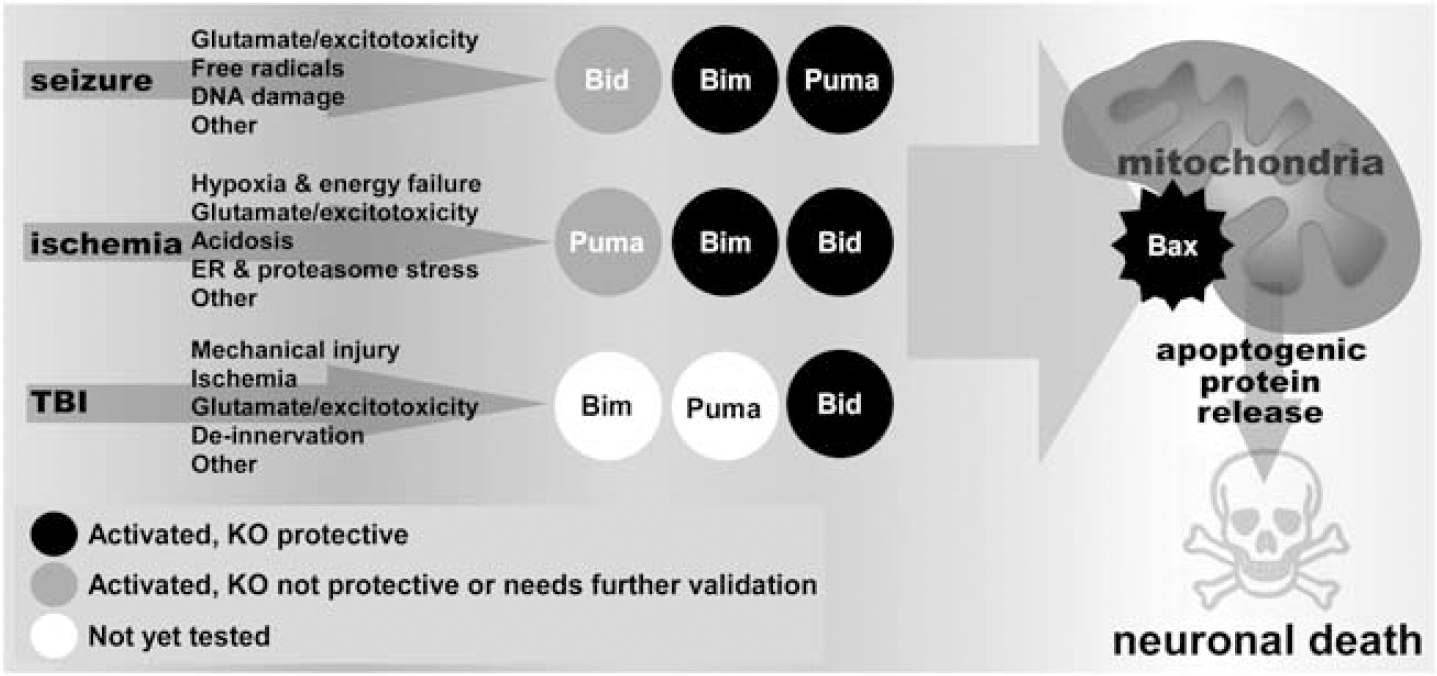
Cartoon representation of the importance of the three potently proapoptotic BcI-2 homology domain 3 (BH3)-only proteins in each model. Bid, Bim, and Puma are activated in ischemia and seizure, although for traumatic brain injury (TBI) there are data only for Bid. Black circle denotes evidence for activation and that the gene-deficient animal (KO) is protected against neuronal death. Grey circle denotes evidence for the BH3-only protein is activated but the knockout mouse was not protected or where further validation will be needed. White circle denotes no evidence yet for activation of the BH3-only protein. ER, endoplasmic reticulum.
Summary of findings on BH3-only mice in seizure, ischemia, and trauma
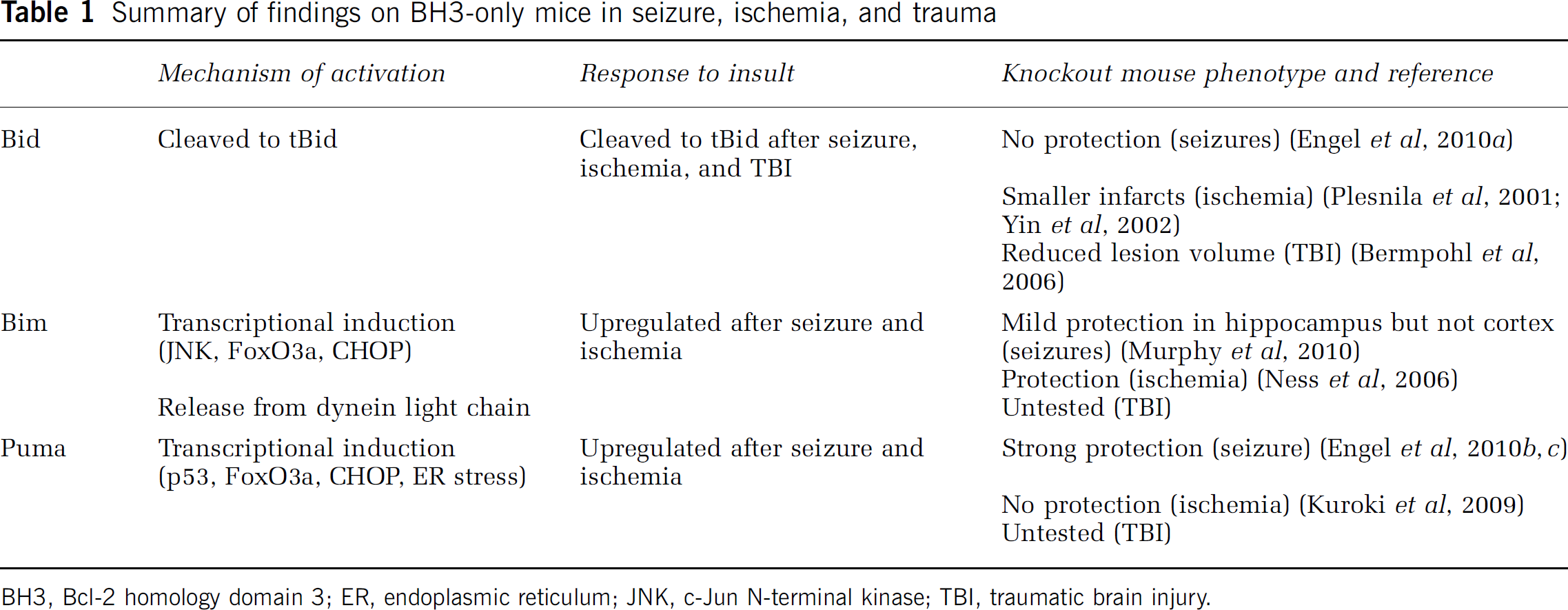
BH3, BcI-2 homology domain 3; ER, endoplasmic reticulum; JNK, c-Jun N-terminal kinase; TBI, traumatic brain injury.
Bid
Bid is a member of the potently proapoptotic class of BH3-only protein. Bid is unusual in that it is expressed in many tissues without an obligatory inhibitory chaperone. In fact, Bid displays little proapoptotic activity until cleaved to a truncated form, termed tBid (Li et al, 1998). Cleavage of Bid can be mediated via caspase-8 and thus serves to link death receptor signaling to the mitochondrial pathway. However, other cell death proteases can cleave Bid, including caspase-2 and calpain (Chen et al, 2001; Upton et al, 2008). Bid is widely expressed in adult brain, including the hippocampus (Krajewska et al, 2002).
Bid Activation and Knockout Phenotype in Status Epilepticus
Bid was the first BH3-only protein to be studied in a model of status epilepticus. Seizures in rats resulted in the rapid appearance of the p15 form (tBid) in the damaged hippocampus (Henshall et al, 2001). Other reports have shown tBid formation after seizures in rats (Li et al, 2006; Wang et al, 2008) and in mice (Engel et al, 2010a). Fractionation analysis has detected both full-length and cleaved Bid in the mitochondrial compartment after seizures (Engel et al, 2010a; Schindler et al, 2004). Pharmacologic studies have supported both caspase-8 and calpain involvement in Bid cleavage after status epilepticus in rats (Henshall et al, 2001; Li et al, 2006; Wang et al, 2008). Calpain has also been proposed to mediate Bid cleavage after seizures in mice (Takano et al, 2005).
Bid-deficient mice were the first BH3-only protein knockouts to be examined for in vivo neurologic injury phenotypes. Bid−/- mice were found to be normal with regard to brain development (Leonard et al, 2001; Plesnila et al, 2001; Yin et al, 1999, 2002). The impact of Bid deficiency on seizure damage was investigated recently, in genetically modified C57BL/6 mice lacking bid, which were developed by Strasser's group (Kaufmann et al, 2007). Bid−/- mice express normal hippocampal levels of various genes including KA receptor subunits and undergo equivalent status epilepticus as wild-type controls (Engel et al, 2010a). Bid-deficient mice were not, however, protected against status epilepticus, and showed similar cell death to wild-type animals in both dorsal and ventral hippocampus (Engel et al, 2010a). This was surprising since glutamate exposure of neuronal cells in vitro, a model of seizure-like injury, was reduced by pharmacologic Bid inhibition or gene silencing (Landshamer et al, 2008). However, Bid-deficient neurons are not resistant to N-methyl-
Bid Activation and Knockout Phenotype in Ischemia and Trauma
Bid cleavage is triggered by both cerebral ischemia (Plesnila et al, 2001; Yin et al, 2002; Zhang et al, 2003) and TBI (Bermpohl et al, 2006; Franz et al, 2002). Caspase-8 has been suggested as a possible cause of Bid cleavage after ischemia (Plesnila et al, 2001) but no direct in vivo evidence has been presented. Plesnila et al, (2001) was the first to report the effects of cerebral ischemia in bid−/- mice, finding that infarct volume after middle cerebral artery occlusion was reduced by 67%. A subsequent paper found a somewhat smaller degree of protection in bid−/- mice (30% reduction) but nevertheless, this supported a causal role for Bid in ischemic infarction in vivo (Yin et al, 2002). One explanation for the differing scale of protection between these studies may have been the use of a milder ischemic insult in the Plesnila study, which could be associated with a greater apoptotic ‘contribution’ (Yin et al, 2002).
Bermpohl et al, (2006) described the histologic and neurologic consequences of TBI in bid−/- mice. Mice subject to controlled cortical impact showed smaller lesion volumes in Bid-deficient animals at 12 days, although this protection was no longer evident at 30-and 40-day follow-up (Bermpohl et al, 2006). Functional deficits were also not mitigated in the bid−/- mice (Bermpohl et al, 2006). Thus, while Bid is activated by each neurologic injury, a different functional contribution to the cell death process is found between the three models.
Bim
Like Bid, Bim is a member of the more potent class of BH3-only protein owing to its ability to bind all antiapoptotic Bcl-2 family proteins and, probably, directly activate Bax/Bak (Chipuk et al, 2010). Alternative splicing gives rise to three main isoforms termed short (S), long (L), and extra-long (EL), with the short form being most potent (O'Connor et al, 1998). BimEL is constitutively expressed in neurons in the mouse brain, including hippocampal neurons, and is not expressed in glia (O'Reilly et al, 2000). The proapoptotic functions of Bim are triggered when its expression is increased or it is released from sequestration by dynein LC8 light chain (Puthalakath et al, 1999). Bim is also posttranslationally regulated by phosphorylation, which can result in activation (Putcha et al, 2003) or targeting to the proteasome for degradation (Meller et al, 2006).
Bim Activation and Knockout Phenotype in Status Epilepticus
Prolonged seizures evoked by intraamygdala KA upregulate Bim protein levels in the hippocampus of rats (Shinoda et al, 2004) and mice (Murphy et al, 2010). The upregulation of Bim is JNK dependent in mice since it is blocked by SP600125 (Murphy et al, 2010), although FoxO1 or 3a may also be important (Murphy et al, 2010; Shinoda et al, 2004). Bim is also upregulated after pilocarpine-induced seizures (Yang et al, 2007), but Bim levels were reported to decline after seizures caused by intraventricular KA (Korhonen et al, 2003). Also, Bim levels did not increase in the neocortex of mice after seizures, despite the presence of cell death, indicting regional importance of this BH3-only protein (Murphy et al, 2010).
Bimr−/- mice display normal brain development and are equally sensitive to the convulsive actions of KA as wild-type animals (Murphy et al, 2010). A causal role was found for Bim in seizure-induced neuronal death in vivo in the intraamygdala KA model (Murphy et al, 2010). Analysis of hippocampal damage 3 days after status epilepticus revealed a modest but nevertheless significant reduction in neuronal death in bimr−/- mice compared with wild-type animals. Hippocampal neurons are also protected in in vitro models of KA- and N-methyl-
Bim in Ischemia and Trauma
Bim is upregulated quickly after focal cerebral ischemia, compatible with a contributory role in mitochondrial release of cytochrome c (Gao et al, 2005; Okuno et al, 2004; Shibata et al, 2002). It does not appear to be induced after global cerebral ischemia (Sanderson et al, 2009). Almost no data are available on Bim in TBI. Bim was reported to be increased in human head injury (Minambres et al, 2008) while it was not changed in experimental traumatic spinal cord injury (Yin et al, 2005).
Only one study has been published to date that examined the effects of ischemia in Bim-deficient mice (Ness et al, 2006). This study found that hippocampal damage was strongly reduced in bimr−/- mice subjected to neonatal hypoxia/ischemia (Ness et al, 2006). There has not yet been a report describing TBI outcome in mice lacking bim.
Puma
Puma was discovered as a p53-induced BH3-only protein but can also be induced in a p53-independent manner (Jeffers et al, 2003; You et al, 2006). Puma is potently proapoptotic, avidly binding all antiapoptotic Bcl-2 family proteins and Puma may also be capable of directly activating Bax/Bak (Jabbour et al, 2009). Puma does not appear to be expressed in normal adult brain.
Puma Activation and Knockout Phenotype in Status Epilepticus
Puma is strongly upregulated following status epilepticus in mice in both whole-cell lysates and mitochondrial fractions, peaking at 8 hours (Engel et al, 2010c). Induction of Puma after status epilepticus appears to be p53 dependent because Puma is not induced after seizures in p53−/- mice or in mice pretreated with p53 inhibitor pifithrin-α (Engel et al, 2010c). Puma−/- mice were found to be potently protected against seizure-induced neuronal death in vivo, displaying reduced DNA damage, neuronal death, and increased surviving NeuN-positive cells compared with puma+/- mice and wild-type animals (Engel et al, 2010c). Puma−/- mice also displayed protection when subjected to more prolonged status epilepticus, indicating that Puma may still be important in strongly necrotic insults (Engel et al, 2010b). Neuroprotection in puma−/- mice is also long lasting and evident at least 2 weeks after the initial status epilepticus (Engel et al, 2010c). This paper also examined the impact of Puma deficiency on the frequency of spontaneous seizures in the model, which develop within a few days of status epilepticus. Continuous EEG recordings of puma+/- and puma−/- mice showed the Puma-deficient animals had > 60% fewer epileptic seizures (Engel et al, 2010c). Thus, Puma seems to be an important regulator of seizure-induced neuronal death in vivo and is capable of influencing the development of epilepsy.
Puma Activation and Knockout Phenotype in Ischemia and Trauma
The first report of Puma in an in vivo neurologic injury model was actually following global ischemia in rats, where it was upregulated (Reimertz et al, 2003). Other laboratories have confirmed Puma is induced after global cerebral ischemia (Niizuma et al, 2008) and following focal cerebral ischemia (Kuroki et al, 2009; Luo et al, 2009). Pifithrin-α blocks upregulation of Puma in these models, supporting p53 involvement (Luo et al, 2009; Niizuma et al, 2008). Whether Puma is required for neuronal death after cerebral ischemia is uncertain. The induction of Puma 4 hours after global cerebral ischemia seems at odds with the long delay before the appearance at 72 hours of CA1 neuronal death (Niizuma et al, 2008). Puma-deficient mice are not protected against cell death caused by N-methyl-
Other BcI-2 Homology Domain 3-only Proteins
Bad
Bad only strongly interacts with antiapoptotic Bcl-xL (Yang et al, 1995); binding to Bcl-2 is weaker and Bad does not bind other Bcl-2 family proteins including proapoptotic Bax/Bak (Yang et al, 1995). Indeed, Bad cannot induce apoptosis in cells lacking the potent subfamily BH3-only proteins Bid, Bim, and Puma (Ren et al, 2010). When phosphorylated, Bad resides complexed to 14-3-3 in the cytoplasm. Dephosphorylation releases Bad from 14-3-3 whereupon it displaces Bax by binding to Bcl-xL (Datta et al, 1997; Kelekar et al, 1997; Zha et al, 1996).
A constitutively active Bad transgenic mouse in which serine phosphorylation sites were mutated was reported to have a reduced apoptotic threshold for certain death signals, but the mice were viable (Datta et al, 2002). Likewise, bad−/- mice grow to adulthood and display normal organ development (Ranger et al, 2003). Some defects are present in these mice, including in the testis, slight resistance of thymocytes to irradiation, and earlier death due to malignancy (Ranger et al, 2003). However, other groups have claimed not to detect some of these Bad-deficient mouse phenotypes (Youle and Strasser, 2008).
Because of its early discovery, the response of Bad after each of the three neurologic insults has been well covered. Bad is activated following status epilepticus in rats and mice, as evidenced by its dissociation from 14-3-3 and increased binding to Bcl-xL in hippocampus (Henshall et al, 2002; Li et al, 2005; Noh et al, 2006). Bad-deficient mice have not been evaluated for their response to status epilepticus. However, FK506, a known inhibitor of this pathway, reduces hippocampal damage after seizures in vivo (Henshall et al, 2002). Both focal cerebral ischemia and TBI also activate Bad, with dephosphorylation, dissociation from 14-3-3, translocation to mitochondria, and binding to Bcl-xL all reported (Kamada et al, 2007; Saito et al, 2003; Yao et al, 2005). Again, a causal role remains uncertain as no study has yet investigated ischemia or TBI in mice deficient or overexpressing Bad.
Hrk/DP5, Noxa, BNIP3, Noxa, and Bmf
Knockout mice data are not available for the remaining BH3-only proteins in status epilepticus, ischemia, or TBI but on the basis of expressional responses major roles for the remainder seem unlikely. BNIP3 may increase in vulnerable neurons after cerebral ischemia (Schmidt-Kastner et al, 2004) but appears with a substantial delay relative to induction of the mitochondrial pathway after focal cerebral ischemia (Althaus et al, 2006). Hrk/DP5 is upregulated after cerebral ischemia in rats and mice, although with a substantial delay relative to induction of the mitochondrial pathway (Gao et al, 2005; Guan et al, 2006). Hrk/DP5 expression appears to decline in hippocampus after seizures (Jimenez-Mateos et al, 2008; Korhonen et al, 2003). A role for Noxa has been suggested in cerebral ischemia (Inta et al, 2006) and motor neuron death (Kiryu-Seo et al, 2005), but has been ruled out in seizure-induced neuronal death (Engel et al, 2010c). A BH3-only protein currently awaiting investigation in any of the three models is Bmf (Bcl-2 modifying factor). It will be interesting to see if any of the remaining BH3-only proteins have causal roles in cell death after acute neurologic injuries.
Mechanistic Basis for Differing Causal Roles of BcI-2 Homology Domain 3-only Proteins in Neurologic Injury Models
The available data show that all three potently proapoptotic BH3-only proteins are activated early during neurologic injury after seizures and ischemia. Bid is also induced after TBI but we await firm data on Bim and examination of Puma. Status epilepticus is the only insult for which all three knockout mice have been evaluated in a single model. Until an equivalent body of work is completed in ischemia and TBI, we cannot properly judge the importance or lack thereof of Bim and Puma in those models. However, it seems likely that each neurologic injury has a common component that broadly induces BH3-only proteins but distinct pathophysiology drives which of the three impacts most on the cell death process. Several factors might underlie this apparent insult-specific tailoring of the BH3-only contribution. (1) The ischemic/hypoxic component may introduce different molecular players less prominent after status epilepticus such as AMPK, hypoxia-inducible factor 1α, and proteasomal stress. (2) A difference in the degree to which KA versus N-methyl-
Caveats Associated with the Use of Knockout Mice
Studies to date have exclusively relied on constitutive knockout (null mutant) mice to elucidate the contribution of BH3-only proteins to neuronal death after seizures, ischemia, and TBI. There are inherent limitations with the use of such animals. These include possible compensatory responses of other genes and the likelihood that complete deficiency of the gene has unexpected effects on brain development or function (Gaveriaux-Ruff and Kieffer, 2007). Very few studies have checked for evidence of compensation among BH3-only proteins and efforts to assuage concerns on the second matter have been restricted to checking cerebrovascular anatomy, blood flow monitoring, and EEG analysis. Some of these experimental concerns can be overcome by employing tissue-specific and inducible knockouts or other methods for gene modulation (Gaveriaux-Ruff and Kieffer, 2007).
Future Challenges
To date only a small number of BH3-only protein-deficient mice have been fully assessed in all three neurologic injury models. A comprehensive analysis requires assessment of neuronal death after focal cerebral ischemia in bim−/- mice and TBI in pumar−/- and bim−/- animals. To properly assess the roles of BH3-only may also require analysis of double- or triple-deficient mice. Bim/Bik and Bim/Puma knockout mice have been generated (Coultas et al, 2005; Erlacher et al, 2006), and now Bid/Bim/Puma triple knockout mice (Ren et al, 2010). Central nervous system-specific knockouts would also offer further advantages. Evaluation of such models may complete our understanding. Whether research on BH3-only proteins can translate into therapeutics for the discussed neurologic disorders is uncertain. The BH3 mimetics are becoming drugs of interest for cancer treatment. Small molecular inhibitors of BH3-only proteins may also be achievable. Indeed, targeting the BH3-only proteins has significant advantages over their upstream transcription factors. For example, loss of p53 has deleterious effects in both ischemia and status epilepticus models (Engel et al, 2010d; Maeda et al, 2001), presumably due to non-cell death functions. In conclusion, BH3-only proteins comprise a novel class of molecule underlying the pathophysiology of status epilepticus, ischemia, and TBI, which might in future be targeted to yield potent neuroprotection.
Footnotes
Acknowledgements
The authors thank the Science Foundation Ireland (08/IN1/B1875, 08/IN1/B1949, 08/RFP/1745, 04/IN3/ B466), the Health Research Board (RP/2005/24, RP/ 2007/37), and the Irish Research Council for Science Engineering and Technology for support.
The authors declare no conflict of interest.